
Application of Lipid-Based Nanocarriers for Antitubercular Drug Delivery: A Review


 ,
,  , , , and
, , , and
Abstract
:1. Introduction
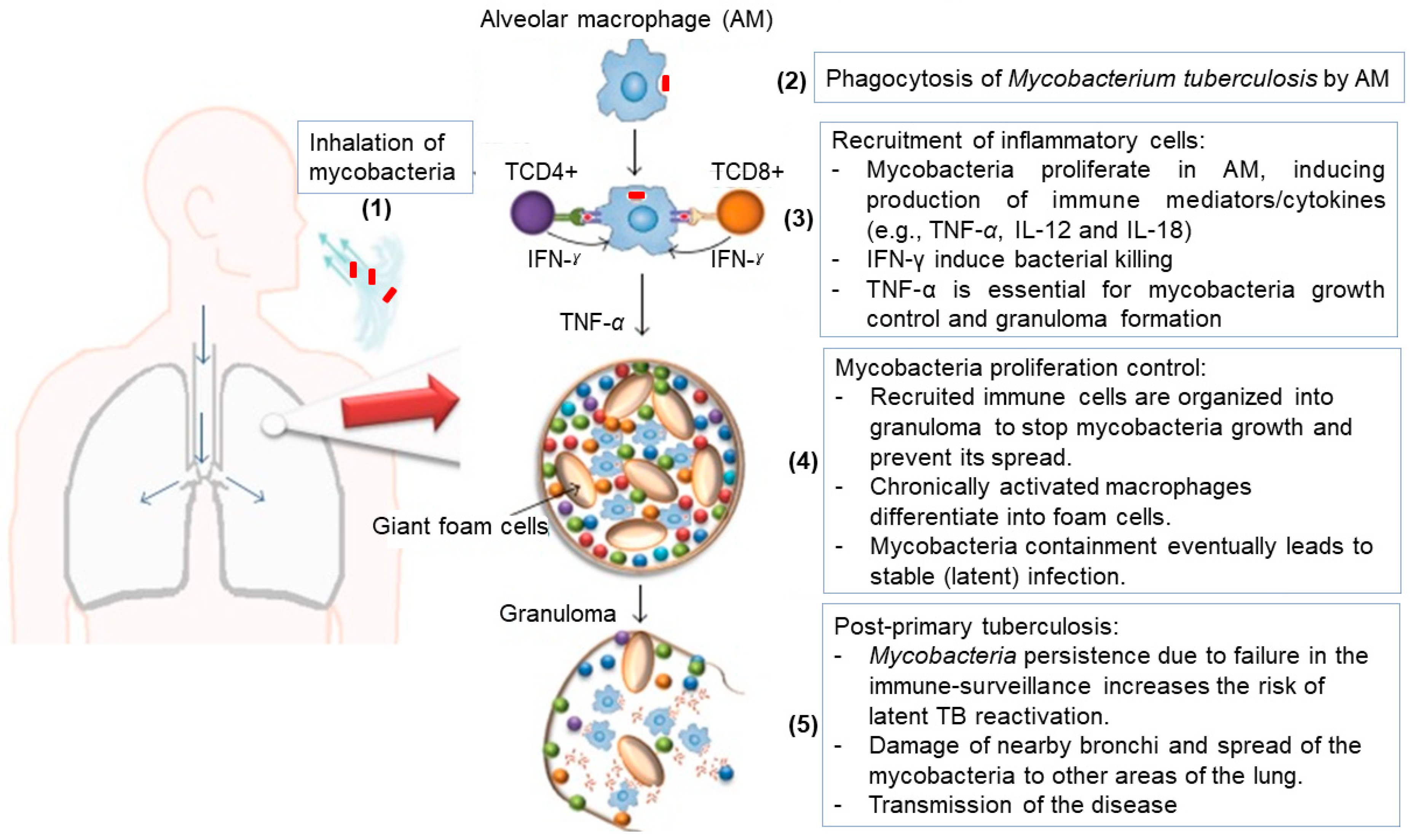
2. Pathogenesis and Management of Tuberculosis
2.1. Pathogenesis
2.2. Therapeutic Management and Limitations
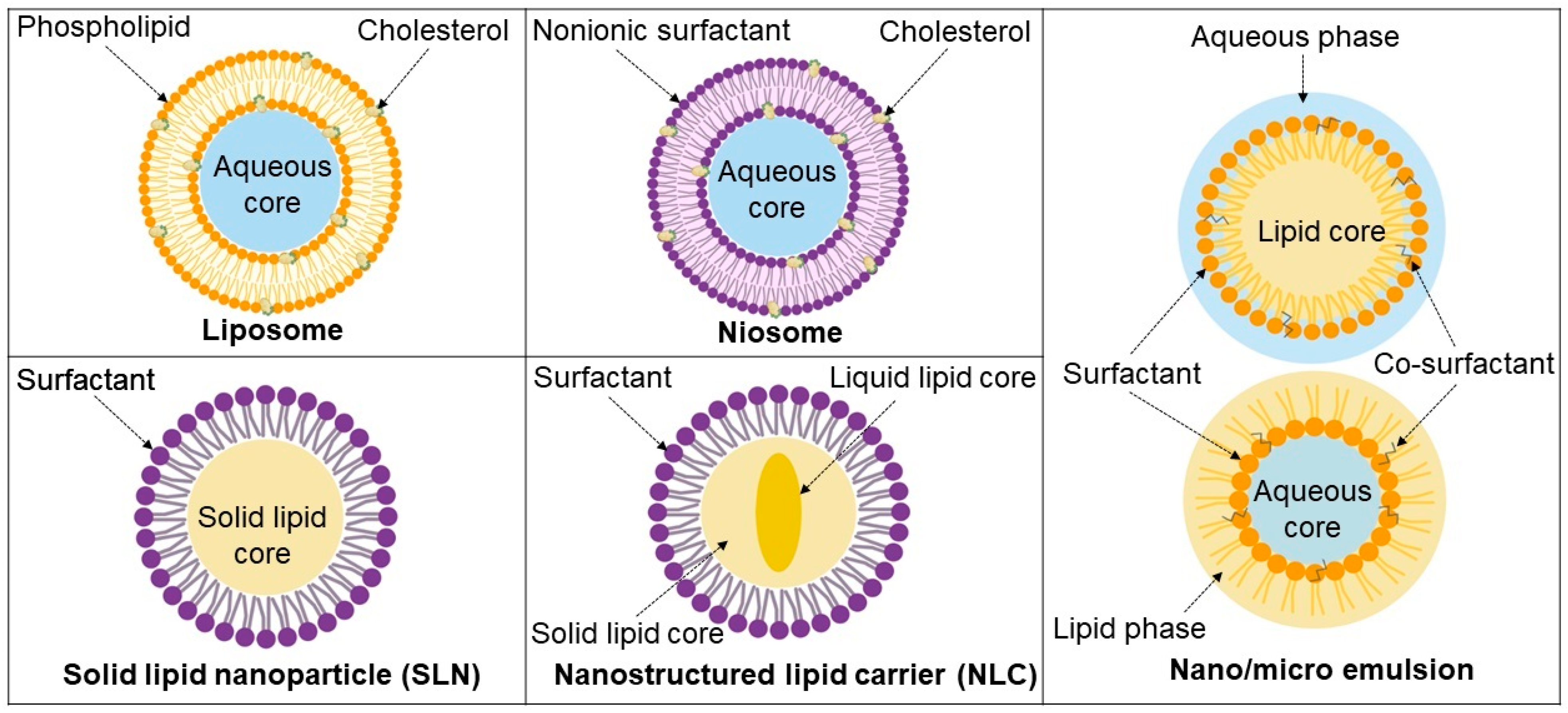
3. Lipid-Based Systems as Carriers of Anti-TB Drugs
3.1. Liposomes
3.2. Niosomes
3.3. Solid Lipid Nanoparticles
3.4. Nanostructured Lipid Carriers
3.5. Emulsions
3.5.1. Nano- and Microemulsions
3.5.2. Self-Emulsifying Drug Delivery Systems
4. Trends in Lipid-Based Drug Delivery Research for TB
4.1. Liposomes
4.2. Niosomes
4.3. Solid Lipid Particles
4.4. Nanostructured Lipid Carriers
4.5. Emulsions
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Baranyai, Z.; Soria-Carrera, H.; Alleva, M.; Millán-Placer, A.C.; Lucía, A.; Martín-Rapún, R.; Aínsa, J.A.; de la Fuente, J.M. Nanotechnology-Based Targeted Drug Delivery: An Emerging Tool to Overcome Tuberculosis. Adv. Ther. 2021, 4, 2000113. [Google Scholar] [CrossRef]
- World Health Organization. Global Tuberculosis Report; WHO: Geneva, Switzerland, 2021. [Google Scholar]
- Seki, M.; Choi, H.; Kim, K.; Whang, J.; Sung, J.; Mitarai, S. Tuberculosis: A persistent unpleasant neighbour of humans. J. Infect. Public Health 2021, 14, 508–513. [Google Scholar] [CrossRef]
- Gao, Y.; Liu, M.; Chen, Y.; Shi, S.; Geng, J.; Tian, J. Association between tuberculosis and COVID-19 severity and mortality: A rapid systematic review and meta-analysis. J. Med. Virol. 2021, 93, 194–196. [Google Scholar] [CrossRef]
- Motta, I.; Centis, R.; D’Ambrosio, L.; García-García, J.M.; Goletti, D.; Gualano, G.; Lipani, F.; Palmieri, F.; Sánchez-Montalvá, A.; Pontali, E.; et al. Tuberculosis, COVID-19 and migrants: Preliminary analysis of deaths occurring in 69 patients from two cohorts. Pulmonology 2020, 26, 233–240. [Google Scholar] [CrossRef]
- Chen, Y.; Wang, Y.; Fleming, J.; Yu, Y.; Gu, Y.; Liu, C.; Fan, L.; Wang, X.; Cheng, M.; Bi, L.; et al. Active or latent tuberculosis increases susceptibility to COVID-19 and disease severity. medRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Mousquer, G.T.; Peres, A.; Fiegenbaum, M. Pathology of TB/COVID-19 Co-Infection: The phantom menace. Tuberculosis 2021, 126, 102020. [Google Scholar] [CrossRef]
- Witika, B.A.; Makoni, P.A.; Mweetwa, L.L.; Ntemi, P.V.; Chikukwa, M.T.R.; Matafwali, S.K.; Mwila, C.; Mudenda, S.; Katandula, J.; Walker, R.B. Nano-Biomimetic Drug Delivery Vehicles: Potential Approaches for COVID-19 Treatment. Molecules 2020, 25, 5952. [Google Scholar] [CrossRef]
- Nkanga, C.I.; Krause, R.W.M. Encapsulation of Isoniazid-conjugated Phthalocyanine-In-Cyclodextrin-In-Liposomes Using Heating Method. Sci. Rep. 2019, 9, 11485. [Google Scholar] [CrossRef]
- Hawn, T.R.; Matheson, A.I.; Maley, S.N.; Vandal, O. Host-Directed Therapeutics for Tuberculosis: Can We Harness the Host? Microbiol. Mol. Biol. Rev. 2013, 77, 608–627. [Google Scholar] [CrossRef] [Green Version]
- Yuen, C.M.; Tolman, A.W.; Cohen, T.; Parr, J.B.; Keshavjee, S.; Becerra, M.C. Isoniazid-Resistant Tuberculosis in Children: A Systematic Review. Pediatr. Infect. Dis. J. 2013, 32, e217. [Google Scholar] [CrossRef]
- Mehanna, M.M.; Mohyeldin, S.M.; Elgindy, N.A. Respirable nanocarriers as a promising strategy for antitubercular drug delivery. J. Control. Release 2014, 187, 183–197. [Google Scholar] [CrossRef]
- Garcia Contreras, L.; Sung, J.; Ibrahim, M.; Elbert, K.; Edwards, D.; Hickey, A. Pharmacokinetics of Inhaled Rifampicin Porous Particles for Tuberculosis Treatment: Insight into Rifampicin Absorption from the Lungs of Guinea Pigs. Mol. Pharm. 2015, 12, 2642–2650. [Google Scholar] [CrossRef]
- Akbarzadeh, A.; Rezaei-Sadabady, R.; Davaran, S.; Joo, S.W.; Zarghami, N.; Hanifehpour, Y.; Samiei, M.; Kouhi, M.; Nejati-Koshki, K. Liposome: Classification, preparation, and applications. Nanoscale Res. Lett. 2013, 8, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Alexandru-flaviu, T.; Cornel, C. Macrophages Targeted Drug Delivery as a Key Therapy in Infectious Disease. Biotechnol. Mol. Biol. Nanomed. 2014, 2, 19–21. [Google Scholar]
- Muttil, P.; Kaur, J.; Kumar, K.; Yadav, A.B.; Sharma, R.; Misra, A. Inhalable microparticles containing large payload of anti-tuberculosis drugs. Eur. J. Pharm. Sci. 2007, 32, 140–150. [Google Scholar] [CrossRef]
- Parikh, R.; Dalwadi, S.; Aboti, P.; Patel, L. Inhaled microparticles of antitubercular antibiotic for in vitro and in vivo alveolar macrophage targeting and activation of phagocytosis. J. Antibiot. 2014, 67, 387–394. [Google Scholar] [CrossRef]
- Ahmad, Z.; Pandey, R.; Sharma, S.; Khuller, G.K. Alginate Nanoparticles as Antituberculosis Drug Carriers: Formulation Development, Pharmacokinetics and Therapeutic Potential. Indian J. Chest Dis. Allied Sci. 2006, 48, 171–176. [Google Scholar]
- Khawbung, J.L.; Nath, D.; Chakraborty, S. Drug resistant Tuberculosis: A review. Comp. Immunol. Microbiol. Infect. Dis. 2021, 74, 101574. [Google Scholar] [CrossRef]
- Cousins, D.; Bastida, R.; Cataldi, A.; Quse, V.; Redrobe, S.; Dow, P.; Duignan, P.; Murray, A.; Dupont, C.; Ahmed, N.; et al. Tuberculosis in seals caused by a novel member of the Mycobacterium tuberculosis complex: Mycobacterium pinnipedii sp. nov. Int. J. Syst. Evol. Microbiol. 2003, 53, 1305–1314. [Google Scholar] [CrossRef] [Green Version]
- Kanabalan, R.D.; Lee, L.J.; Lee, T.Y.; Chong, P.P.; Hassan, L.; Ismail, R.; Chin, V.K. Human tuberculosis and Mycobacterium tuberculosis complex: A review on genetic diversity, pathogenesis and omics approaches in host biomarkers discovery. Microbiol. Res. 2021, 246, 126674. [Google Scholar] [CrossRef]
- Parish, T.; Stoker, N.G. Mycobacteria: Bugs and bugbears (two steps forward and one step back). Appl. Biochem. Biotechnol. Part B Mol. Biotechnol. 1999, 13, 191–200. [Google Scholar] [CrossRef]
- BM, M. Application of stains in clinical microbiology. Biotech. Histochem. 2001, 76, 119–125. [Google Scholar] [CrossRef]
- Kumar, V.; Abbas, A.K.; Fausto, N.; Mitchell, R.N. Robbins Basic Pathology, 8th ed.; Saunders Elsevier: Philadelphia, PA, USA, 2007; pp. 516–522. [Google Scholar]
- Arch, G. Mainous III and Claire Pomoroy. In Management of Antimicrobials in Infectious Diseases, 2nd ed.; Humana Press: Totowa, NJ, USA, 2010. [Google Scholar]
- Zuñiga, J.; Torres-García, D.; Santos-Mendoza, T.; Rodriguez-Reyna, T.S.; Granados, J.; Yunis, E.J. Cellular and Humoral Mechanisms Involved in the Control of Tuberculosis. Clin. Dev. Immunol. 2012, 2012, 1–18. [Google Scholar] [CrossRef]
- Nuermberger, E.L.; Spigelman, M.K.; Yew, W.W. Current development and future prospects in chemotherapy of tuberculosis. Respirology 2010, 15, 764–778. [Google Scholar] [CrossRef] [Green Version]
- Piccini, P.; Chiappini, E.; Tortoli, E.; de Martino, M.; Galli, L. Clinical peculiarities of tuberculosis. BMC Infect. Dis. 2014, 14, S4. [Google Scholar] [CrossRef] [Green Version]
- Kumar, N.; Kumar, P.; Kumar, P.; Kumar, M.; Kumar, R. Nanotechnology: A focus on treatment of Tuberculosis. Int. J. Drug Deliv. 2011, 3, 25–42. [Google Scholar] [CrossRef]
- Eker, B.; Ortmann, J.; Migliori, G.B.; Sotgiu, G.; Muetterlein, R.; Centis, R.; Hoffmann, H.; Kirsten, D.; Schaberg, T.; Ruesch-Gerdes, S.; et al. Multidrug- and extensively drug-resistant tuberculosis, Germany. Emerg. Infect. Dis. 2008, 14, 1700–1706. [Google Scholar] [CrossRef]
- Mitnick, C.D.; Shin, S.S.; Seung, K.J.; Rich, M.L.; Atwood, S.S.; Furin, J.J.; Fitzmaurice, G.M.; Alcantara Viru, F.A.; Appleton, S.C.; Bayona, J.N.; et al. Comprehensive Treatment of Extensively Drug-Resistant Tuberculosis. N. Engl. J. Med. 2008, 359, 563–574. [Google Scholar] [CrossRef] [Green Version]
- Caminero, J.A.; Sotgiu, G.; Zumla, A.; Migliori, G.B. Best drug treatment for multidrug-resistant and extensively drug-resistant tuberculosis. Lancet Infect. Dis. 2010, 10, 621–629. [Google Scholar] [CrossRef]
- Chan, E.D.; Laurel, V.; Strand, M.J.; Chan, J.F.; Huynh, M.L.N.; Goble, M.; Iseman, M.D. Treatment and outcome analysis of 205 patients with multidrug-resistant tuberculosis. Am. J. Respir. Crit. Care Med. 2004, 169, 1103–1109. [Google Scholar] [CrossRef]
- Venturini, E.; Turkova, A.; Chiappini, E.; Galli, L.; de Martino, M.; Thorne, C. Tuberculosis and HIV co-infection in children. BMC Infect. Dis. 2014, 14, S5. [Google Scholar] [CrossRef] [Green Version]
- Eleraky, N.E.; Allam, A.; Hassan, S.B.; Omar, M.M. Nanomedicine Fight against Antibacterial Resistance: An Overview of the Recent Pharmaceutical Innovations. Pharmaceutics 2020, 12, 142. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Michalowski, C.B.; Beloqui, A. Advances in lipid carriers for drug delivery to the gastrointestinal tract. Curr. Opin. Colloid Interface Sci. 2021, 52, 101414. [Google Scholar] [CrossRef]
- Pattni, B.S.; Chupin, V.V.; Torchilin, V.P. New Developments in Liposomal Drug Delivery. Chem. Rev. 2015, 115, 10938–10966. [Google Scholar] [CrossRef]
- Euliss, L.E.; DuPont, J.A.; Gratton, S.; DeSimone, J. Imparting size, shape, and composition control of materials for nanomedicine. Chem. Soc. Rev. 2006, 35, 1095–1104. [Google Scholar] [CrossRef]
- Kumar, S.; Dutta, J.; Dutta, P.K.; Koh, J. A systematic study on chitosan-liposome based systems for biomedical applications. Int. J. Biol. Macromol. 2020, 160, 470–481. [Google Scholar] [CrossRef]
- Isalomboto Nkanga, C.; Murhimalika Bapolisi, A.; Ikemefuna Okafor, N.; Werner Maçedo Krause, R. General Perception of Liposomes: Formation, Manufacturing and Applications. In Liposomes—Advances and Perspectives; Catala, A., Ed.; IntechOpen: London, UK, 2019; pp. 1–24. [Google Scholar]
- Weber, C.; Voigt, M.; Simon, J.; Danner, A.K.; Frey, H.; Mailänder, V.; Helm, M.; Morsbach, S.; Landfester, K. Functionalization of Liposomes with Hydrophilic Polymers Results in Macrophage Uptake Independent of the Protein Corona. Biomacromolecules 2019, 20, 2989–2999. [Google Scholar] [CrossRef] [Green Version]
- Bhardwaj, P.; Tripathi, P.; Gupta, R.; Pandey, S. Niosomes: A review on niosomal research in the last decade. J. Drug Deliv. Sci. Technol. 2020, 56, 101581. [Google Scholar] [CrossRef]
- Babadi, D.; Dadashzadeh, S.; Osouli, M.; Daryabari, M.S.; Haeri, A. Nanoformulation strategies for improving intestinal permeability of drugs: A more precise look at permeability assessment methods and pharmacokinetic properties changes. J. Control. Release 2020, 321, 669–709. [Google Scholar] [CrossRef]
- Masjedi, M.; Montahaei, T. An illustrated review on nonionic surfactant vesicles (niosomes) as an approach in modern drug delivery: Fabrication, characterization, pharmaceutical, and cosmetic applications. J. Drug Deliv. Sci. Technol. 2021, 61, 102234. [Google Scholar] [CrossRef]
- Witika, B.A.; Walker, R.B. Development, manufacture and characterization of niosomes for the delivery for nevirapine. Pharmazie 2019, 74, 91–96. [Google Scholar] [CrossRef]
- Mishra, D.K.; Shandilya, R.; Mishra, P.K. Lipid based nanocarriers: A translational perspective. Nanomed. Nanotechnol. Biol. Med. 2018, 14, 2023–2050. [Google Scholar] [CrossRef]
- Kumar, R. Lipid-Based Nanoparticles for Drug-Delivery Systems. Nanocarriers Drug Deliv. 2019, 249–284. [Google Scholar] [CrossRef]
- Witika, B.A. The Development, Manufacture and Characterisation of Niosomes Intended to Deliver Nevirapine to the Brain. Master’s Thesis, Rhodes University, Grahamstown, South Africa, 2017. [Google Scholar]
- Makoni, P.A.; Kasongo, K.W.; Walker, R.B. Short Term Stability Testing of Efavirenz-Loaded Solid Lipid Nanoparticle (SLN) and Nanostructured Lipid Carrier (NLC) Dispersions. Pharmaceutics 2019, 11, 397. [Google Scholar] [CrossRef] [Green Version]
- Dumont, C.; Bourgeois, S.; Fessi, H.; Jannin, V. Lipid-based nanosuspensions for oral delivery of peptides, a critical review. Int. J. Pharm. 2018, 541, 117–135. [Google Scholar] [CrossRef]
- Barroso, L.; Viegas, C.; Vieira, J.; Ferreira-Pêgo, C.; Costa, J.; Fonte, P. Lipid-based carriers for food ingredients delivery. J. Food Eng. 2021, 295, 110451. [Google Scholar] [CrossRef]
- Mehnert, W.; Mäder, K. Solid lipid nanoparticles: Production, characterization and applications. Adv. Drug Deliv. Rev. 2012, 64, 83–101. [Google Scholar] [CrossRef]
- Abrishami, M.; Abrishami, M.; Mahmoudi, A.; Mosallaei, N.; Vakili Ahrari Roodi, M.; Malaekeh-Nikouei, B. Solid Lipid Nanoparticles Improve the Diclofenac Availability in Vitreous after Intraocular Injection. J. Drug Deliv. 2016, 2016, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Beloqui, A.; Solinís, M.Á.; Rodríguez-Gascón, A.; Almeida, A.J.; Préat, V. Nanostructured lipid carriers: Promising drug delivery systems for future clinics. Nanomed. Nanotechnol. Biol. Med. 2016, 12, 143–161. [Google Scholar] [CrossRef]
- Makoni, P.A.; Khamanga, S.M.; Walker, R.B. Muco-adhesive clarithromycin-loaded nanostructured lipid carriers for ocular delivery: Formulation, characterization, cytotoxicity and stability. J. Drug Deliv. Sci. Technol. 2020, 61, 102171. [Google Scholar] [CrossRef]
- Weber, S.; Zimmer, A.; Pardeike, J. Solid Lipid Nanoparticles (SLN) and Nanostructured Lipid Carriers (NLC) for pulmonary application: A review of the state of the art. Eur. J. Pharm. Biopharm. 2014, 86, 7–22. [Google Scholar] [CrossRef]
- Vieira, A.C.C.; Magalhães, J.; Rocha, S.; Cardoso, M.S.; Santos, S.G.; Borges, M.; Pinheiro, M.; Reis, S. Targeted macrophages delivery of rifampicin-loaded lipid nanoparticles to improve tuberculosis treatment. Nanomedicine 2017, 12, 2721–2736. [Google Scholar] [CrossRef]
- Patil, T.S.; Deshpande, A.S. Nanostructured lipid carriers-based drug delivery for treating various lung diseases: A State-of-the-Art Review. Int. J. Pharm. 2018, 547, 209–225. [Google Scholar] [CrossRef]
- Callender, S.P.; Mathews, J.A.; Kobernyk, K.; Wettig, S.D. Microemulsion utility in pharmaceuticals: Implications for multi-drug delivery. Int. J. Pharm. 2017, 526, 425–442. [Google Scholar] [CrossRef]
- Singh, Y.; Meher, J.G.; Raval, K.; Khan, F.A.; Chaurasia, M.; Jain, N.K.; Chourasia, M.K. Nanoemulsion: Concepts, development and applications in drug delivery. J. Control. Release 2017, 252, 28–49. [Google Scholar] [CrossRef]
- Rehman, F.U.; Shah, K.U.; Shah, S.U.; Khan, I.U.; Khan, G.M.; Khan, A. From nanoemulsions to self-nanoemulsions, with recent advances in self-nanoemulsifying drug delivery systems (SNEDDS). Expert Opin. Drug Deliv. 2017, 14, 1325–1340. [Google Scholar] [CrossRef]
- Sonneville-Aubrun, O.; Simonnet, J.T.; L’Alloret, F. Nanoemulsions: A new vehicle for skincare products. Adv. Colloid Interface Sci. 2004, 108–109, 145–149. [Google Scholar] [CrossRef]
- Anton, N.; Vandamme, T.F. Nano-emulsions and micro-emulsions: Clarifications of the critical differences. Pharm. Res. 2011, 28, 978–985. [Google Scholar] [CrossRef]
- Wennerström, H.; Balogh, J.; Olsson, U. Interfacial tensions in microemulsions. Colloids Surf. A Physicochem. Eng. Asp. 2006, 291, 69–77. [Google Scholar] [CrossRef]
- Gutiérrez, J.M.; González, C.; Maestro, A.; Solè, I.; Pey, C.M.; Nolla, J. Nano-emulsions: New applications and optimization of their preparation. Curr. Opin. Colloid Interface Sci. 2008, 13, 245–251. [Google Scholar] [CrossRef]
- Anton, N.; Vandamme, T.F. The universality of low-energy nano-emulsification. Int. J. Pharm. 2009, 377, 142–147. [Google Scholar] [CrossRef]
- Tadros, T.; Izquierdo, P.; Esquena, J.; Solans, C. Formation and stability of nano-emulsions. Adv. Colloid Interface Sci. 2004, 108–109, 303–318. [Google Scholar] [CrossRef]
- Ligório Fialho, S.; da Silva-Cunha, A. New vehicle based on a microemulsion for topical ocular administration of dexamethasone. Clin. Exp. Ophthalmol. 2004, 32, 626–632. [Google Scholar] [CrossRef]
- Rai, V.K.; Mishra, N.; Yadav, K.S.; Yadav, N.P. Nanoemulsion as pharmaceutical carrier for dermal and transdermal drug delivery: Formulation development, stability issues, basic considerations and applications. J. Control. Release 2018, 270, 203–225. [Google Scholar] [CrossRef]
- Agrawal, V.; Patel, R.; Patel, M.; Thanki, K.; Mishra, S. Design and evaluation of microemulsion-based efinaconazole formulations for targeted treatment of onychomycosis through transungual route: Ex vivo and nail clipping studies. Colloids Surf. B Biointerfaces 2021, 201, 111652. [Google Scholar] [CrossRef]
- Nardin, I.; Köllner, S. Successful development of oral SEDDS: Screening of excipients from the industrial point of view. Adv. Drug Deliv. Rev. 2019, 142, 128–140. [Google Scholar] [CrossRef]
- Justo, O.R.; Moraes, Â.M. Incorporation of antibiotics in liposomes designed for tuberculosis therapy by inhalation. Drug Deliv. 2003, 10, 201–207. [Google Scholar] [CrossRef]
- Vyas, S.P.; Kannan, M.E.; Jain, S.; Mishra, V.; Singh, P. Design of liposomal aerosols for improved delivery of rifampicin to alveolar macrophages. Int. J. Pharm. 2004, 269, 37–49. [Google Scholar] [CrossRef]
- Pandey, R.; Sharma, S.; Khuller, G.K. Nebulization of liposome encapsulated antitubercular drugs in guinea pigs. Int. J. Antimicrob. Agents 2004, 24, 93–94. [Google Scholar] [CrossRef]
- Gürsoy, A.; Kut, E.; Özkirimli, S. Co-encapsulation of isoniazid and rifampicin in liposomes and characterization of liposomes by derivative spectroscopy. Int. J. Pharm. 2004, 271, 115–123. [Google Scholar] [CrossRef]
- Chimote, G.; Banerjee, R. In vitro evaluation of inhalable isoniazid-loaded surfactant liposomes as an adjunct therapy in pulmonary tuberculosis. J. Biomed. Mater. Res. Part B Appl. Biomater. 2010, 94, 1–10. [Google Scholar] [CrossRef]
- Rojanarat, W.; Changsan, N.; Tawithong, E.; Pinsuwan, S.; Chan, H.K.; Srichana, T. Isoniazid proliposome powders for inhalation-preparation, characterization and cell culture studies. Int. J. Mol. Sci. 2011, 12, 4414–4434. [Google Scholar] [CrossRef] [Green Version]
- Manca, M.L.; Sinico, C.; Maccioni, A.M.; Diez, O.; Fadda, A.M.; Manconi, M. Composition influence on pulmonary delivery of rifampicin liposomes. Pharmaceutics 2012, 4, 590–606. [Google Scholar] [CrossRef]
- Patil-Gadhe, A.A.; Kyadarkunte, A.Y.; Pereira, M.; Jejurikar, G.; Patole, M.S.; Risbud, A.; Pokharkar, V.B. Rifapentine-proliposomes for inhalation: In vitro and in vivo toxicity. Toxicol. Int. 2014, 21, 275–282. [Google Scholar] [CrossRef] [Green Version]
- Singh, C.; Koduri, L.V.S.K.; Singh, A.; Suresh, S. Novel potential for optimization of antitubercular therapy: Pulmonary delivery of rifampicin lipospheres. Asian J. Pharm. Sci. 2015, 10, 549–562. [Google Scholar] [CrossRef] [Green Version]
- Patil, J.; Devi, V.; Devi, K.; Sarasija, S. A novel approach for lung delivery of rifampicin-loaded liposomes in dry powder form for the treatment of tuberculosis. Lung India 2015, 32, 331–338. [Google Scholar] [CrossRef]
- Bhardwaj, A.; Grobler, A.; Rath, G.; Kumar Goyal, A.; Kumar Jain, A.; Mehta, A. Pulmonary Delivery of Anti-Tubercular Drugs Using Ligand Anchored pH Sensitive Liposomes for the Treatment of Pulmonary Tuberculosis. Curr. Drug Deliv. 2016, 13, 909–922. [Google Scholar] [CrossRef]
- Kaur, M.; Garg, T.; Narang, R.K. A review of emerging trends in the treatment of tuberculosis. Artif. Cells Nanomed. Biotechnol. 2016, 44, 478–484. [Google Scholar] [CrossRef]
- Poerio, N.; Bugli, F.; Taus, F.; Santucci, M.B.; Rodolfo, C.; Cecconi, F.; Torelli, R.; Varone, F.; Inchingolo, R.; Majo, F.; et al. Liposomes loaded with bioactive lipids enhance antibacterial innate immunity irrespective of drug resistance. Sci. Rep. 2017, 7, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Nkanga, C.I.; Krause, R.W.; Noundou, X.S.; Walker, R.B. Preparation and characterization of isoniazid-loaded crude soybean lecithin liposomes. Int. J. Pharm. 2017, 526, 466–473. [Google Scholar] [CrossRef]
- Tian, M.; Zhou, Z.; Tan, S.; Fan, X.; Li, L.; Ullah, N. Formulation in DDA-MPLA-TDB liposome enhances the immunogenicity and protective efficacy of a DNA vaccine against Mycobacterium tuberculosis infection. Front. Immunol. 2018, 9, 1–13. [Google Scholar] [CrossRef]
- Nkanga, C.I.; Walker, R.B.; Krause, R.W. pH-Dependent release of isoniazid from isonicotinic acid (4-hydroxy-benzylidene)-hydrazide loaded liposomes. J. Drug Deliv. Sci. Technol. 2018, 45, 264–271. [Google Scholar] [CrossRef]
- Miretti, M.; Juri, L.; Cosiansi, M.C.; Tempesti, T.C.; Baumgartner, M.T. Antimicrobial Effects of ZnPc Delivered into Liposomes on Multidrug Resistant (MDR)-Mycobacterium tuberculosis. ChemistrySelect 2019, 4, 9726–9730. [Google Scholar] [CrossRef]
- Nkanga, C.I.; Noundou, X.S.; Walker, R.B.; Krause, R.W.M. Co-encapsulation of Rifampicin and Isoniazid in Crude Soybean Lecithin Liposomes. S. Afr. J. Chem. 2019, 72, 80–87. [Google Scholar] [CrossRef] [Green Version]
- Nkanga, C.I.; Roth, M.; Walker, R.B.; Noundou, X.S.; Krause, R.W.M. Co-loading of isoniazid-grafted phthalocyanine-in-cyclodextrin and rifampicin in crude soybean lecithin liposomes: Formulation, spectroscopic and biological characterization. J. Biomed. Nanotechnol. 2020, 16, 14–28. [Google Scholar] [CrossRef]
- Jain, C.P.; Vyas, S.P. Preparation and characterization of niosomes containing rifampicin for lung targeting. J. Microencapsul. 1995, 12, 401–407. [Google Scholar] [CrossRef]
- Karki, R.; Mamatha, G.C.; Subramanya, G.; Udupa, N. Preparation, characterization and tissue disposition of niosomes containing isoniazid. Rasayan J. Chem. 2008, 1, 224–227. [Google Scholar]
- El-Ridy, M.S.; Abdelbary, A.; Nasr, E.A.; Khalil, R.M.; Mostafa, D.M.; El-Batal, A.I.; Abd El-Alim, S.H. Niosomal encapsulation of the antitubercular drug, pyrazinamide. Drug Dev. Ind. Pharm. 2011, 37, 1110–1118. [Google Scholar] [CrossRef]
- Singh, G.; Dwivedi, H.; Saraf, S.K.; Saraf, S.A. Niosomal delivery of isoniazid—Development and characterization. Trop. J. Pharm. Res. 2011, 10, 203–210. [Google Scholar] [CrossRef]
- Mehta, S.K.; Jindal, N.; Kaur, G. Quantitative investigation, stability and in vitro release studies of anti-TB drugs in Triton niosomes. Colloids Surf. B Biointerfaces 2011, 87, 173–179. [Google Scholar] [CrossRef]
- Mehta, S.K.; Jindal, N. Formulation of Tyloxapol niosomes for encapsulation, stabilization and dissolution of anti-tubercular drugs. Colloids Surf. B Biointerfaces 2013, 101, 434–441. [Google Scholar] [CrossRef]
- El-Ridy, M.S.; Yehia, S.A.; Kassem, M.A.E.M.; Mostafa, D.M.; Nasr, E.A.; Asfour, M.H. Niosomal encapsulation of ethambutol hydrochloride for increasing its efficacy and safety. Drug Deliv. 2015, 22, 21–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pandey, R.; Khuller, G.K. Solid lipid particle-based inhalable sustained drug delivery system against experimental tuberculosis. Tuberculosis 2005, 85, 227–234. [Google Scholar] [CrossRef]
- Aboutaleb, E.; Noori, M.; Gandomi, N.; Atyabi, F.; Fazeli, M.R.; Jamalifar, H.; Dinarvand, R. Improved antimycobacterial activity of rifampin using solid lipid nanoparticles. Int. Nano Lett. 2012, 2, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Maretti, E.; Rossi, T.; Bondi, M.; Croce, M.A.; Hanuskova, M.; Leo, E.; Sacchetti, F.; Iannuccelli, V. Inhaled Solid Lipid Microparticles to target alveolar macrophages for tuberculosis. Int. J. Pharm. 2014, 462, 74–82. [Google Scholar] [CrossRef]
- Maretti, E.; Rustichelli, C.; Romagnoli, M.; Balducci, A.G.; Buttini, F.; Sacchetti, F.; Leo, E.; Iannuccelli, V. Solid Lipid Nanoparticle assemblies (SLNas) for an anti-TB inhalation treatment—A Design of Experiments approach to investigate the influence of pre-freezing conditions on the powder respirability. Int. J. Pharm. 2016, 511, 669–679. [Google Scholar] [CrossRef]
- Gaspar, D.P.; Faria, V.; Gonçalves, L.M.D.; Taboada, P.; Remuñán-López, C.; Almeida, A.J. Rifabutin-loaded solid lipid nanoparticles for inhaled antitubercular therapy: Physicochemical and in vitro studies. Int. J. Pharm. 2016, 497, 199–209. [Google Scholar] [CrossRef]
- Vieira, A.C.C.; Chaves, L.L.; Pinheiro, S.; Pinto, S.; Pinheiro, M.; Lima, S.C.; Ferreira, D.; Sarmento, B.; Reis, S. Mucoadhesive chitosan-coated solid lipid nanoparticles for better management of tuberculosis. Int. J. Pharm. 2018, 536, 478–485. [Google Scholar] [CrossRef] [PubMed]
- Nemati, E.; Mokhtarzadeh, A.; Panahi-Azar, V.; Mohammadi, A.; Hamishehkar, H.; Mesgari-Abbasi, M.; Ezzati Nazhad Dolatabadi, J.; de la Guardia, M. Ethambutol-Loaded Solid Lipid Nanoparticles as Dry Powder Inhalable Formulation for Tuberculosis Therapy. AAPS PharmSciTech 2019, 20, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Lin, Q.; Guo, L.; Fu, Y.; Han, J.; Ke, H.; Sun, X.; Gong, T.; Zhang, Z. Rifampicin loaded mannosylated cationic nanostructured lipid carriers for alveolar macrophage-specific delivery. Pharm. Res. 2015, 32, 1741–1751. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.; Roy, S.; Bhaumik, K.N.; Pillai, J. Mechanisms of the effectiveness of lipid nanoparticle formulations loaded with anti-tubercular drugs combinations toward overcoming drug bioavailability in tuberculosis. J. Drug Target. 2020, 28, 55–69. [Google Scholar] [CrossRef] [PubMed]
- Mehta, S.K.; Kaur, G.; Bhasin, K.K. Analysis of Tween based microemulsion in the presence of TB drug rifampicin. Colloids Surf. B Biointerfaces 2007, 60, 95–104. [Google Scholar] [CrossRef]
- Kaur, G.; Mehta, S.K. Probing location of anti-TB drugs loaded in brij 96 microemulsions using thermoanalytical and photophysical approach. J. Pharm. Sci. 2014, 103, 937–944. [Google Scholar] [CrossRef]
- Kaur, G.; Mehta, S.K.; Kumar, S.; Bhanjana, G.; Dilbaghi, N. Coencapsulation of Hydrophobic and Hydrophilic Antituberculosis Drugs in Synergistic Brij 96 Microemulsions: A Biophysical Characterization. J. Pharm. Sci. 2015, 104, 2203–2212. [Google Scholar] [CrossRef]
- Hussain, A.; Shakeel, F.; Singh, S.K.; Alsarra, I.A.; Faruk, A.; Alanazi, F.K.; Peter Christoper, G.V. Solidified SNEDDS for the oral delivery of rifampicin: Evaluation, proof of concept, in vivo kinetics, and in silico GastroPlusTM simulation. Int. J. Pharm. 2019, 566, 203–217. [Google Scholar] [CrossRef]
- Bulbake, U.; Doppalapudi, S.; Kommineni, N.; Khan, W. Liposomal Formulations in Clinical Use: An Updated Review. Pharmaceutics 2017, 9, 12. [Google Scholar] [CrossRef]
- Lila, A.S.A.; Ishida, T. Liposomal delivery systems: Design optimization and current applications. Biol. Pharm. Bull. 2017, 40, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Rudokas, M.; Najlah, M.; Alhnan, M.A.; Elhissi, A. Liposome Delivery Systems for Inhalation: A Critical Review Highlighting Formulation Issues and Anticancer Applications. Med. Princ. Pract. 2016, 25, 60–72. [Google Scholar] [CrossRef]
- Kaul, A.; Chaturvedi, S.; Attri, A.; Kalra, M.; Mishra, A.K. Targeted theranostic liposomes: Rifampicin and ofloxacin loaded pegylated liposomes for theranostic application in mycobacterial infections. RSC Adv. 2016, 6, 28919–28926. [Google Scholar] [CrossRef]
- Drummond, D.C.; Zignani, M.; Leroux, J.C. Current status of pH-sensitive liposomes in drug delivery. Prog. Lipid Res. 2000, 39, 409–460. [Google Scholar] [CrossRef]
- Karanth, H.; Murthy, R.S.R. pH-Sensitive liposomes-principle and application in cancer therapy. J. Pharm. Pharmacol. 2007, 59, 469–483. [Google Scholar] [CrossRef] [PubMed]
- Yokota, D.; Moraes, M.; Pinho, S.C. Characterization of lyophilized liposomes produced with non-purified soy lecithin: A case study of casein hydrolysate microencapsulation. Brazilian J. Chem. Eng. 2012, 29, 325–335. [Google Scholar] [CrossRef] [Green Version]
- Nkanga, C.I.; Krause, R.W.M. Conjugation of isoniazid to a zinc phthalocyanine via hydrazone linkage for pH-dependent liposomal controlled release. Appl. Nanosci. 2018, 8, 1313–1323. [Google Scholar] [CrossRef]
- Kelly, C.; Jefferies, C.; Cryan, S.-A. Targeted Liposomal Drug Delivery to Monocytes and Macrophages. J. Drug Deliv. 2011, 2011, 1–11. [Google Scholar] [CrossRef]
- Bermudez, L.E.; Goodman, J. Mycobacterium tuberculosis invades and replicates within type II alveolar cells. Infect. Immun. 1996, 64, 1400–1406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinheiro, M.; Ribeiro, R.; Vieira, A.; Andrade, F.; Reis, S. Design of a nanostructured lipid carrier intended to improve the treatment of tuberculosis. Drug Des. Devel. Ther. 2016, 10, 2467–2475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carneiro, S.P.; Carvalho, K.V.; de Oliveira Aguiar Soares, R.D.; Carneiro, C.M.; de Andrade, M.H.G.; Duarte, R.S.; dos Santos, O.D.H. Functionalized rifampicin-loaded nanostructured lipid carriers enhance macrophages uptake and antimycobacterial activity. Colloids Surf. B Biointerfaces 2019, 175, 306–313. [Google Scholar] [CrossRef]
- Suciati, T.; Rachmawati, P.; Soraya, E.; Mahardhika, A.B.; Satrialdi, R.; Hartarti, R.; Anggadiredja, K. A novel acemannan-chitosan modified lipid nanoparticles as intracellular delivery vehicles of antibiotic. J. Appl. Pharm. Sci. 2018, 8, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Kanwar, R.; Gradzielski, M.; Prevost, S.; Appavou, M.; Mehta, S. Experimental validation of biocompatible nanostructured lipid carriers of sophorolipid: Optimization, characterization and in-vitro evaluation. Colloids Surf. B. Biointerfaces 2019, 181, 845–855. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.; Roy, S.; Nath Bhaumik, K.; Kshetrapal, P.; Pillai, J. Comparative study of oral lipid nanoparticle formulations (LNFs) for chemical stabilization of antitubercular drugs: Physicochemical and cellular evaluation. Artif. Cells Nanomed. Biotechnol. 2018, 46, 540–558. [Google Scholar] [CrossRef] [Green Version]
- Da Silva, P.B.; Bonifácio, B.; Frem, R.; Godoy Netto, A.; Mauro, A.; Ferreira, A.; Lopes Ede, O.; Raddi, M.; Bauab, T.; Pavan, F.; et al. A Nanostructured Lipid System as a Strategy to Improve the in Vitro Antibacterial Activity of Copper(II) Complexes. Molecules 2015, 20, 22534–22545. [Google Scholar] [CrossRef]
- Sato, M.R.; Junior, J.A.O.; Machado, R.T.; de Souza, P.C.; Campos, D.L.; Pavan, F.R.; da Silva, P.B.; Chorilli, M. Nanostructured lipid carriers for incorporation of copper(II) complexes to be used against Mycobacterium tuberculosis. Drug Des. Dev. Ther. 2017, 11, 909–921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, M.R.; Oshiro-Junior, J.A.; Souza, P.C.; Campos, D.L.; Pereira-da-Silva, M.A.; Pavan, F.R.; da Silva, P.B.; Chorilli, M. Copper(II) complex-loaded castor oil-based nanostructured lipid carriers used against Mycobacterium tuberculosis: Development, characterisation, in vitro and in vivo biological assays. Pharmazie 2019, 74, 715–720. [Google Scholar] [CrossRef]
- Mehta, S.K.; Kaur, G.; Bhasin, K.K. Incorporation of antitubercular drug isoniazid in pharmaceutically accepted microemulsion: Effect on microstructure and physical parameters. Pharm. Res. 2008, 25, 227–236. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Vehicles | Composition | Drug Molecules | System Specificity and Functionality | Reference |
|---|---|---|---|---|
| Liposomes | Phosphatidylcholine, cholesterol | Isoniazid, pyrazinamide, rifampicin, ethionamide, streptomycin | Attempt for multiple drug encapsulation in liposomes; co-encapsulation of isoniazid and pyrazinamide was successful whereas rifampicin, ethionamide, and streptomycin was not substantial | [72] |
| Egg phosphatidylcholine, cholesterol, maleylated bovine serum albumin, O-steroyl amylopectin/dicetylphosphate | Rifampicin | Enhanced drug concentration in alveolar macrophages, a higher clearance rate of M. smegmatis in the rat macrophage, improved efficiency with aerosol formulation | [73] | |
| Phosphatidylcholine, cholesterol | Rifampicin Isoniazid | Sustained drug release in alveolar macrophages by pulmonary administration to guinea pigs | [74] | |
| Egg yolk phosphatidylcholine type XI-E, dipalmitoylphosphatidylcholine, cholesterol | Rifampicin Isoniazid | Co-loading increased the encapsulation and extended the release of both drugs | [75] | |
| Dipalmitoyl phosphatidylcholine (DPPC) | Isoniazid | Deep lung deposition (27%), effective delivery of isoniazid, lung surfactant mimic action | [76] | |
| Soybean phosphatidylcholine, cholesterol, mannitol | Isoniazid | Proliposomes with attractive flowability, powder performance, and promising biological effect | [77] | |
| Soy phosphatidylcholine/hydrogenated derivative, cholesterol, oleic acid | Rifampicin | Good cellular uptake and less toxicity towards alveolar epithelium for the formulation without oleic acid | [78] | |
| Hydrogenated soy phosphatidylcholine, cholesterol | Rifampetine | Antimicrobial efficacy without cytotoxicity in A549 cells | [79] | |
| Phospholipid (Lipoid S-75), sulfphobutyl ether P-cyclodextrin, vitamin C | Rifampicin | Good flowability, aerodynamic diameter for pulmonary delivery, good in vitro antitubercular activity | [80] | |
| Soy lecithin, cholesterol | Rifampicin | Controlled and sustained release behavior, better pharmacokinetic profile | [81] | |
| 4-aminophenyl-a-D mannopyranoside as a macrophage-targeting agent. Cholesteryl hemisuccinate (CHEMS) and 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE) | Isoniazid Ciprofloxacin | pH stimuli release optimal at macrophage acidic conditions, high alveolar macrophage uptake, and pulmonary delivery of drug achieved | [82] | |
| Hydrogenated phosphatidylcholine from soybean, cholesterol, α-tocopherol, and folate-MPEG2000-DSPE | Rifampicin Ofloxacin | Efficient antimicrobial activity in vitro and in murine models, enhanced pharmacokinetic profiles, macrophage-targeting activity, and particulates endowed with radiolabeling properties for visualization | [83] | |
| D-erythro-sphingosine-1-phosphate (S1P); lysobisphosphatidic acid (LBPA) or arachidonic acid, L-α-phosphatidylserine | Phosphatidic acid Phosphatidylinositol 3-phosphate Phosphatidylinositol 5-phosphate | Increased intracellular death of Mycobacteria BCG and Pseudomonas aeruginosa by phagosome acidification and ROS generation | [84] | |
| Crude soybean lecithin and cholesterol | Isoniazid | Crude soybean lecithin liposomes exhibited much higher encapsulation efficiency for isoniazid than purified soybean lecithin liposomes, introducing the crude product for cost-effective drug encapsulation | [85] | |
| Dimethyldioctadecylammonium (DDA), monophosphoryl lipid A (MPLA), trehalose 6,6′-dibehenate (TDB) | DNA vaccine | Slow and prolonged release of DNA, enhanced and persistent protection against TB, increased storage stability of the vaccine | [86] | |
| Crude soybean lecithin and cholesterol | Isonicotinic acid (4-hydroxy-benzylidene)-hydrazide | Crude soybean lecithin liposomes showed high encapsulation efficiency for hydrazone–drug conjugates and controlled release of isoniazid at different pH | [87] | |
| Crude soybean lecithin | Isoniazid-grafted zinc phthalocyanine | The conjugation of chemotherapeutics to phthalocyanines as a potential strategy for liposomal controlled release was successfully established | [87] | |
| Dipalmitoylphosphatidylcholine, cholesterol | Zinc phthalocyanine | Inactivation of sensible and multidrug-resistant strains of M. tuberculosis by photodynamic activity | [88] | |
| Crude soybean lecithin | Inclusion complexes of cyclodextrin with isoniazid-grafted zinc phthalocyanine | The use of cyclodextrin complexation to facilitate liposomal encapsulation of hydrophobic compounds under organic, solvent-free conditions was introduced | [9] | |
| Crude soybean lecithin | Rifampicin Isoniazid | The feasibility of using crude soybean lecithin for preparation of combination products for liposomal dual delivery was demonstrated | [89] | |
| Crude soybean lecithin | Rifampicin and isoniazid-grafted zinc phthalocyanine | The prepared liposomes demonstrated pH-dependent controlled dual delivery of the two drugs, good biocompatibility, and marked uptake by the lung fibroblasts and epithelial cells | [90] | |
| Niosomes | Span® 85, cholesterol | Rifampicin | Good distribution with lung affinity of approximately 65% due to the controlled size of particles | [91] |
| Span® 60, cholesterol | Isoniazid | Low accumulation of drugs in visceral organs (lung, kidney, spleen) | [92] | |
| Span® 60/85, cholesterol, dicetyl phosphate/stearyl amine | Pyrazinamide | Improved drug efficacy in guinea pigs infected with M. tuberculosis | [93] | |
| Span® 20/60, cholesterol, di-cetylphosphate | Isoniazid | Prolonged delivery in treated sites and high macrophage uptake of negatively charged particles | [94] | |
| Triton X 100, polyethylene glycol (PEG) 2000, Span® 80 | Rifampicin Isoniazid Pyrazinamide | Stability and compatibility of drugs in niosomes, release of rifampicin and isoniazid by a Fickian mechanism, and a non-Fickian release observed for pyrazinamide | [95] | |
| Tyloxapol, PEG 2000 | Rifampicin Isoniazid Pyrazinamide | Stability of the formulation, isoniazid released by a Fickian diffusion, rifampicin and pyrazinamide by a non-Fickian mechanism | [96] | |
| Span® 60/85, cholesterol, dicetyl phosphate/stearyl amine | Ethambutol | Good stability for neutral and positively charged niosomes | [97] | |
| Solid lipid nanoparticles | Stearic acid | Rifampicin Isoniazid Pyrazinamide | Good aerodynamic size for broncho-alveolar delivery, bioavailability, greater activity in M. tuberculosis infected guinea pigs, and no hepatotoxicity induced | [98] |
| Cetyl palmitate, Tween® 80/Poloxamer 188 | Rifampin | Improved antitubercular activity and sustained release of rifampin | [99] | |
| Stearic acid, sodium taurocholate | Rifampicin | Appropriate aerodynamic size for pulmonary delivery to alveolar epithelium, with good respirability fraction (>50%) and activity against Bacillus subtilis strains | [100,101] | |
| Glyceryl dibehenate/glyceryl tristearate, Tween® 80 | Rifabutin | The macrophage uptake of 46% for nanoparticles made with glyceryl dibehenate and low cytotoxicity effect on lung cell lines | [102] | |
| Cetyl palmitate, chitosan | Rifampicin | Higher in vitro mucoadhesive properties and permeability in alveolar epithelial cells | [103] | |
| Comptitol, Tween ® 80 | Ethambutol | Biocompatible, non-toxic particles, dry powder inhaler suitable for pulmonary delivery | [104] | |
| Nanostructured lipid carriers | Polyoxyethylene 40 stearate, caprylic/capric triglyceride, and polyoxyl 40 hydrogenated castor oil, Poloxamer 407, cetyltrimethylammonium bromide | Rifampicin | Improved uptake of drug in alveolar macrophages | [105] |
| Precirol®ATO 5, polysorbate 60, miglyol-812, mannose | Rifampicin | Efficient uptake by bone-marrow-derived macrophages and decrease in the intracellular growth of the mycobacteria | [57] | |
| Lipoid S-75, Tween 80, Poloxamer 188, Precirol® ATO-5, glyceryl distearate, squalene | Rifampicin | Enhancement of pharmacokinetic parameters and improvement of drug bioavailability | [106] | |
| Emulsions | Oleic acid, phosphate buffer, Tween 80, ethanol | Rifampicin | Controlled release of rifampicin achieved | [107] |
| Oleic acid, phosphate buffer, Tween 80, ethanol | Isoniazid | Stable formulation, isoniazid release by a non-Fickian release mechanism | [107] | |
| Ethyl oleate, Brij 96, Butanol | Rifampicin Isoniazid Pyrazinamide | Isoniazid and rifampicin located at the interface toward oil side, pyrazinamide remained in free water Isoniazid and pyrazinamide released by Fickian mechanism and rifampicin exhibited anomalous release | [108,109] | |
| Capmul MCM C8, Labrasol, Cremophor-EL | Rifampicin | Intestinal permeation of rifampicin facilitated, improved pharmacokinetic profile | [110] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Buya, A.B.; Witika, B.A.; Bapolisi, A.M.; Mwila, C.; Mukubwa, G.K.; Memvanga, P.B.; Makoni, P.A.; Nkanga, C.I. Application of Lipid-Based Nanocarriers for Antitubercular Drug Delivery: A Review. Pharmaceutics 2021, 13, 2041. https://doi.org/10.3390/pharmaceutics13122041
Buya AB, Witika BA, Bapolisi AM, Mwila C, Mukubwa GK, Memvanga PB, Makoni PA, Nkanga CI. Application of Lipid-Based Nanocarriers for Antitubercular Drug Delivery: A Review. Pharmaceutics. 2021; 13(12):2041. https://doi.org/10.3390/pharmaceutics13122041
Chicago/Turabian StyleBuya, Aristote B., Bwalya A. Witika, Alain M. Bapolisi, Chiluba Mwila, Grady K. Mukubwa, Patrick B. Memvanga, Pedzisai A. Makoni, and Christian I. Nkanga. 2021. "Application of Lipid-Based Nanocarriers for Antitubercular Drug Delivery: A Review" Pharmaceutics 13, no. 12: 2041. https://doi.org/10.3390/pharmaceutics13122041