
Protease-Resistant Peptides for Targeting and Intracellular Delivery of Therapeutics


Abstract
:1. Introduction
2. Peptide Resistance to Proteases May Be Enhanced through a Variety of Chemical Modifications
2.1. N- and C-Termini
2.2. Backbone
2.3. Side Chains
2.4. Cyclization
2.5. Conjugation
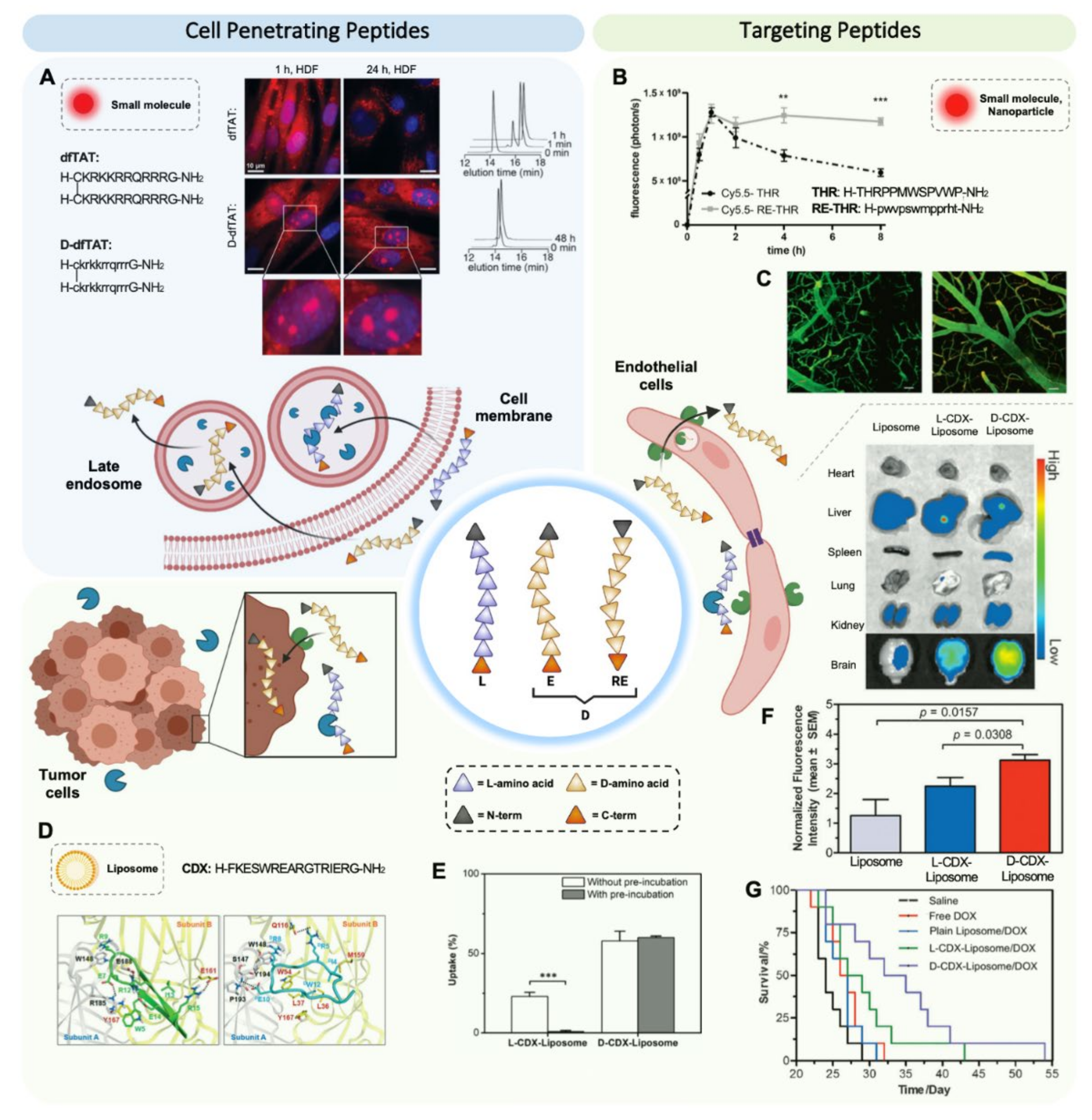
3. Enantio and Retro-Enantio Isomerizations Enhance Targeting Efficiency by Increasing Resistance against Proteolytic Degradation
3.1. Enantio/Retro-Enantio Cell Penetrating Peptides
3.2. Enantio/Retro-Enantio Targeting Peptides
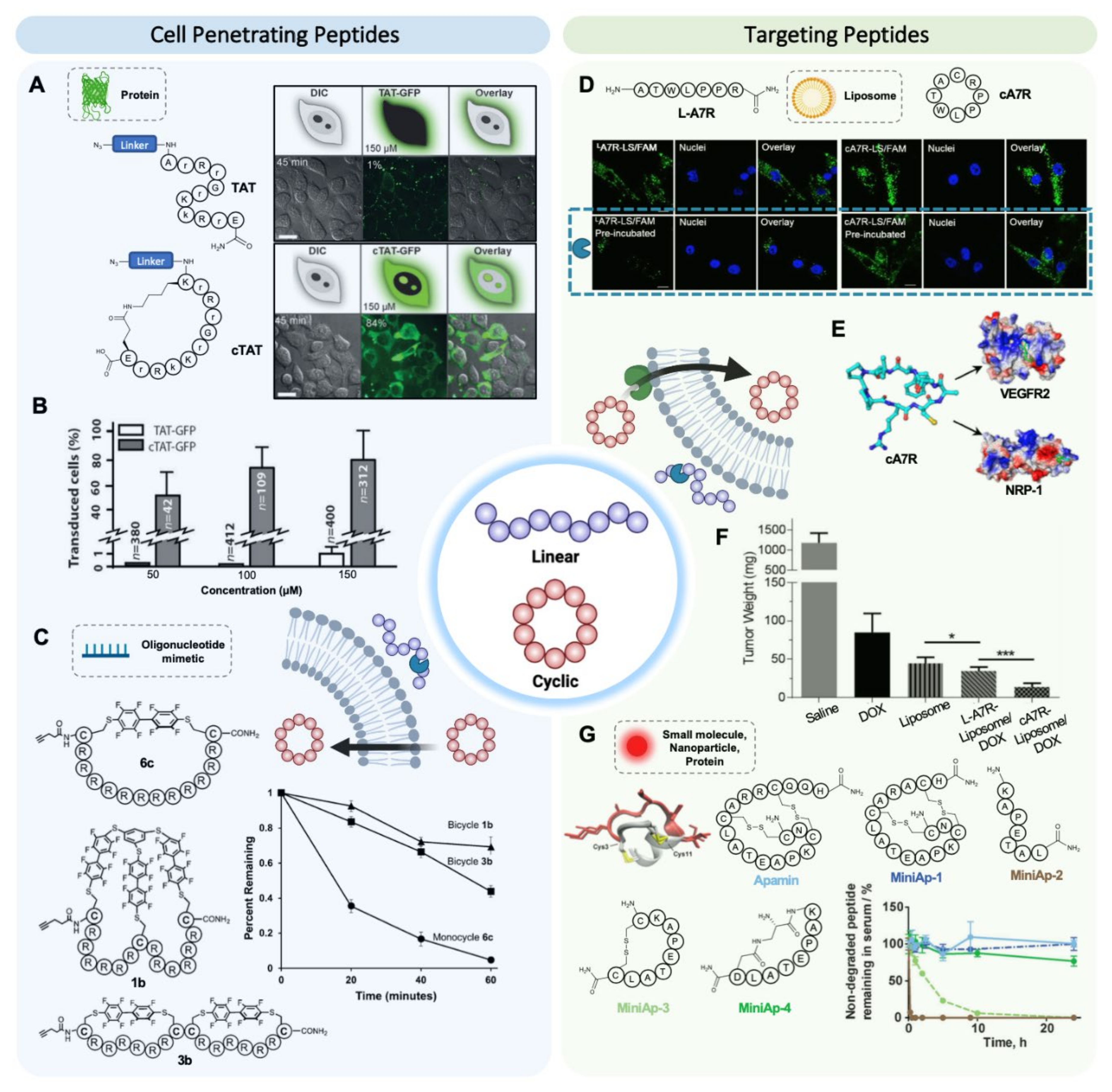
4. Cyclization May Enhance Targeting Efficiency Both by Providing High Protease Resistance and High Binding Affinity
4.1. Cyclic Cell Penetrating Peptides
4.2. Cyclic Targeting Peptides
5. Conclusions and Outlook
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| BBB | Blood–brain barrier |
| CPP | Cell penetrating peptide |
| DOX | Doxorubicin |
| EGFR | Epidermal growth factor receptor |
| EGFRvIII | Epidermal growth factor receptor mutation variant III |
| FDA | Food and Drug Administration |
| FTC | 2’,3’-dideoxy-5-fluoro-3’-thiacytidine (Emtricitabine) |
| GFP | Green Fluorescent Protein |
| HIV-1 | Human immunodeficiency virus type I |
| HPLC | High-performance liquid chromatography |
| LS | Liposomes |
| MMAE | Monomethyl auristatin E |
| nAchRs | Nicotine acetylcholine receptors |
| NRP-1 | Neuropilin-1 receptor |
| PEG | Polyethylene glycol |
| PMO | Phosphorodiamidate morpholino oligonucleotide |
| QD | Quantum dot |
| RCM | Ring-closing metathesis |
| RE | Retro-enantio |
| siRNA | Small interfering RNA |
| TAM | Tumor-associated macrophages |
| TfR1 | Transferrin receptor |
| VEGFR2 | Vascular endothelial growth factor receptor 2 |
References
- Davenport, A.P.; Scully, C.C.G.; de Graaf, C.; Brown, A.J.H.; Maguire, J.J. Advances in therapeutic peptides targeting G protein-coupled receptors. Nat. Rev. Drug Discov. 2020, 19, 389–413. [Google Scholar] [CrossRef]
- Brayden, D.J.; Hill, T.A.; Fairlie, D.P.; Maher, S.; Mrsny, R.J. Systemic delivery of peptides by the oral route: Formulation and medicinal chemistry approaches. Adv. Drug Deliv. Rev. 2020, 157, 2–36. [Google Scholar] [CrossRef] [PubMed]
- Mathur, D.; Prakash, S.; Anand, P.; Kaur, H.; Agrawal, P.; Mehta, A.; Kumar, R.; Singh, S.; Raghava, G.P.S. PEPlife: A Repository of the Half-life of Peptides. Sci. Rep. 2016, 6, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Dissanayake, S.; Denny, W.A.; Gamage, S.; Sarojini, V. Recent developments in anticancer drug delivery using cell penetrating and tumor targeting peptides. J. Control. Release 2017, 250, 62–76. [Google Scholar] [CrossRef] [PubMed]
- Khatri, B.; Nuthakki, V.R.; Chatterjee, J. Strategies to Enhance Metabolic Stabilities. Methods Mol. Biol. 2019, 2001, 17–40. [Google Scholar] [CrossRef] [PubMed]
- Cavaco, M.; Andreu, D.; Castanho, M.A.R.B. The Challenge of Peptide Proteolytic Stability Studies: Scarce Data, Difficult Readability, and the Need for Harmonization. Angew. Chemie Int. Ed. 2021, 60, 1686–1688. [Google Scholar] [CrossRef]
- Pelay-gimeno, M.; Glas, A.; Koch, O.; Grossmann, T.N. Structure-Based Design of Inhibitors of Protein—Protein Interactions: Mimicking Peptide Binding Epitopes. Angewandte 2015, 54, 8896–8927. [Google Scholar] [CrossRef]
- Di, L. Strategic Approaches to Optimizing Peptide ADME Properties. AAPS J. 2015, 17, 134–143. [Google Scholar] [CrossRef]
- Lenci, E.; Trabocchi, A. Peptidomimetic toolbox for drug discovery. Chem. Soc. Rev. 2020, 49, 3262–3277. [Google Scholar] [CrossRef] [PubMed]
- Avan, I.; Hall, C.D.; Katritzky, A.R. Peptidomimetics via modifications of amino acids and peptide bonds. Chem. Soc. Rev. 2014, 43, 3575. [Google Scholar] [CrossRef]
- Chatterjee, J.; Rechenmacher, F.; Kessler, H. N -Methylation of Peptides and Proteins: An Important Element for Modulating Biological Functions. Angew. Chemie Int. Ed. 2013, 52, 254–269. [Google Scholar] [CrossRef]
- Niehaus, E.-M.; Janevska, S.; von Bargen, K.W.; Sieber, C.M.K.; Harrer, H.; Humpf, H.-U.; Tudzynski, B. Apicidin F: Characterization and Genetic Manipulation of a New Secondary Metabolite Gene Cluster in the Rice Pathogen Fusarium fujikuroi. PLoS ONE 2014, 9, e103336. [Google Scholar] [CrossRef] [Green Version]
- Laird, D.W.; LaBarbera, D.V.; Feng, X.; Bugni, T.S.; Harper, M.K.; Ireland, C.M. Halogenated cyclic peptides isolated from the sponge Corticium sp. J. Nat. Prod. 2007, 70, 741–746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, H.; Kumar, K. Antimicrobial activity and protease stability of peptides containing fluorinated amino acids. J. Am. Chem. Soc. 2007, 129, 15615–15622. [Google Scholar] [CrossRef]
- Wei, X.; Zhan, C.; Shen, Q.; Fu, W.; Xie, C.; Gao, J.; Peng, C.; Zheng, P.; Lu, W. A D-peptide ligand of nicotine acetylcholine receptors for brain-targeted drug delivery. Angew. Chemie Int. Ed. 2015, 54, 3023–3027. [Google Scholar] [CrossRef]
- Li, Z.; Xie, J.; Peng, S.; Liu, S.; Wang, Y.; Lu, W.; Shen, J.; Li, C. Novel Strategy Utilizing Extracellular Cysteine-Rich Domain of Membrane Receptor for Constructing d -Peptide Mediated Targeted Drug Delivery Systems: A Case Study on Fn14. Bioconjug. Chem. 2017, 28, 2167–2179. [Google Scholar] [CrossRef]
- Hervé, M.; Maillère, B.; Mourier, G.; Texier, C.; Leroy, S.; Ménez, A. On the immunogenic properties of retro-inverso peptides. Total retro- inversion of T-cell epitopes causes a loss of binding to MHC II molecules. Mol. Immunol. 1997, 34, 157–163. [Google Scholar] [CrossRef]
- Chow, H.Y.; Zhang, Y.; Matheson, E.; Li, X. Ligation Technologies for the Synthesis of Cyclic Peptides. Chem. Rev. 2019, 119, 9971–10001. [Google Scholar] [CrossRef] [PubMed]
- Dougherty, P.G.; Sahni, A.; Pei, D. Understanding Cell Penetration of Cyclic Peptides. Chem. Rev. 2019, 119, 10241–10287. [Google Scholar] [CrossRef]
- Malde, A.K.; Hill, T.A.; Iyer, A.; Fairlie, D.P. Crystal Structures of Protein-Bound Cyclic Peptides. Chem. Rev. 2019, 119, 9861–9914. [Google Scholar] [CrossRef] [PubMed]
- Reguera, L.; Rivera, D.G. Multicomponent Reaction Toolbox for Peptide Macrocyclization and Stapling. Chem. Rev. 2019, 119, 9836–9860. [Google Scholar] [CrossRef]
- Tyndall, J.D.A.; Nall, T.; Fairlie, D.P. Proteases Universally Recognize? Strands in Their Active Sites. ChemInform 2005, 36, 973–1000. [Google Scholar] [CrossRef]
- Joo, S.-H. Cyclic Peptides as Therapeutic Agents and Biochemical Tools. Biomol. Ther. 2012, 20, 19–26. [Google Scholar] [CrossRef] [Green Version]
- Bird, G.H.; Christian Crannell, W.; Walensky, L.D. Chemical Synthesis of Hydrocarbon-Stapled Peptides for Protein Interaction Research and Therapeutic Targeting. Curr. Protoc. Chem. Biol. 2011, 3, 99–117. [Google Scholar] [CrossRef] [Green Version]
- Qian, Z.; Rhodes, C.A.; McCroskey, L.C.; Wen, J.; Appiah-Kubi, G.; Wang, D.J.; Guttridge, D.C.; Pei, D. Enhancing the Cell Permeability and Metabolic Stability of Peptidyl Drugs by Reversible Bicyclization. Angew. Chemie Int. Ed. 2017, 56, 1525–1529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.; Lu, D.; Jiang, Y.; Jin, J.; Liu, S.; Chen, L.; Zhang, H.; Zhou, Y.; Chen, H.; Nagle, D.G.; et al. Stapled Wasp Venom-Derived Oncolytic Peptides with Side Chains Induce Rapid Membrane Lysis and Prolonged Immune Responses in Melanoma. J. Med. Chem. 2021, 64, 5802–5815. [Google Scholar] [CrossRef] [PubMed]
- Montgomery, J.E.; Donnelly, J.A.; Fanning, S.W.; Speltz, T.E.; Shangguan, X.; Coukos, J.S.; Greene, G.L.; Moellering, R.E. Versatile Peptide Macrocyclization with Diels-Alder Cycloadditions. J. Am. Chem. Soc. 2019, 141, 16374–16381. [Google Scholar] [CrossRef] [PubMed]
- Bechtler, C.; Lamers, C. Macrocyclization strategies for cyclic peptides and peptidomimetics. RSC Med. Chem. 2021, 12, 1325–1351. [Google Scholar] [CrossRef] [PubMed]
- Draper, S.R.E.; Lawrence, P.B.; Billings, W.M.; Xiao, Q.; Brown, N.P.; Bécar, N.A.; Matheson, D.J.; Stephens, A.R.; Price, J.L. Polyethylene Glycol Based Changes to β-Sheet Protein Conformational and Proteolytic Stability Depend on Conjugation Strategy and Location. Bioconjug. Chem. 2017, 28, 2507–2513. [Google Scholar] [CrossRef] [PubMed]
- Kalimuthu, K.; Lubin, B.C.; Bazylevich, A.; Gellerman, G.; Shpilberg, O.; Luboshits, G.; Firer, M.A. Gold nanoparticles stabilize peptide-drug-conjugates for sustained targeted drug delivery to cancer cells. J. Nanobiotechnology 2018, 16, 1–13. [Google Scholar] [CrossRef]
- Niu, F.; Yan, J.; Ma, B.; Li, S.; Shao, Y.; He, P.; Zhang, W.; He, W.; Ma, P.X.; Lu, W. Lanthanide-doped nanoparticles conjugated with an anti-CD33 antibody and a p53-activating peptide for acute myeloid leukemia therapy. Biomaterials 2018, 167, 132–142. [Google Scholar] [CrossRef]
- Yan, J.; He, W.; Yan, S.; Niu, F.; Liu, T.; Ma, B.; Shao, Y.; Yan, Y.; Yang, G.; Lu, W.; et al. Self-Assembled Peptide-Lanthanide Nanoclusters for Safe Tumor Therapy: Overcoming and Utilizing Biological Barriers to Peptide Drug Delivery. ACS Nano 2018, 12, 2017–2026. [Google Scholar] [CrossRef] [PubMed]
- Mascarin, A.; Valverde, I.E.; Mindt, T.L. Radiolabeled analogs of neurotensin (8-13) containing multiple 1,2,3-triazoles as stable amide bond mimics in the backbone. Medchemcomm 2016, 7, 1640–1646. [Google Scholar] [CrossRef]
- Wender, P.A.; Mitchell, D.J.; Pattabiraman, K.; Pelkey, E.T.; Steinman, L.; Rothbard, J.B. The design, synthesis, and evaluation of molecules that enable or enhance cellular uptake: Peptoid molecular transporters. Proc. Natl. Acad. Sci. USA 2000, 97, 13003–13008. [Google Scholar] [CrossRef] [Green Version]
- Snyder, E.L.; Meade, B.R.; Saenz, C.C.; Dowdy, S.F. Treatment of terminal peritoneal carcinomatosis by a transducible p53-activating peptide. PLoS Biol. 2004, 2, 186–193. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Xie, H.J.; Mu, L.M.; Liu, R.; Su, Z.B.; Cui, Y.N.; Xie, Y.; Lu, W.L. Functional chlorin gold nanorods enable to treat breast cancer by photothermal/photodynamic therapy. Int. J. Nanomedicine 2018, 13, 8119–8135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Najjar, K.; Erazo-Oliveras, A.; Brock, D.J.; Wang, T.Y.; Pellois, J.P. An L- to D-amino acid conversion in an endosomolytic analog of the cell-penetrating peptide TAT influences proteolytic stability, endocytic uptake, and endosomal escape. J. Biol. Chem. 2017, 292, 847–861. [Google Scholar] [CrossRef] [Green Version]
- Youngblood, D.S.; Hatlevig, S.A.; Hassinger, J.N.; Iversen, P.L.; Moulton, H.M. Stability of cell-penetrating peptide-morpholino oligomer conjugates in human serum and in cells. Bioconjug. Chem. 2007, 18, 50–60. [Google Scholar] [CrossRef]
- Prades, R.; Oller-Salvia, B.; Schwarzmaier, S.M.; Selva, J.; Moros, M.; Balbi, M.; Grazú, V.; De La Fuente, J.M.; Egea, G.; Plesnila, N.; et al. Applying the retro-enantio approach to obtain a peptide capable of overcoming the blood-brain barrier. Angew. Chemie Int. Ed. 2015, 54, 3967–3972. [Google Scholar] [CrossRef] [PubMed]
- Bukchin, A.; Sanchez-Navarro, M.; Carrera, A.; Teixidó, M.; Carcaboso, A.M.; Giralt, E.; Sosnik, A. Amphiphilic Polymeric Nanoparticles Modified with a Retro-Enantio Peptide Shuttle Target the Brain of Mice. Chem. Mater. 2020, 32, 7679–7693. [Google Scholar] [CrossRef]
- Wei, X.; Zhan, C.; Chen, X.; Hou, J.; Xie, C.; Lu, W. Retro-inverso isomer of angiopep-2: A stable d -peptide ligand inspires brain-targeted drug delivery. Mol. Pharm. 2014, 11, 3261–3268. [Google Scholar] [CrossRef]
- Ying, M.; Shen, Q.; Liu, Y.; Yan, Z.; Wei, X.; Zhan, C.; Gao, J.; Xie, C.; Yao, B.; Lu, W. Stabilized Heptapeptide A7R for Enhanced Multifunctional Liposome-Based Tumor-Targeted Drug Delivery. ACS Appl. Mater. Interfaces 2016, 8, 13232–13241. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.; Zhan, C.; Gao, J.; Zhang, M.; Wei, X.; Ying, M.; Liu, Z.; Lu, W. A d -Peptide Ligand of Integrins for Simultaneously Targeting Angiogenic Blood Vasculature and Glioma Cells. Mol. Pharm. 2017, 15, 592–601. [Google Scholar] [CrossRef] [PubMed]
- Nischan, N.; Herce, H.D.; Natale, F.; Bohlke, N.; Budisa, N.; Cardoso, M.C.; Hackenberger, C.P.R. Covalent attachment of cyclic TAT peptides to GFP results in protein delivery into live cells with immediate bioavailability. Angew. Chemie Int. Ed. 2014, 54, 1950–1953. [Google Scholar] [CrossRef]
- Shirazi, A.N.; Mozaffari, S.; Sherpa, R.T.; Tiwari, R.; Parang, K. Efficient intracellular delivery of cell-impermeable cargo molecules by peptides containing tryptophan and histidine. Molecules 2018, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolfe, J.M.; Fadzen, C.M.; Holden, R.L.; Yao, M.; Hanson, G.J.; Pentelute, B.L. Perfluoroaryl Bicyclic Cell-Penetrating Peptides for Delivery of Antisense Oligonucleotides. Angew. Chemie 2018, 130, 4846–4849. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Cheng, X.; Tian, X.; Guan, D.; Ao, J.; Wu, Z.; Huang, W.; Le, Z. Design and synthesis of novel dual-cyclic RGD peptides for α v β 3 integrin targeting. Bioorganic Med. Chem. Lett. 2019, 29, 896–900. [Google Scholar] [CrossRef]
- Moore, S.J.; Hayden Gephart, M.G.; Bergen, J.M.; Su, Y.R.S.; Rayburn, H.; Scott, M.P.; Cochran, J.R. Engineered knottin peptide enables noninvasive optical imaging of intracranial medulloblastoma. Proc. Natl. Acad. Sci. USA 2013, 110, 14598–14603. [Google Scholar] [CrossRef] [Green Version]
- Negussie, A.H.; Miller, J.L.; Reddy, G.; Drake, S.K.; Wood, B.J.; Dreher, M.R. Synthesis and in vitro evaluation of cyclic NGR peptide targeted thermally sensitive liposome. J. Control. Release 2010, 143, 265–273. [Google Scholar] [CrossRef] [Green Version]
- Ying, M.; Shen, Q.; Zhan, C.; Wei, X.; Gao, J.; Xie, C.; Yao, B.; Lu, W. A stabilized peptide ligand for multifunctional glioma targeted drug delivery. J. Control. Release 2016, 243, 86–98. [Google Scholar] [CrossRef]
- Ngambenjawong, C.; Gustafson, H.H.; Pineda, J.M.; Kacherovsky, N.A.; Cieslewicz, M.; Pun, S.H. Serum stability and affinity optimization of an M2 macrophage-targeting peptide (M2pep). Theranostics 2016, 6, 1403–1414. [Google Scholar] [CrossRef]
- Oller-Salvia, B.; Sánchez-Navarro, M.; Ciudad, S.; Guiu, M.; Arranz-Gibert, P.; Garcia, C.; Gomis, R.R.; Cecchelli, R.; García, J.; Giralt, E.; et al. MiniAp-4: A Venom-Inspired Peptidomimetic for Brain Delivery. Angew. Chemie 2016, 55, 572–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spengler, J.; Jiménez, J.C.; Burger, K.; Giralt, E.; Albericio, F. Abbreviated nomenclature for cyclic and branched homo- and hetero-detic peptides. J. Pept. Res. 2005, 65, 550–555. [Google Scholar] [CrossRef] [PubMed]
- Derossi, D.; Calvet, S.; Trembleau, A.; Brunissen, A.; Chassaing, G.; Prochiantz, A. Cell internalization of the third helix of the antennapedia homeodomain is receptor-independent. J. Biol. Chem. 1996, 271, 18188–18193. [Google Scholar] [CrossRef] [Green Version]
- Futaki, S.; Suzuki, T.; Ohashi, W.; Yagami, T.; Tanaka, S.; Ueda, K.; Sugiura, Y. Arginine-rich peptides. An abundant source of membrane-permeable peptides having potential as carriers for intracellular protein delivery. J. Biol. Chem. 2001, 276, 5836–5840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oller-Salvia, B.; Sánchez-Navarro, M.; Giralt, E.; Teixidó, M. Blood-brain barrier shuttle peptides: An emerging paradigm for brain delivery. Chem. Soc. Rev. 2016, 45, 4690–4707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prades, R.; Guerrero, S.; Araya, E.; Molina, C.; Salas, E.; Zurita, E.; Selva, J.; Egea, G.; López-Iglesias, C.; Teixidó, M.; et al. Delivery of gold nanoparticles to the brain by conjugation with a peptide that recognizes the transferrin receptor. Biomaterials 2012, 33, 7194–7205. [Google Scholar] [CrossRef] [PubMed]
- Ji, X.; Wang, H.; Chen, Y.; Zhou, J.; Liu, Y. Recombinant expressing angiopep-2 fused anti-VEGF single chain Fab (scFab) could cross blood–brain barrier and target glioma. AMB Express 2019, 9, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Ying, M.; Zhan, C.; Wang, S.; Yao, B.; Hu, X.; Song, X.; Zhang, M.; Wei, X.; Xiong, Y.; Lu, W. Liposome-Based Systemic Glioma-Targeted Drug Delivery Enabled by All- d Peptides. ACS Appl. Mater. Interfaces 2016, 8, 29977–29985. [Google Scholar] [CrossRef] [PubMed]
- Mao, J.; Ran, D.; Xie, C.; Shen, Q.; Wang, S.; Lu, W. EGFR/EGFRvIII Dual-Targeting Peptide-Mediated Drug Delivery for Enhanced Glioma Therapy. ACS Appl. Mater. Interfaces 2017, 9, 24462–24475. [Google Scholar] [CrossRef] [PubMed]
- Habault, J.; Poyet, J.L. Recent advances in cell penetrating peptide-based anticancer therapies. Molecules 2019, 24, 927. [Google Scholar] [CrossRef] [Green Version]
- Herce, H.D.; Schumacher, D.; Schneider, A.F.L.; Ludwig, A.K.; Mann, F.A.; Fillies, M.; Kasper, M.A.; Reinke, S.; Krause, E.; Leonhardt, H.; et al. Cell-permeable nanobodies for targeted immunolabelling and antigen manipulation in living cells. Nat. Chem. 2017, 9, 762–771. [Google Scholar] [CrossRef]
- Cox, D.; Brennan, M.; Moran, N. Integrins as therapeutic targets: Lessons and opportunities. Nat. Rev. Drug Discov. 2010, 9, 804–820. [Google Scholar] [CrossRef]
- Asati, S.; Pandey, V.; Soni, V. RGD Peptide as a Targeting Moiety for Theranostic Purpose: An Update Study. Int. J. Pept. Res. Ther. 2019, 25, 49–65. [Google Scholar] [CrossRef]
- Pierschbacher, M.D.; Ruoslahti, E. Cell attachment activity of fibronectin can be duplicated by small synthetic fragments of the molecule. Nature 1984, 309, 30–33. [Google Scholar] [CrossRef] [PubMed]
- Haubner, R.; Gratias, R.; Diefenbach, B.; Goodman, S.L.; Jonczyk, A.; Kessler, H. Structural and functional aspects of RGD-containing cyclic pentapeptides as highly potent and selective integrin α(v)β3 antagonists. J. Am. Chem. Soc. 1996, 118, 7461–7472. [Google Scholar] [CrossRef]
- Cox, N.; Kintzing, J.R.; Smith, M.; Grant, G.A.; Cochran, J.R. Integrin-Targeting Knottin Peptide–Drug Conjugates Are Potent Inhibitors of Tumor Cell Proliferation. Angew. Chemie Int. Ed. 2016, 55, 9894–9897. [Google Scholar] [CrossRef] [PubMed]
- Colombo, G.; Curnis, F.; De Mori, G.M.S.; Gasparri, A.; Longoni, C.; Sacchi, A.; Longhi, R.; Corti, A. Structure-activity relationships of linear and cyclic peptides containing the NGR tumor-homing motif. J. Biol. Chem. 2002, 277, 47891–47897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhabekar, G.; Dandekar, R.; Kingaonkar, A. Role of macrophages in malignancy. Ann. Maxillofac. Surg. 2011, 1, 150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cieslewicz, M.; Tang, J.; Yu, J.L.; Cao, H.; Zaèaljeèski, M.; Motoyama, K.; Lieber, A.; Raines, E.W.; Pun, S.H. Targeted delivery of proapoptotic peptides to tumor-associated macrophages improves survival. Proc. Natl. Acad. Sci. USA 2013, 110, 15919–15924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oller-Salvia, B.; Teixidó, M.; Giralt, E. From venoms to BBB shuttles: Synthesis and blood-brain barrier transport assessment of apamin and a nontoxic analog. Biopolymers 2013, 100, 675–686. [Google Scholar] [CrossRef] [PubMed]
- Teixidó, M.; Zurita, E.; Mendieta, L.; Oller-Salvia, B.; Prades, R.; Tarragó, T.; Giralt, E. Dual system for the central nervous system targeting and blood-brain barrier transport of a selective prolyl oligopeptidase inhibitor. Biopolymers 2013, 100, 662–674. [Google Scholar] [CrossRef]
- Wu, J.; Jiang, H.; Bi, Q.; Luo, Q.; Li, J.; Zhang, Y.; Chen, Z.; Li, C. Apamin-Mediated Actively Targeted Drug Delivery for Treatment of Spinal Cord Injury: More Than Just a Concept. Mol. Pharm. 2014, 11, 3210–3222. [Google Scholar] [CrossRef] [PubMed]
- Díaz-Perlas, C.; Varese, M.; Guardiola, S.; García, J.; Sánchez-Navarro, M.; Giralt, E.; Teixidó, M. From venoms to BBB-shuttles. MiniCTX3: A molecular vector derived from scorpion venom. Chem. Commun. 2018, 54, 12738–12741. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Name | Sequence | Modification | Application | Cargoes | Reference |
|---|---|---|---|---|---|
| d-Tat 49–57 d-Tat57-49 | rkkrrqrrr rrrqrrkkr | Enantio Retro-enantio | Cell internalization | Small molecules, nanoparticles, proteins, oligonucleotides | [34,35,36] |
| D-dfTAT |  | Enantio | Cell internalization | Small molecules | [37] |
| D-R9F2C | rrrrrrrrrffc | Enantio | Cell internalization | Oligonucleotides | [38] |
| THRre | pwvpswmpprht | Retro-enantio | BBB-shuttle | Small molecules, nanoparticles | [39,40] |
| DAngiopep | cyeetkfnnrkGrsGGyfft | Retro-enantio | Brain tumor targeting | Micelles | [41] |
| DA7R | rpplwta | Retro-enantio | Brain tumor targeting | Liposomes | [42] |
| DVS | svafpsyrhrsfwsv | Retro-enantio | Brain tumor targeting | Micelles | [43] |
| DCDX | GreirtGraerwsekf | Retro-enantio | BBB shuttle and brain tumor targeting | Liposomes | [15] |
| D-FNB | eGakhGltfsGG | Retro-enantio | Tumor targeting | Liposomes | [16] |
| cTAT | K(&)rRrGrKkRrE(&) | Cyclization | Cell internalization | Proteins | [44] |
| (WH)5 | &WHWHWHWHWH& | Cyclization | Cell internalization | Peptides and small molecules | [45] |
| Arginine rich peptide (1b) | C(&)RRRRRRC(&)RRRRRRC(&)* | Cyclization | Cell internalization | Oligonucleotides | [46] |
| Cyclo(RGDfK) | &RGDfK& | Cyclization | Tumor targeting | Cytotoxic drug monomethyl auristatin E (MMAE) | [47] |
| EETI 2.5F | GC(&1)PRPRGDNPLTC(&2)SQDSDC(&3)LAGC(&1)VC(&2)GPNGFC(&3)G | Cyclization | Tumor targeting | Small molecules | [48] |
| cKNGRE | K(&)NGRE(&) | Cyclization | Tumor targeting | Proteins, liposomes | [49] |
| cA7R | &CATWLPPR& | Cyclization | Brain tumor targeting | Liposomes | [50] |
| Cyclic M2pep(RY) | C(&)GYEQDPWGVRYWYGC(&)kkk | Cyclization | Targeting of tumor-associated macrophages | Small molecules | [51] |
| MiniAp-4 | (Dap)(&)KAPETALD(&) | Cyclization | BBB shuttle | Proteins, nanoparticles, small molecules | [52] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lucana, M.C.; Arruga, Y.; Petrachi, E.; Roig, A.; Lucchi, R.; Oller-Salvia, B. Protease-Resistant Peptides for Targeting and Intracellular Delivery of Therapeutics. Pharmaceutics 2021, 13, 2065. https://doi.org/10.3390/pharmaceutics13122065
Lucana MC, Arruga Y, Petrachi E, Roig A, Lucchi R, Oller-Salvia B. Protease-Resistant Peptides for Targeting and Intracellular Delivery of Therapeutics. Pharmaceutics. 2021; 13(12):2065. https://doi.org/10.3390/pharmaceutics13122065
Chicago/Turabian StyleLucana, Maria C., Yolanda Arruga, Emilia Petrachi, Albert Roig, Roberta Lucchi, and Benjamí Oller-Salvia. 2021. "Protease-Resistant Peptides for Targeting and Intracellular Delivery of Therapeutics" Pharmaceutics 13, no. 12: 2065. https://doi.org/10.3390/pharmaceutics13122065