
Preparation and Characterization of Fenofibrate Microparticles with Surface-Active Additives: Application of a Supercritical Fluid-Assisted Spray-Drying Process


Abstract
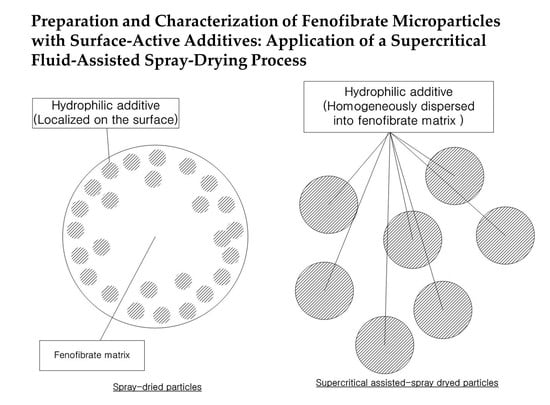
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Preparation of Fenofibrate Microparticles
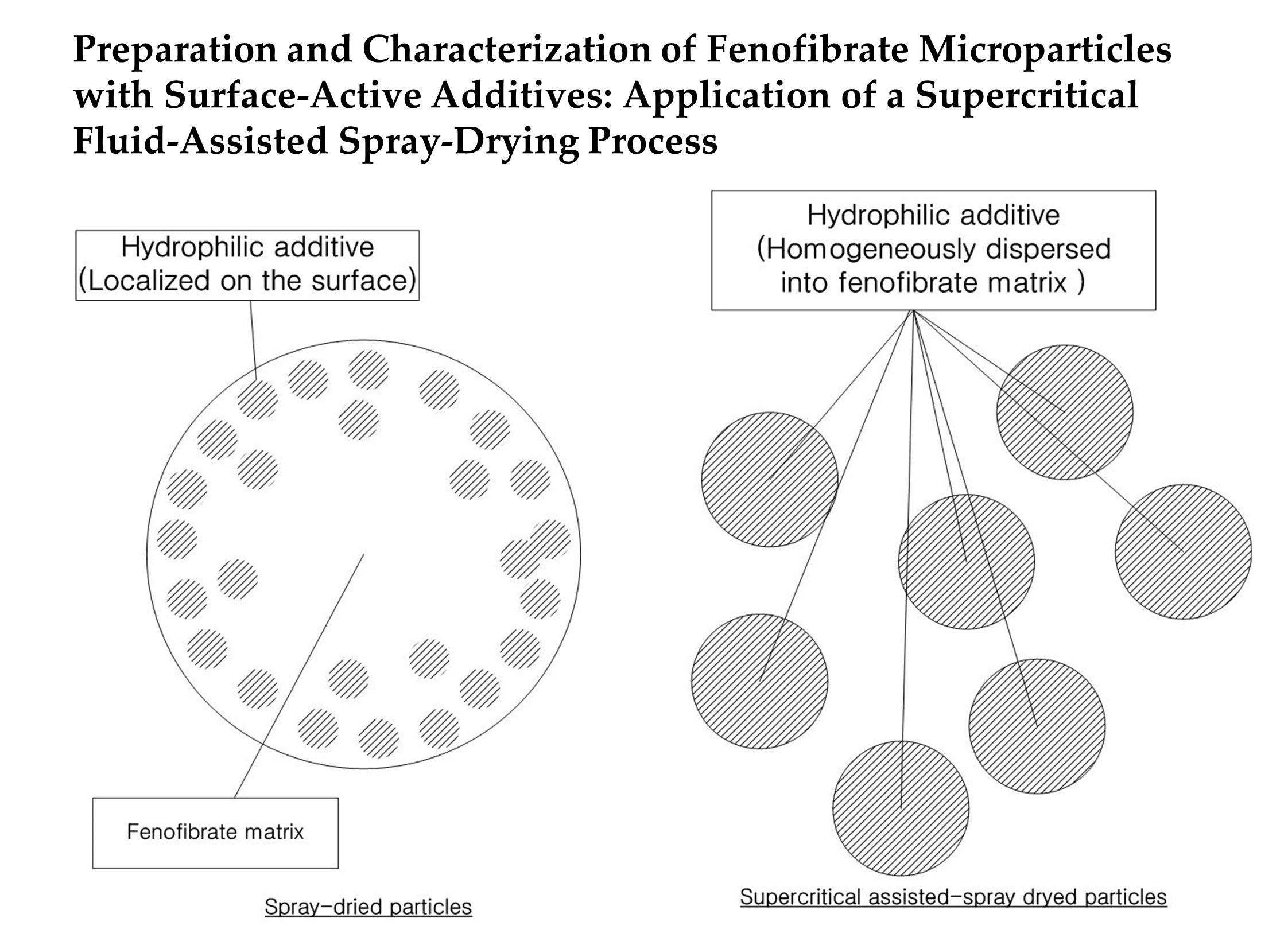
2.2.1. SA-SD Process
2.2.2. Box–Behnken Design (BBD)
2.2.3. Preparation of Fenofibrate Microcomposite Particles under Optimized Conditions
2.2.4. Conventional Spray-Drying (SD) Process
2.3. Physicochemical Characterization of Fenofibrate Microparticles
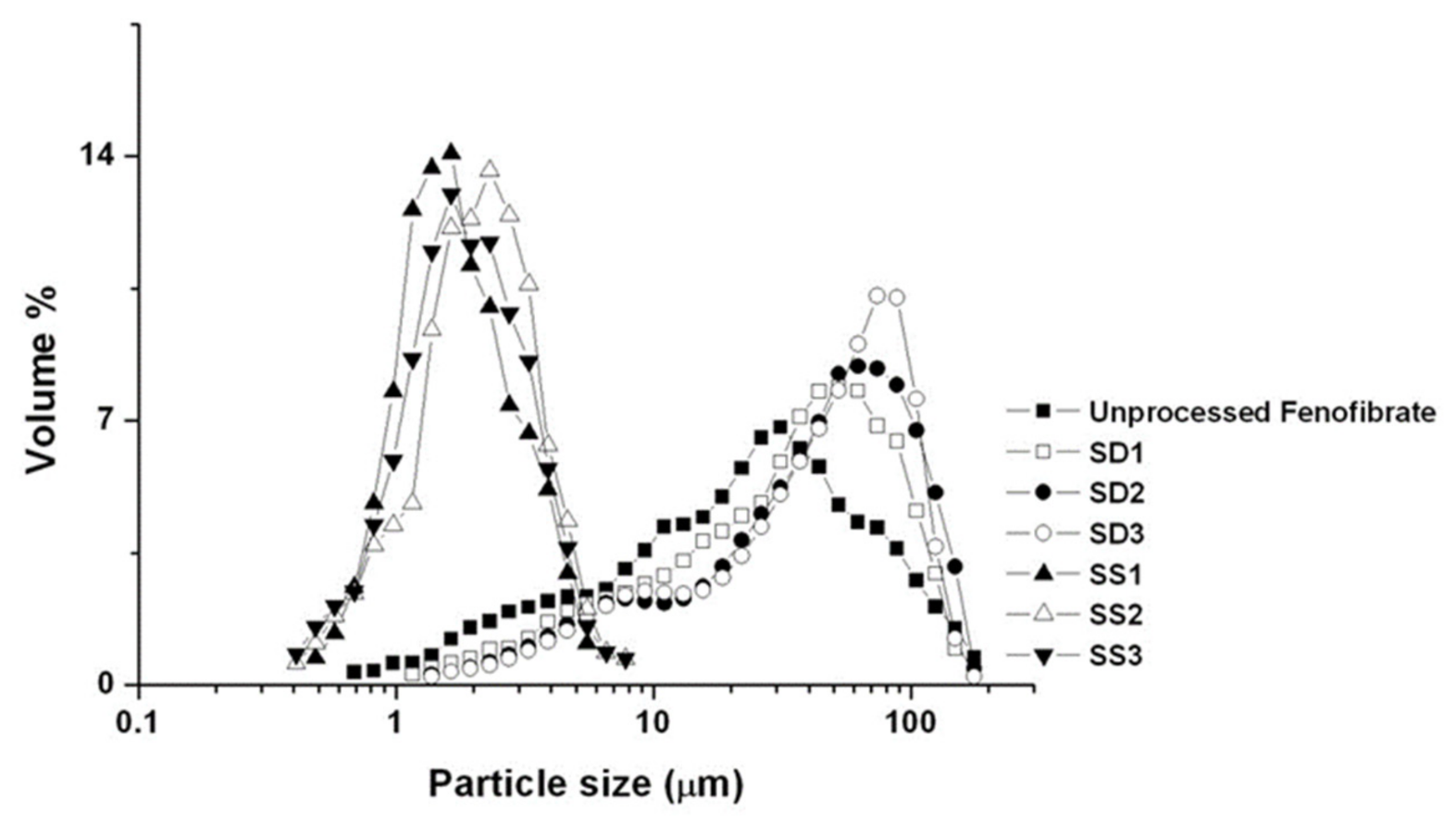
2.3.1. Particle Size Analysis
2.3.2. Powder X-ray Diffraction (PXRD)
2.3.3. Differential Scanning Calorimetry (DSC)
2.3.4. Scanning Electron Microscopy (SEM)
2.3.5. X-ray Photoelectron Spectroscopy (XPS)
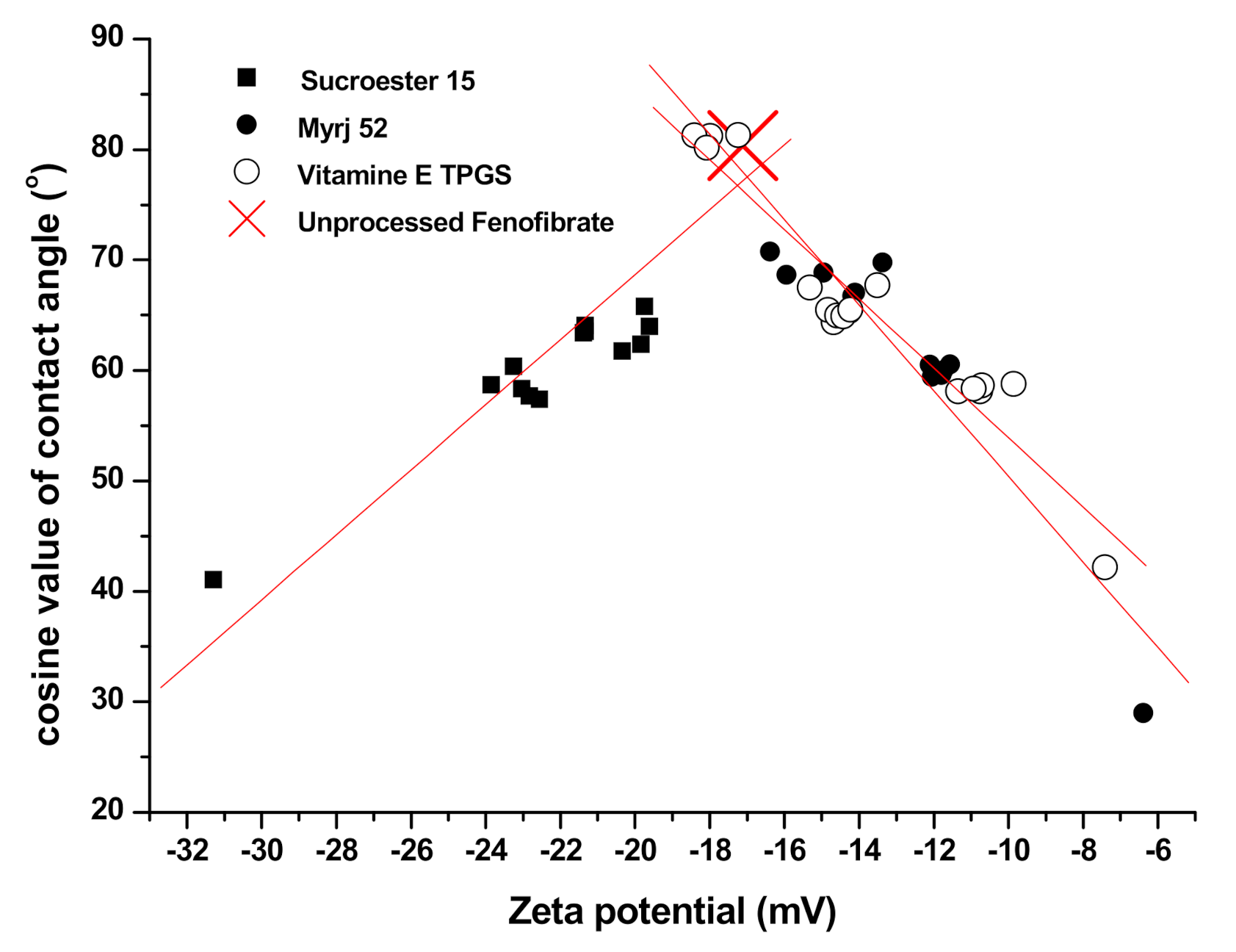
2.3.6. Zeta Potential Measurement
2.3.7. Contact Angle Measurement
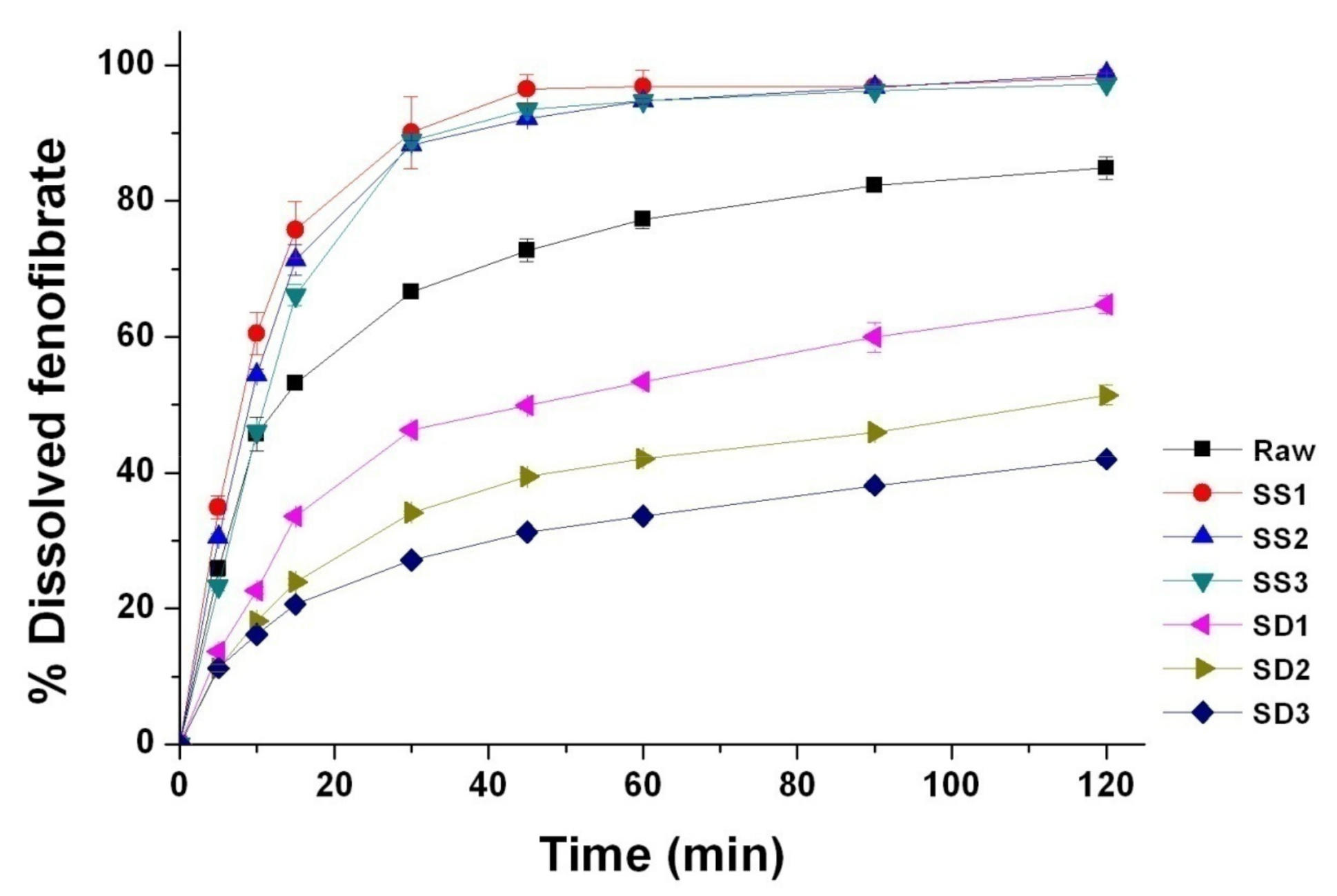
2.3.8. Dissolution Test
2.4. In Vivo Studies Using Sprague–Dawley Rats
2.4.1. Pharmacokinetic (PK) Study
2.4.2. Pharmacodynamic (PD) Studies in Sprague–Dawley Rats
3. Results and Discussion
3.1. Optimization of Fenofibrate Microparticle Formation Using Box–Behnken Design (BBD)
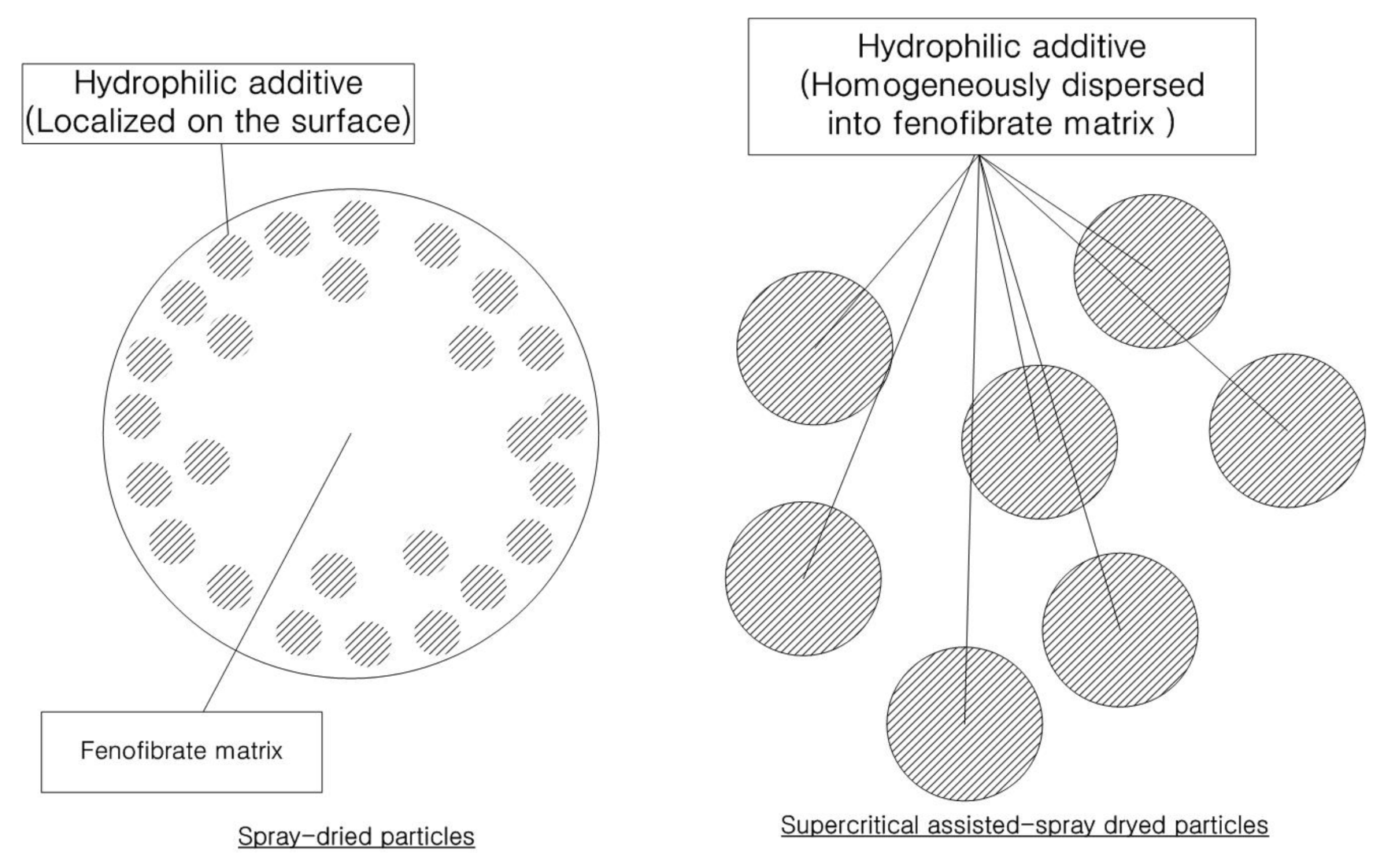
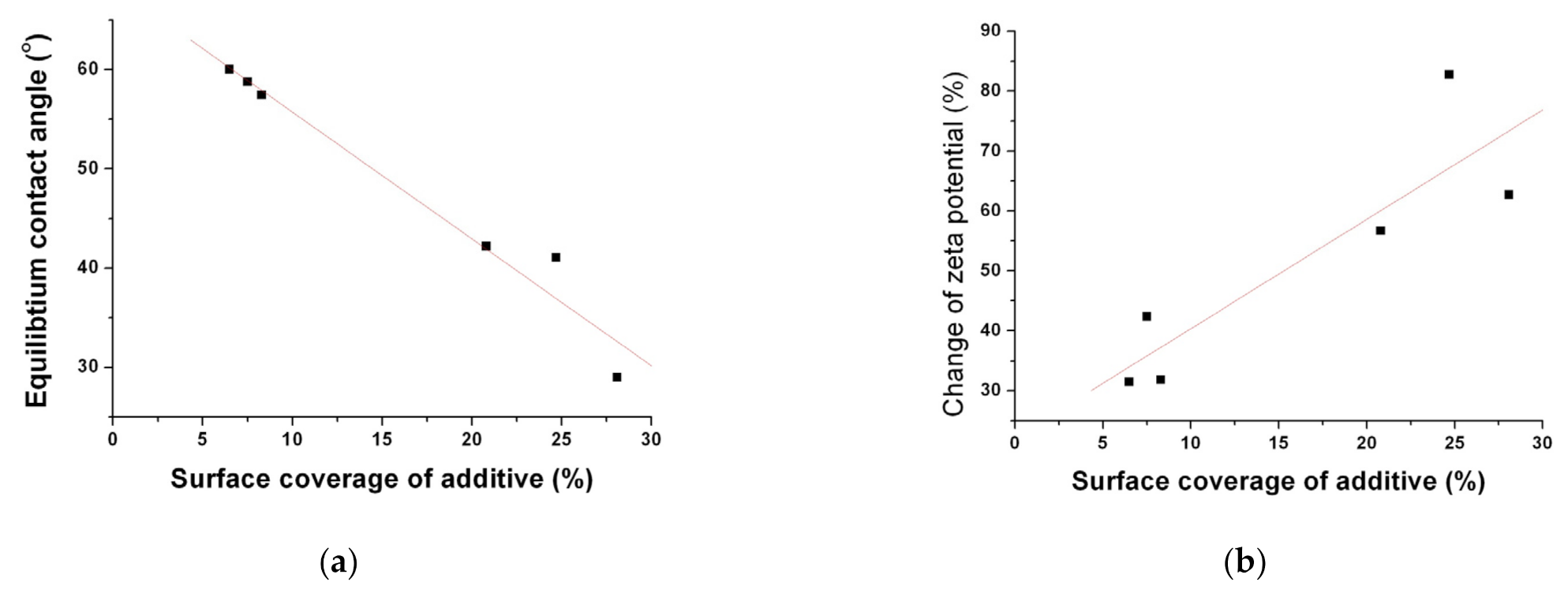
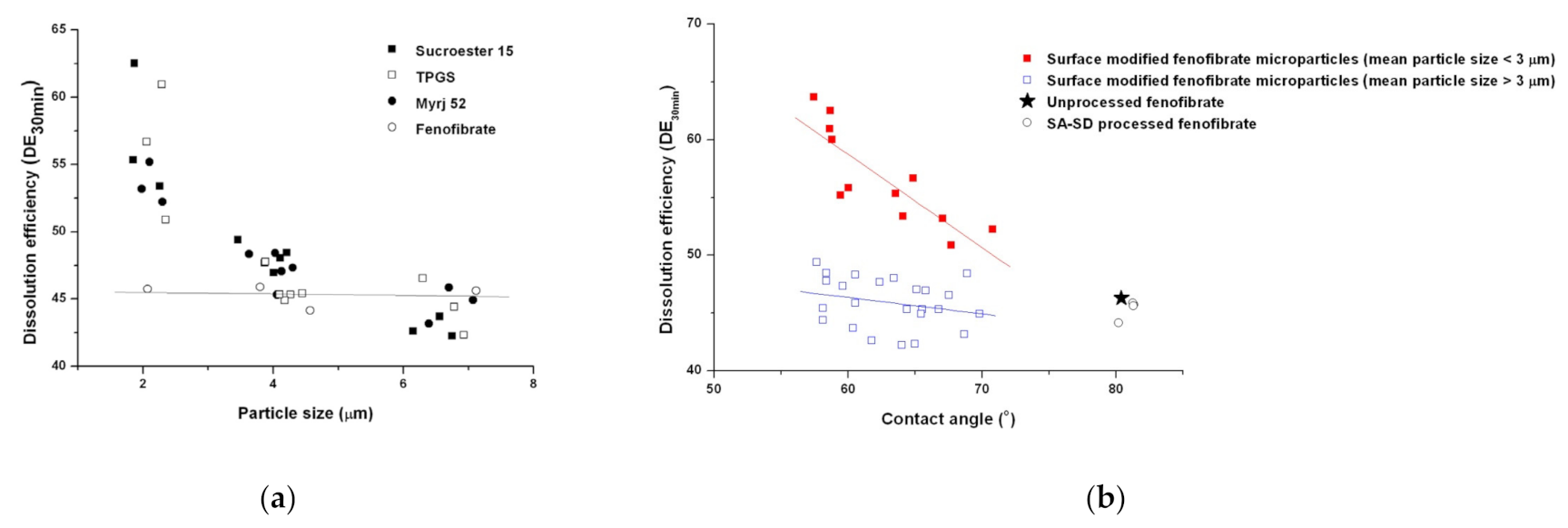
3.2. Physicochemical Characterization of Fenofibrate-Additive Microcomposite Particles
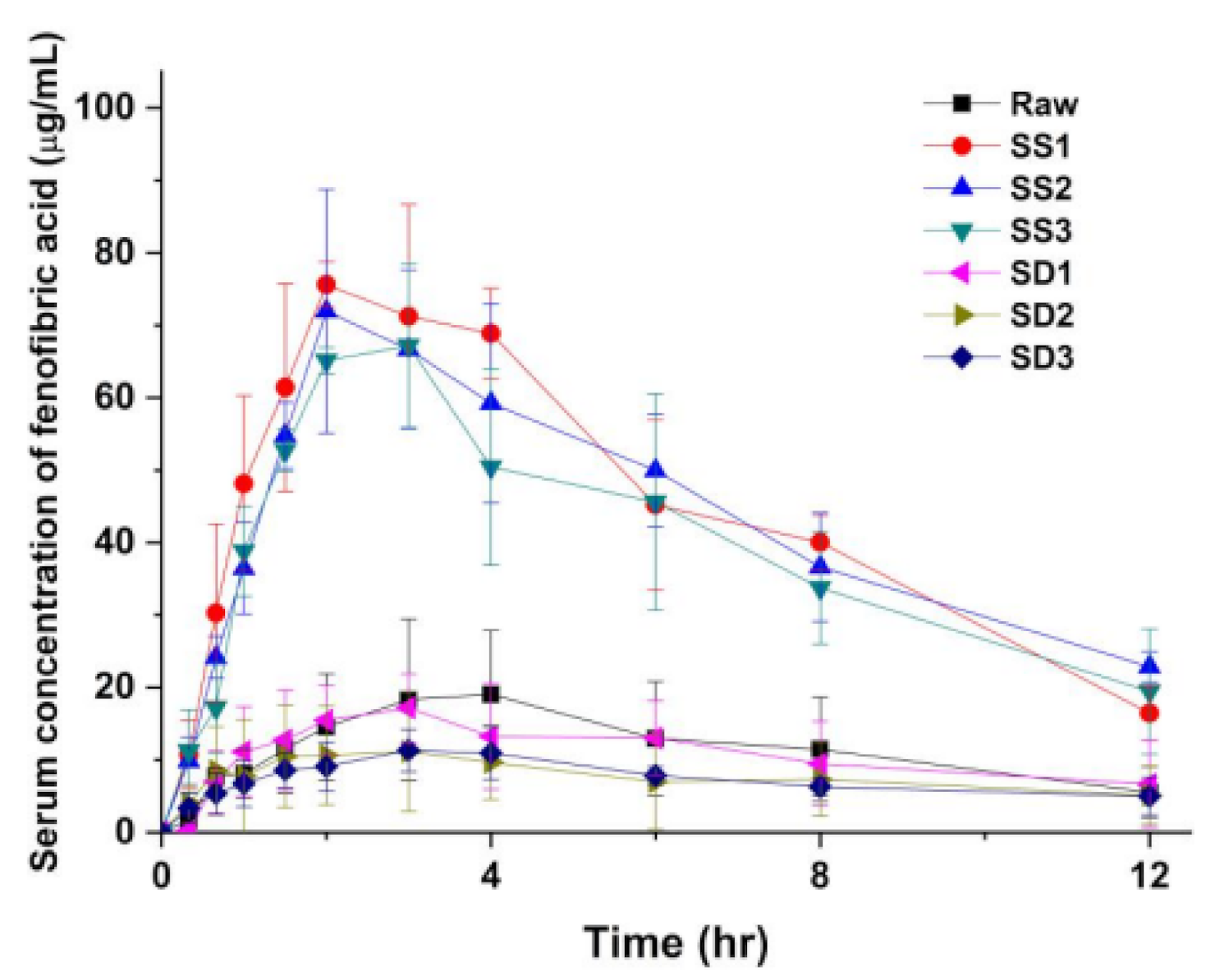
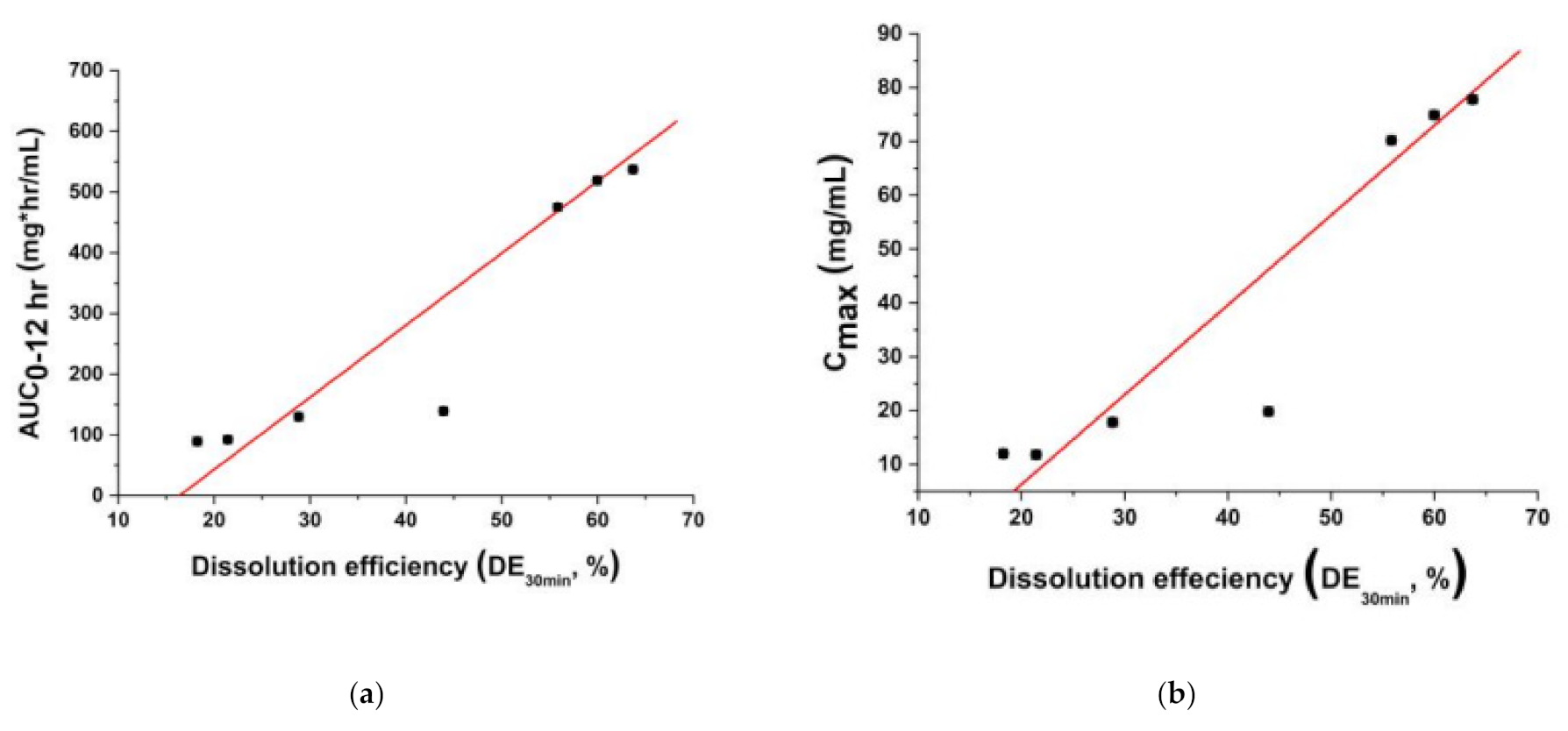
3.3. Pharmacokinetic (PK) Profile of Fenofibrate Microparticles in Sprague–Dawley Rats
3.4. Pharmacodynamic (PD) Therapeutic Efficacy in Sprague–Dawley Rats
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Conflicts of Interest
References
- Cunha, M.S.S.; Martinez-Pacheco, R.; Landin, M. Dissolution rate enhancement of the novel antitumoral beta-lapachone by solvent change precipitation of microparticles. Eur. J. Pharm. Biopharm. 2008, 69, 871–877. [Google Scholar] [CrossRef]
- Huang, Q.P.; Wang, J.X.; Chen, G.Z.; Shen, Z.G.; Chen, J.F.; Yun, J. Micronization of gemfibrozil by reactive precipitation process. Int. J. Pharm. 2008, 360, 58–64. [Google Scholar] [CrossRef]
- Kiio, T.M.; Park, S. Physical properties of nanoparticles do matter. J. Pharm. Investig. 2021, 51, 35–51. [Google Scholar] [CrossRef]
- Kim, N.A.; Oh, H.K.; Lee, J.C.; Choi, Y.H.; Jeong, S.H. Comparison of solubility enhancement by solid dispersion and micronized butein and its correlation with in vivo study. J. Pharm. Investig. 2021, 51, 53–60. [Google Scholar] [CrossRef]
- Erdoğar, N.; Akkın, S.; Nielsen, T.T.; Özçelebi, E.; Erdoğdu, B.; Nemutlu, E.; İskit, A.B.; Bilensoy, E. Development of oral aprepitant-loaded chitosan–polyethylene glycol-coated cyclodextrin nanocapsules: Formulation, characterization, and pharmacokinetic evaluation. J. Pharm. Investig. 2021, 51, 297–310. [Google Scholar] [CrossRef]
- Gautam, M.; Kim, J.O.; Yong, C.S. Fabrication of aerosol-based nanoparticles and their applications in biomedical fields. J. Pharm. Investig. 2021, 51, 1–15. [Google Scholar] [CrossRef]
- Kim, D.-H.; Lee, S.-E.; Pyo, Y.-C.; Tran, P.; Park, J.-S. Solubility enhancement and application of cyclodextrins in local drug delivery. J. Pharm. Investig. 2020, 50, 17–27. [Google Scholar] [CrossRef]
- Ha, E.-S.; Lee, S.-K.; Choi, D.H.; Jeong, S.H.; Hwang, S.-J.; Kim, M.-S. Application of diethylene glycol monoethyl ether in solubilization of poorly water-soluble drugs. J. Pharm. Investig. 2020, 50, 231–250. [Google Scholar] [CrossRef]
- Porta, G.; Reverchon, E. Engineering powder properties by supercritical fluid for optimum drug delivery. Part two: Supercritical-assisted atomisation. BioProcess. Imt. 2005, 3, 54–60. [Google Scholar]
- Park, H.; Ha, E.-S.; Kim, M.-S. Surface modification strategies for high-dose dry powder inhalers. J. Pharm. Investig. 2021, 51, 1–34. [Google Scholar] [CrossRef]
- Krukonis, V. Supercritical fluid nucleation of difficult-to-comminute solids. In Proceedings of the Annual Meeting AIChE, San Francisco, CA, USA, 25–30 November, 25–30 November 1984. [Google Scholar]
- Phillips, E.; Stella, V. Rapid expansion from supercritical solutions: Application to pharmaceutical processes. Int. J. Pharm. 1993, 94, 1–10. [Google Scholar] [CrossRef]
- Matson, D.W.; Fulton, J.L.; Petersen, R.C.; Smith, R.D. Rapid expansion of supercritical fluid solution: Solute formation of powders. Thin films and fibers. Ind. Eng. Chem. Res. 1987, 26, 2298–2306. [Google Scholar] [CrossRef]
- Jung, J.; Perrut, M. Particle design using supercritical fluids: Literature and patent survey. J. Supercrit. Fluids 2001, 20, 179–219. [Google Scholar] [CrossRef]
- Sane, A.; Thies, M.C. Effect of material properties and processing conditions on RESS of poly(l-lactide). J. Supercrit. Fluids 2007, 40, 134–143. [Google Scholar] [CrossRef]
- Matson, D.W.; Fulton, J.L.; Petersen, R.C.; Smith, R.D. Production of powders and Films from Supercritical Solutions. J. Mater. Sci. 1987, 22, 1919–1928. [Google Scholar] [CrossRef]
- Petersen, R.C.; Matson, D.W.; Smith, R.D. The Formation of Polymer Fibers from the Rapid Expansion of Supercritical Fluid Solutions. Polym. Eng. Sci. 1987, 27, 1963–1967. [Google Scholar] [CrossRef]
- Yang, T.M.; Su, C.S.; Li, J.S.; Lu, K.T.; Yeh, T.F. Recrystallization and micronization of p-toluenesulfonamide using the rapid expansion of supercritical solution (RESS) process. Crystals 2019, 9, 449. [Google Scholar] [CrossRef] [Green Version]
- Meziani, M.; Pathak, P.; Beacham, F.; Allard, L.; Sun, Y. Nanoparticle formation in rapid expansion of water-in-supercritical carbon dioxide microemulsion into liquid solution. J. Supercrit. Fluids 2005, 34, 91–97. [Google Scholar] [CrossRef]
- Young, T.; Johnston, K.; Pace, G.; Mishra, A. Phospholipid-stabilized nanoparticles of cyclosporine a by rapid expansion from supercritical to aqueous solution. AAPS PharmSciTech 2004, 5, 70–85. [Google Scholar]
- Young, T.J.; Mawson, S.; Johnston, K.P.; Henriksen, I.B.; Pace, G.W.; Mishra, A.K. Rapid expansion from supercritical to aqueous solution to produce submicron suspensions of water-insoluble drugs. Biotechnol. Prog. 2000, 16, 402–407. [Google Scholar] [CrossRef]
- Chattopadhyay, P.; Gupta, R.B. Supercritical CO2 based production of magnetically responsive micro- and nano-particles for drug targeting. Ind. Eng. Chem. Res. 2002, 41, 6049–6058. [Google Scholar] [CrossRef]
- Seckner, A.J.; McClellan, A.K.; McHugh, M.A. High solution behavior of the polymer-toluene-ethane system. AIChE 1988, 34, 9–16. [Google Scholar] [CrossRef]
- McHugh, M.A.; McClellan, T.L. Seperating polymer solutions with supercritical fluids. Macromolecules 1985, 18, 674–681. [Google Scholar] [CrossRef]
- Hanna, M.; York, P. Method And Apparatus For The Formation Of Particles. WO Patent 1995/001221, 12 January 1995. [Google Scholar]
- Moshashaee, S.; Bisrat, M.; Forbes, R.; Quinn, E.; Nyqvist, H.; York, P. Supercritical fluid processing of proteins: Lysozyme precipitation from aqueous solution. J. Pharm. Pharmacol. 2003, 55, 185–192. [Google Scholar] [CrossRef]
- Graser, F.; Wickenhaeuser, G. Conditioning of finely divided crude organic pigments. U.S. Patent 4,451,654, 20 September 1982. [Google Scholar]
- Kazarian, S. Polymer processing with supercritical fluids. Polym. Sci. Ser. C 2000, 42, 78–101. [Google Scholar]
- Pasquali, I.; Comi, L.; Pucciarelli, F.; Bettini, R. Swelling, melting point reduction, and solubility of PEG 1500 in supercritical CO2. Int. J. Pharm. 2008, 356, 76–81. [Google Scholar] [CrossRef]
- Ventosa, N.; Sala, S.; Veciana, J. DELOS process: A crystallization technique using compressed fluids: 1. Comparison to the GAS crystallization method. J. Supercrit. Fluids 2003, 26, 33–45. [Google Scholar] [CrossRef]
- Weidner, E.; Petermann, M.; Blatter, K.; Rekowski, V. Manufacture of powder coatings by spraying of gas-enriched melts. Chem. Eng. Technol. 2001, 24, 529–533. [Google Scholar] [CrossRef]
- Sievers, R.; Huang, E.; Villa, J.; Engling, G.; Brauer, P. Micronization of water-soluble or alcohol-soluble pharmaceuticals and model compounds with a low-temperature Bubble Dryer®. J. Supercrit. Fluids 2003, 26, 9–16. [Google Scholar] [CrossRef]
- Reverchon, E. Supercritical-assisted atomization to produce micro-and/or nanoparticles of controlled size and distribution. Ind. Eng. Chem. Res. 2002, 41, 2405–2411. [Google Scholar] [CrossRef]
- Cho, W.; Kim, M.-S.; Jung, M.-S.; Park, J.; Cha, K.-H.; Kim, J.-S.; Park, H.J.; Alhalaweh, A.; Velaga, S.P.; Hwang, S.-J. Design of salmon calcitonin particles for nasal delivery using spray-drying and novel supercritical fluid-assisted spray-drying processes. Int. J. Pharm. 2015, 478, 288–296. [Google Scholar] [CrossRef]
- Hwang, S.J.; Kim, M.S.; Kim, J.S.; Cha, K.H.; Cho, W.K.; Park, J.S.; Seo, S.J. Manufacturing Method and Apparatus of Ultrafine Particles Having Uniform Particle Size Distribution. U.S. Patent 8,734,830, 27 May 2014. [Google Scholar]
- Kim, J.A.; Paxton, T.E.; Tomasko, D.L. Microencapsulation of naproxen using rapid expansion of supercritical solutions. Biotechnol. Prog. 1996, 12, 650–661. [Google Scholar] [CrossRef]
- Falk, R.F.; Randolph, T.W. Process variable implications for residual solvent removal and polymer morphology in the formation of Gentamycin-loaded poly(l-lactide) microparticles. Pharm. Res. 1998, 15, 1233–1237. [Google Scholar] [CrossRef]
- Ghaderi, R.; Artursson, P.; Carlfors, J. Preparation of biodegradable microparticles using solution-enhanced dispersion by supercritical fluids (SEDS). Pharm. Res. 1999, 16, 676–681. [Google Scholar] [CrossRef]
- Elvassore, N.; Baggio, M.; Pallado, P.; Bertucco, A. Production of insulin-loaded poly(ethylene glycol)/poly(l-lactide)(PEG/PLA) nanoparticles by gas antisolvent techniques. J. Pharm. Sci. 2001, 90, 1628–1636. [Google Scholar] [CrossRef]
- Corrigan, O.; Crean, A. Comparative physicochemical properties of hydrocortisone-PVP composites prepared using supercritical carbon dioxide by the GAS anti-solvent recrystallization process, by coprecipitation and by spray drying. Int. J. Pharm. 2002, 245, 75–82. [Google Scholar] [CrossRef]
- Sethia, S.; Squillante, E. Physicochemical characterization of solid dispersion of carbamazepine formulated by supercritical carbon dioxide and conventional solvent evaporation method. J. Pharm. Sci. 2002, 91, 1948–1957. [Google Scholar] [CrossRef]
- Snavely, W.; Subramaniam, B.; Rajewski, R.; Defelippis, M. Micronization of insulin from halogenated alcohol solution using supercritical carbon dioxide as an antisolvent. J. Pharm. Sci. 2002, 91, 2026–2038. [Google Scholar] [CrossRef]
- Wang, Y.; Wei, D.; Dave, R.; Pfeffer, R.; Sauceau, M.; Letourneau, J.; Fages, J. Extraction and precipitation particles coating using supercritical CO2. Powder Technol. 2002, 127, 32–44. [Google Scholar] [CrossRef] [Green Version]
- Meziani, M.J.; Pathak, P.; Hurezeanu, R.; Thies, M.C.; Enick, R.M.; Sun, Y.P. Supercritical-fluid processing technique for nanoscale polymer particles. Angew. Chem. Int. 2004, 3, 704–707. [Google Scholar] [CrossRef]
- Pathak, P.; Meziani, M.J.; Desai, T.; Sun, Y.P. Nanosizing drug particles in supercritical fluid processing. J. Am. Chem. Soc. 2004, 126, 10842–10843. [Google Scholar] [CrossRef] [PubMed]
- Reverchon, E.; Marco, I. Supercritical antisolvent micronization of Cefonicid: Thermodynamic interpretation of results. J. Supercrit. Fluids 2004, 31, 207–215. [Google Scholar] [CrossRef]
- Rodier, E.; Lochard, G.; Sauceau, M.; Letourneau, J.; Freiss, B.; Fages, J. A three step supercritical process to improve the dissolution rate of Eflucimibe. Eur. J. Pharm. Sci 2005, 26, 184–193. [Google Scholar] [CrossRef] [Green Version]
- Duarte, A.R.C.; Costa, M.S.; Simplicio, A.L.; Cardoso, M.M.; Duarte, C.M.M. Preparation of controlled release microspheres using supercritical fluid technology for delivery of anti-inflammatory drugs. Int. J. Pharm. 2006, 308, 168–174. [Google Scholar] [CrossRef] [Green Version]
- Moura, C.; Casimiro, T.; Costa, E.; Aguiar-Ricardo, A. Optimization of supercritical CO2-assisted spray drying technology for the production of inhalable composite particles using quality-by-design principles. Powder Technol. 2019, 357, 387–397. [Google Scholar] [CrossRef]
- Tabernero, A.; del Valle, E.M.M.; Galán, M.A. Supercritical fluids for pharmaceutical particle engineering: Methods, basic fundamentals and modelling. Chem. Eng. Process. Process. Intensif. 2012, 60, 9–25. [Google Scholar] [CrossRef]
- Cabral, R.; Sousa, A.; Silva, A.; Paninho, A.; Temtem, M.; Costa, E.; Casimiro, T.; Aguiar-Ricardo, A. Design of experiments approach on the preparation of dry inhaler chitosan composite formulations by supercritical CO2-assisted spray-drying. J. Supercrit. Fluids 2016, 116, 26–35. [Google Scholar] [CrossRef]
- Costa, C.; Nobre, B.; Matos, A.S.; Silva, A.S.; Casimiro, T.; Corvo, M.L.; Aguiar-Ricardo, A. Inhalable hydrophilic molecule-loaded liposomal dry powder formulations using supercritical CO2–assisted spray-drying. J. CO2 Util. 2021, 53, 101709. [Google Scholar] [CrossRef]
- Long, B.; Walker, G.M.; Ryan, K.M.; Padrela, L. Controlling polymorphism of carbamazepine nanoparticles in a continuous supercritical-CO2-assisted spray drying process. Cryst. Growth Des. 2019, 19, 3755–3767. [Google Scholar] [CrossRef]
- Adeoye, O.; Costa, C.; Casimiro, T.; Aguiar-Ricardo, A.; Cabral-Marques, H. Preparation of ibuprofen/hydroxypropyl-γ-cyclodextrin inclusion complexes using supercritical CO2-assisted spray drying. J. Supercrit. Fluids 2018, 133, 479–485. [Google Scholar] [CrossRef]
- Silva, M.C.; Silva, A.S.; Fernandez-Lodeiro, J.; Casimiro, T.; Lodeiro, C.; Aguiar-Ricardo, A. Supercritical CO2-assisted spray drying of strawberry-like gold-coated magnetite nanocomposites in chitosan powders for inhalation. Materials 2017, 10, 74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tavares, M.; Cabral, R.P.; Costa, C.; Martins, P.; Fernandes, A.R.; Casimiro, T.; Aguiar-Ricardo, A. Development of PLGA dry powder microparticles by supercritical CO2-assisted spray-drying for potential vaccine delivery to the lungs. J. Supercrit. Fluids 2017, 128, 235–243. [Google Scholar] [CrossRef]
- Franco, P.; De Marco, I. Nanoparticles and Nanocrystals by Supercritical CO2-Assisted Techniques for Pharmaceutical Applications: A Review. Appl. Sci. 2021, 11, 1476. [Google Scholar] [CrossRef]
- Briggs, D.; Seah, M. (Eds.) Practical Surface Analysis, Auger and X-ray Photoelectron Spectroscopy, 2nd ed.; Wiley: Hoboken, NJ, USA, 1990; Volume 1, pp. 635–638. [Google Scholar]
- Dahlberg, C.; Millqvist-Fureby, A.; Schuleit, M. Surface composition and contact angle relationships for differently prepared solid dispersions. Eur. J. Pharm. Biopharm. 2008, 70, 478–485. [Google Scholar] [CrossRef] [PubMed]
- Kadav, A.; Vora, D. Stability indicating UPLC method for simultaneous determination of atorvastatin, fenofibrate and their degradation products in tablets. J. Pharm. Biomed. Anal. 2008, 48, 120–126. [Google Scholar] [CrossRef] [PubMed]
- Cha, K.-H.; Cho, K.-J.; Kim, M.-S.; Kim, J.-S.; Park, H.J.; Park, J.; Cho, W.; Park, J.-S.; Hwang, S.-J. Enhancement of the dissolution rate and bioavailability of fenofibrate by a melt-adsorption method using supercritical carbon dioxide. Int. J. Nanomed. 2012, 7, 5565. [Google Scholar]
- Khan, K.A.; Rhodes, C.T. The concept of disolution efficiency. Pharm. Acta Helv. 1972, 47, 594–607. [Google Scholar] [PubMed]
- Dollery, C. (Ed.) Therapeutic Drugs, 2nd ed.; Elsevier Health Sciences: Amsterdam, The Netherlands, 1999. [Google Scholar]
- Patel, A.; Vavia, P. Preparation and in vivo evaluation of SMEDDS (Self-Microemulsifying Drug Delivery System) containing fenofibrate. AAPS J. 2007, 9, E344–E352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nutting, D.; Tso, P. Hypolipidemic effects of intravenous pluronic L-81 in fasted rats treated with Triton WR-1449: Possible inhibition of hepatic lipoprotein secretion. Horm. Metab. Res. 1989, 21, 113–115. [Google Scholar] [CrossRef] [PubMed]
- Cho, E.; Cho, W.; Cha, K.-H.; Park, J.; Kim, M.-S.; Kim, J.-S.; Park, H.J.; Hwang, S.-J. Enhanced dissolution of megestrol acetate microcrystals prepared by antisolvent precipitation process using hydrophilic additives. Int. J. Pharm. 2010, 396, 91–98. [Google Scholar] [CrossRef]
- Abuzar, S.M.; Hyun, S.-M.; Kim, J.-H.; Park, H.J.; Kim, M.-S.; Park, J.-S.; Hwang, S.-J. Enhancing the solubility and bioavailability of poorly water-soluble drugs using supercritical antisolvent (SAS) process. Int. J. Pharm. 2018, 538, 1–13. [Google Scholar] [CrossRef]
- Kim, J.-S.; Kim, M.-S.; Park, H.J.; Jin, S.-J.; Lee, S.; Hwang, S.-J. Physicochemical properties and oral bioavailability of amorphous atorvastatin hemi-calcium using spray-drying and SAS process. Int. J. Pharm. 2008, 359, 211–219. [Google Scholar] [CrossRef]
- Shetty, N.; Ahn, P.; Park, H.; Bhujbal, S.; Zemlyanov, D.; Cavallaro, A.; Mangal, S.; Li, J.; Zhou, Q.T. Improved physical stability and aerosolization of inhalable amorphous ciprofloxacin powder formulations by incorporating synergistic colistin. Mol. Pharm. 2018, 15, 4004–4020. [Google Scholar] [CrossRef]
- Shetty, N.; Park, H.; Zemlyanov, D.; Mangal, S.; Bhujbal, S.; Zhou, Q.T. Influence of excipients on physical and aerosolization stability of spray dried high-dose powder formulations for inhalation. Int. J. Pharm. 2018, 544, 222–234. [Google Scholar] [CrossRef]
- Corrigan, I. Retardation of polymeric carrier dissolution by dispersed drugs: Factors influencing the dissolution of solid dispersions containing polyethylene glycols. Drug. Dev. Ind. Pharm. 1986, 12, 1777–1793. [Google Scholar] [CrossRef]
- Higuchi, W.; Mir, N.; Desai, S. Dissolution rates of polyphase mixtures. J. Pharm. Sci. 1965, 54, 1405–1410. [Google Scholar] [CrossRef]
- Higuchi, W. Diffusional models useful in biopharmaceutics. J. Pharm. Sci. 1967, 54, 645. [Google Scholar] [CrossRef]
- Faldt, P.; Bergenstahl; Carlsson, G. The surface coverage of fat on food powders analyzed by ESCA (Electron Spectroscopy for Chemical Analysis). Food Struct. 1993, 12, 225–234. [Google Scholar]
- Kasim, N.; Whitehouse, M.; Ramachandran, C. Molecular properties of WHO essential drugs and provisional biopharmaceutical classification. Mol. Pharm. 2004, 1, 85–96. [Google Scholar] [CrossRef]
- Hayakawa, T.; Shimoyama, K.; Sekiya, S.; Sekiguchi, M.; Inotsume, N. Pharmacoeconomomic evaluation of anti-hyperlipidemic agent fenofibrate. Yakugaku Zasshi 2002, 122, 1145–1151. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formula | Surface-Active Additive | Content (w/w, %) of Additive in Solid Formula | Particle Formation Process |
|---|---|---|---|
| SS1 | Sucroester 15 | 5 | Supercritical assisted spray-drying (SA-SD) |
| SS2 | TPGS | 5 | SA-SD |
| SS3 | Myrj 52 | 5 | SA-SD |
| SD1 | Sucroester 15 | 5 | Spray-drying (SD) |
| SD2 | TPGS | 5 | SD |
| SD3 | Myrj 52 | 5 | SD |
| Formula | Mean Particle Size (μm) | SPAN a | Zeta Potential (mV) | Contact Angle (°) | |||
|---|---|---|---|---|---|---|---|
| Raw b | 23.80 | ±0.64 | 4.25 | −17.12 | ±1.13 | 80.38 | ±2.83 |
| SS1 | 1.86 | ±0.21 | 1.27 | −22.56 | ±1.74 | 57.42 | ±4.12 |
| SS2 | 2.17 | ±0.12 | 1.18 | −9.87 | ±1.35 | 58.78 | ±3.37 |
| SS3 | 2.04 | ±0.25 | 1.22 | −11.74 | ±2.03 | 60.01 | ±2.81 |
| SD1 | 33.98 | ±1.21 | 3.84 | −31.29 | ±1.96 | 41.05 | ±3.24 |
| SD2 | 43.76 | ±1.53 | 3.93 | −7.42 | ±2.14 | 42.17 | ±4.56 |
| SD3 | 49.16 | ±1.37 | 3.60 | −6.39 | ±1.94 | 28.97 | ±3.89 |
| Numbers of Atom | Atomic Concentration (%) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Substance | C | O | Cl | C1s | O1s | Cl2p | |||
| Fenofibrate | 20 | 4 | 1 | 80.4 | ±0.8 | 15.0 | ±0.7 | 4.6 | ±0.4 |
| Theory a | - | - | - | 80.0 | 16.0 | 4.0 | |||
| Sucroester 15 | 28 | 12 | - | 70.3 | ±0.7 | 29.7 | ±0.5 | - | |
| Theory a | - | - | - | 70.0 | 30.0 | - | |||
| TPGS | 57 | 28 | - | 68.1 | ±0.9 | 31.9 | ±0.6 | - | |
| Theory a | - | - | - | 67.1 | 32.9 | - | |||
| Myrj 52 | 122 | 54 | - | 69.9 | ±0.7 | 20.1 | ±0.4 | - | |
| Theory a | - | - | - | 69.3 | 30.7 | - | |||
| Formula | Atomic Concentration (%) | Surface Coverage of Additive (%) | Surface Excess of Additive a | |||||
|---|---|---|---|---|---|---|---|---|
| C1s | O1s | Cl2p | ||||||
| SS1 | 77.5 | ±0.9 | 16.8 | ±0.6 | 3.7 | ±0.6 | 8.3 | 1.7 |
| SS2 | 78.7 | ±0.6 | 17.2 | ±0.4 | 3.6 | ±0.3 | 7.5 | 1.5 |
| SS3 | 78.1 | ±0.5 | 16.7 | ±0.5 | 4.1 | ±0.7 | 6.5 | 1.3 |
| SD1 | 77.7 | ±0.4 | 19.5 | ±0.7 | 2.8 | ±0.8 | 24.7 | 4.9 |
| SD2 | 77.9 | ±0.8 | 19.7 | ±0.3 | 2.4 | ±0.9 | 20.8 | 4.2 |
| SD3 | 77.2 | ±0.7 | 20.2 | ±0.6 | 2.6 | ±0.7 | 28.1 | 5.6 |
| AUC0–12 h (μg∙h/mL) | Cmax (μg/mL) | Tmax (h) | ||||
|---|---|---|---|---|---|---|
| Unprocessed Fenofibrate | 139.1 | ±74.4 | 19.8 | ±10.1 | 3.5 | ±0.6 |
| SS1 | 537.4 | ±90.1 | 77.8 | ±6.2 | 2.5 | ±0.6 |
| SS2 | 519.0 | ±65.5 | 74.9 | ±14.3 | 2.8 | ±1.0 |
| SS3 | 475.1 | ±96.8 | 70.2 | ±8.9 | 2.5 | ±0.6 |
| SD1 | 129.8 | ±59.0 | 17.8 | ±5.3 | 3.5 | ±1.0 |
| SD2 | 92.3 | ±56.6 | 11.8 | ±6.7 | 3.3 | ±0.6 |
| SD3 | 89.2 | ±24.9 | 12.0 | ±3.6 | 3.5 | ±0.6 |
| Phase I (After 24 h) | Phase II (After 48 h) | |||||||
|---|---|---|---|---|---|---|---|---|
| Group | Total Cholesterol (mg/dL) | Triglycerides (mg/dL) | Total Cholesterol (mg/dL) | Triglycerides (mg/dL) | ||||
| Control | 70.4 | ±9.5 | 131.7 | ±6.7 | 68.9 | ±10.6 | 109.2 | ±4.9 |
| Triton | 256.7 | ±9.0 | 370.1 | ±7.5 | 141.1 | ±6.2 | 221.1 | ±4.5 |
| Unprocessed | 124.7 | ±24.5 (70.8) | 241.4 | ±3.7 (54.0) | 88.7 | ±9.2 (72.6) | 159.8 | ±7.1 (54.8) |
| SS1 | 86.1 | ±8.6 (91.6) | 152.4 | ±2.5 (91.3) | 73.9 | ±4.0 (93.0) | 127.5 | ±3.9 (83.7) |
| SS2 | 89.6 | ±10.8 (89.7) | 173.9 | ±10.5 (82.3) | 74.1 | ±5.9 (92.8) | 139.6 | ±12.2 (72.9) |
| SS3 | 96.7 | ±4.7 (85.9) | 170.8 | ±4.0 (83.6) | 78.3 | ±9.2 (86.9) | 135.4 | ±3.3 (76.6) |
| SD1 | 138.1 | ±1.3 (63.6) | 231.9 | ±5.3 (58.0) | 93.6 | ±9.8 (65.8) | 148.7 | ±5.8 (64.7) |
| SD2 | 146.1 | ±2.3 (59.3) | 243.9 | ±4.1 (52.9) | 91.7 | ±14.0 (68.4) | 165.8 | ±7.6 (49.5) |
| SD3 | 146.3 | ±23.8 (59.2) | 249.7 | ±6.4 (50.5) | 102.9 | ±8.5 (52.9) | 159.8 | ±5.1 (54.8) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, J.-S.; Park, H.; Ha, E.-S.; Kang, K.-T.; Kim, M.-S.; Hwang, S.-J. Preparation and Characterization of Fenofibrate Microparticles with Surface-Active Additives: Application of a Supercritical Fluid-Assisted Spray-Drying Process. Pharmaceutics 2021, 13, 2061. https://doi.org/10.3390/pharmaceutics13122061
Kim J-S, Park H, Ha E-S, Kang K-T, Kim M-S, Hwang S-J. Preparation and Characterization of Fenofibrate Microparticles with Surface-Active Additives: Application of a Supercritical Fluid-Assisted Spray-Drying Process. Pharmaceutics. 2021; 13(12):2061. https://doi.org/10.3390/pharmaceutics13122061
Chicago/Turabian StyleKim, Jeong-Soo, Heejun Park, Eun-Sol Ha, Kyu-Tae Kang, Min-Soo Kim, and Sung-Joo Hwang. 2021. "Preparation and Characterization of Fenofibrate Microparticles with Surface-Active Additives: Application of a Supercritical Fluid-Assisted Spray-Drying Process" Pharmaceutics 13, no. 12: 2061. https://doi.org/10.3390/pharmaceutics13122061