
New Antifungal Compound: Impact of Cosolvency, Micellization and Complexation on Solubility and Permeability Processes


Abstract
:1. Introduction
2. Materials and Methods
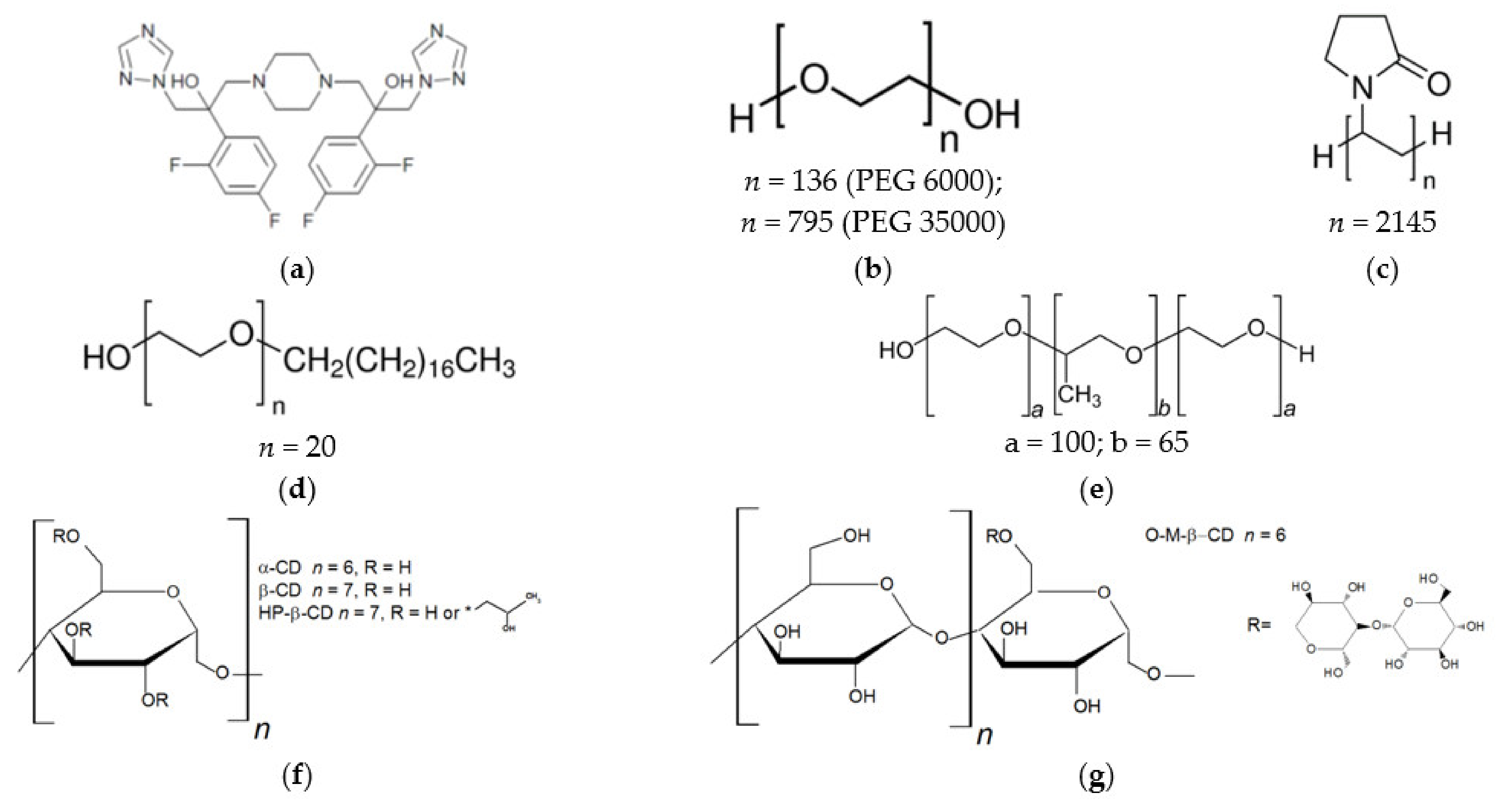
2.1. Materials
2.2. Methods
2.2.1. Phase Solubility Study
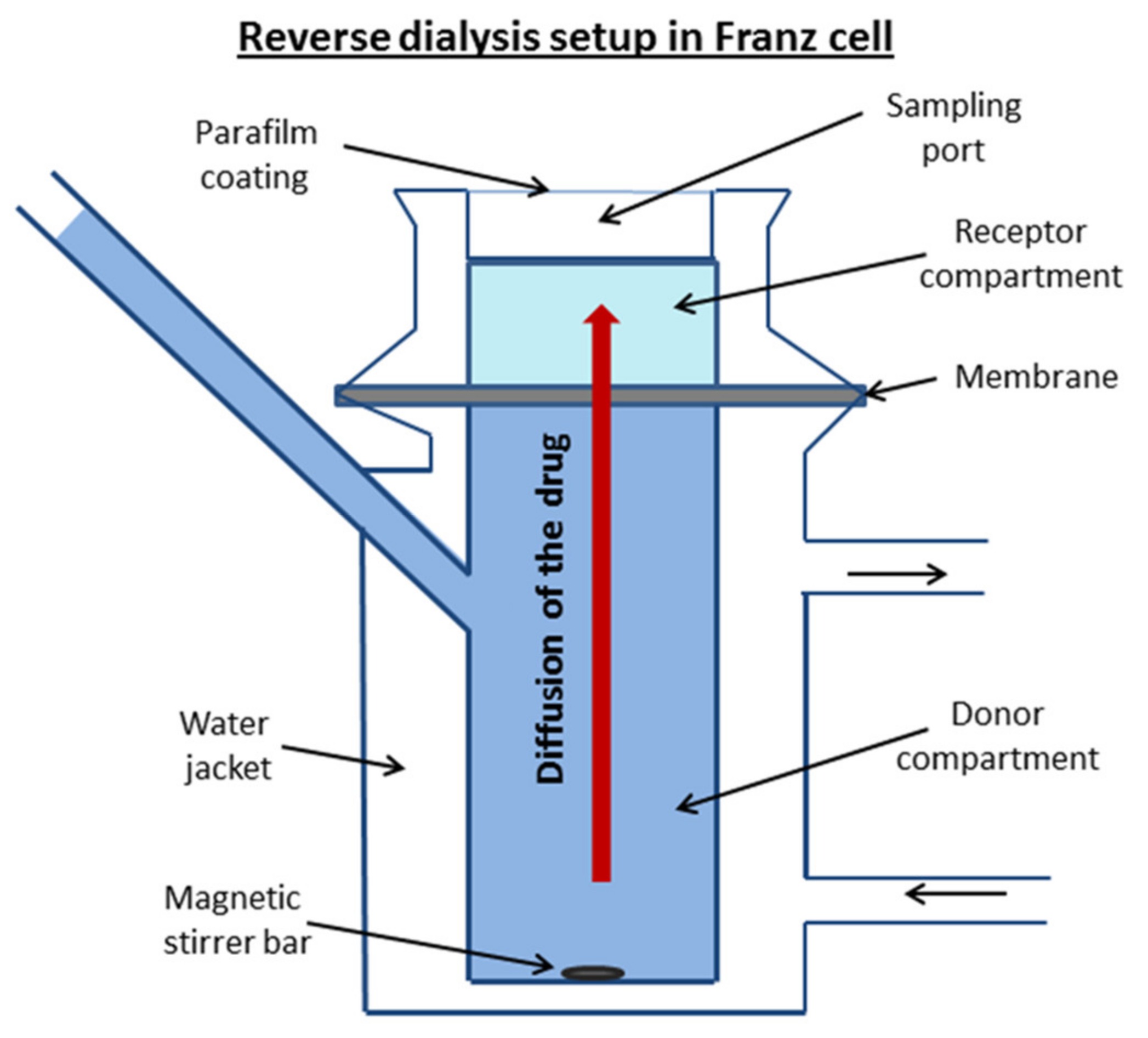
2.2.2. In Vitro Permeability Assay
3. Results
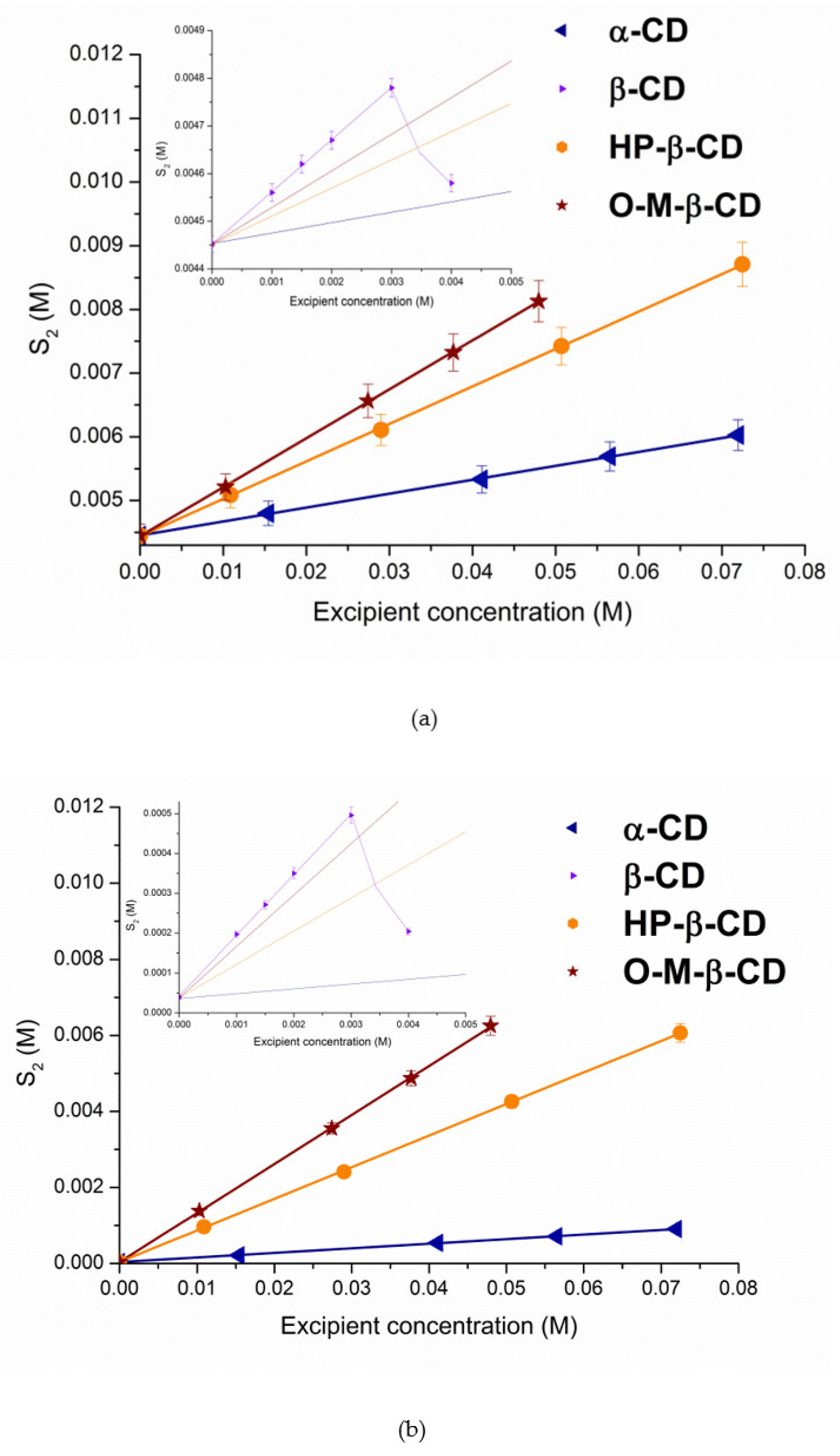
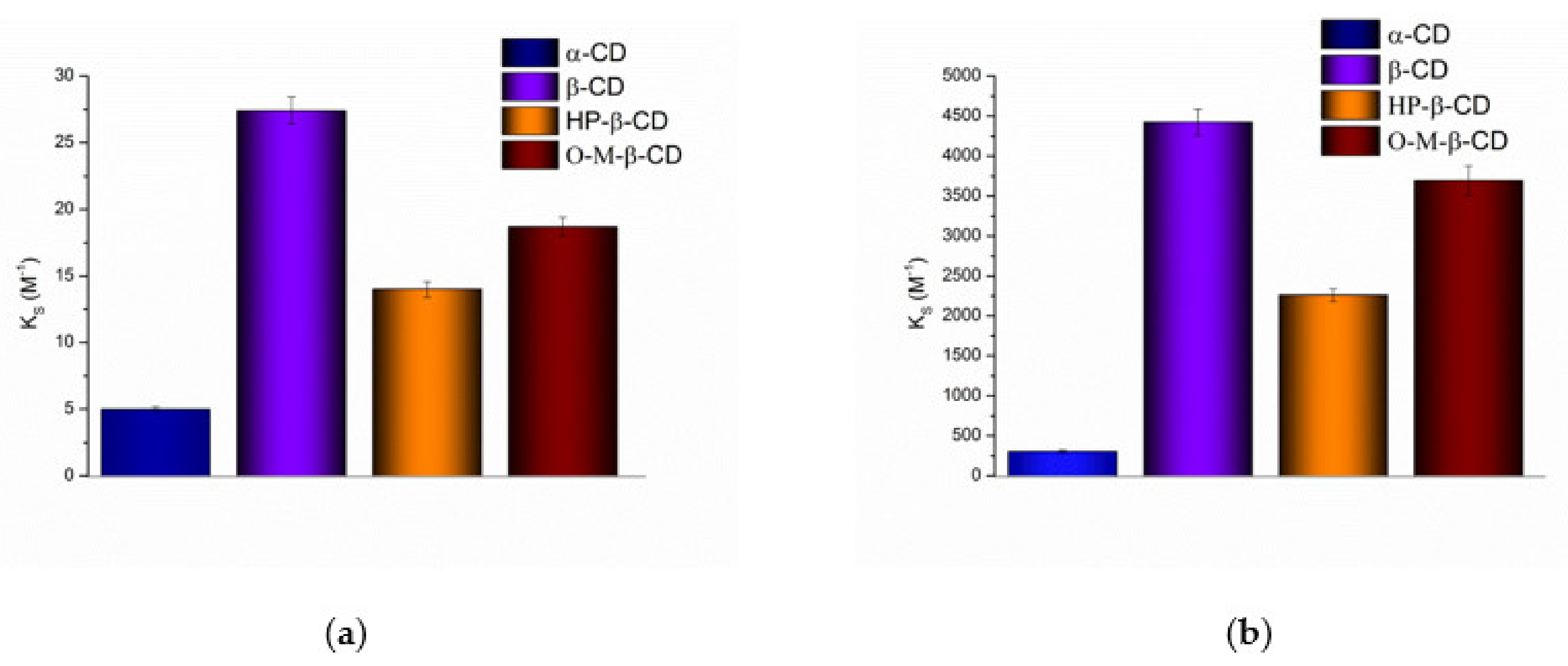
3.1. Phase Solubility Study
3.1.1. Solubilization in Cyclodextrins Solutions
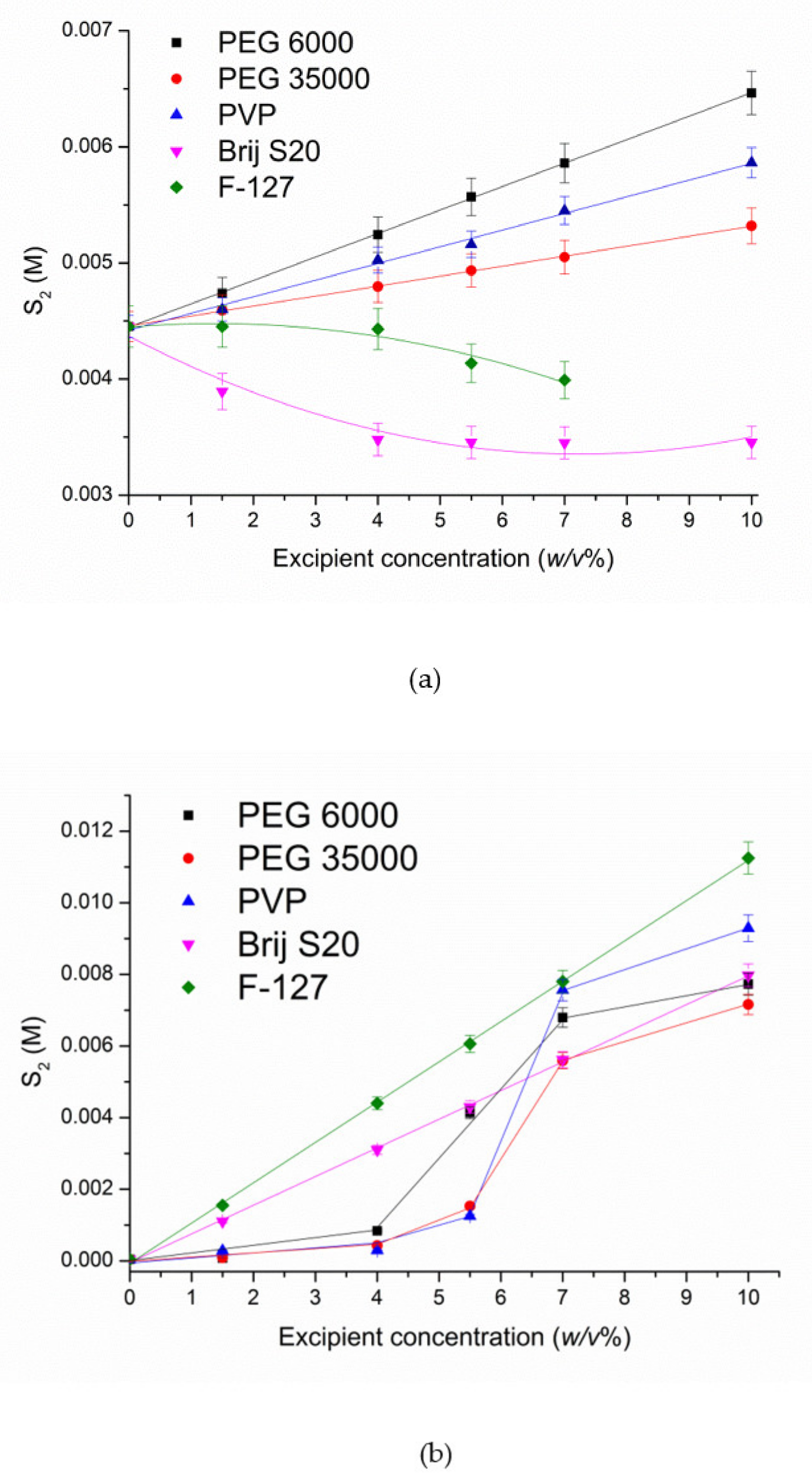
3.1.2. Solubilization by Biopolymers
3.1.3. Solubilization with Non-Ionic Surfactants
3.2. Permeability Study
3.3. Solubilization, Micelle Formation and Complexation Processes: Thermodynamic Considerations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bhogireddy, R.; Krishnamurthy, V.; Jabaris, S.S.L.; Pullaiah, C.P.; Manohar, S. Is Mucormycosis an inevitable complication of Covid-19 in India? Braz. J. Infect. Dis. 2021, 25, 101597. [Google Scholar] [CrossRef] [PubMed]
- Kharb, R.; Sharma, P.C.; Yar, M.S. Pharmacological significance of triazole scaffold. J. Enzym. Inhib. Med. Chem. 2010, 26, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Inkmann, E.; Holzgrabe, U. 1H and 13C nuclear magnetic resonance studies of the sites of protonation in itraconazole and fluconazole. J. Pharm. Biomed. Anal. 1999, 20, 297–307. [Google Scholar] [CrossRef]
- Bozorov, K.; Zhao, J.; Aisa, H.A. 1,2,3-Triazole-containing hybrids as leads in medicinal chemistry: A recent overview. Bioorg. Med. Chem. 2019, 27, 3511–3531. [Google Scholar] [CrossRef] [PubMed]
- Carrillo-Muñoz, A.J.; Giusiano, G.; Ezkurra, P.A.; Quindós, G. Antifungal agents: Mode of action inyeast cells. Rev. Esp. Quimioter. 2006, 19, 130–139. [Google Scholar] [PubMed]
- Takagi, T.; Ramachandran, C.; Bermejo, M.; Yamashita, S.; Yu, L.X.; Amidon, G.L. A provisional biopharmaceutical classi-fication of the top 200 oral drug products in the United States, Great Britain, Spain, and Japan. Mol. Pharm. 2006, 3, 631–643. [Google Scholar] [CrossRef]
- Lipinski, C.A. Drug-like properties and the causes of poor solubility and poor permeability. J. Pharmacol. Toxicol. Methods 2000, 44, 235–249. [Google Scholar] [CrossRef]
- Basavaraj, S.; Betageri, G.V. Can formulation and drug delivery reduce attrition during drug discovery and development—Review of feasibility, benefits and challenges. Acta Pharm. Sin. B 2014, 4, 3–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, L.; Liu, G.; Ma, J.; Wang, X.; Zhou, L.; Li, X. Drug nanocrystals: In vivo performances. J. Control. Release 2012, 160, 418–430. [Google Scholar] [CrossRef]
- Semalty, A. Cyclodextrin and phospholipid complexation in solubility and dissolution enhancement: A critical and me-ta-analysis. Expert Opin. Drug Deliv. 2014, 11, 1255–1272. [Google Scholar] [CrossRef] [PubMed]
- Geng, N.; Chen, J.-M.; Li, Z.-J.; Jiang, L.; Lu, T.-B. Approach of Cocrystallization to Improve the Solubility and Photostability of Tranilast. Cryst. Growth Des. 2013, 13, 3546–3553. [Google Scholar] [CrossRef]
- Sarfraz, R.M.; Bashir, S.; Mahmood, A.; Ahsan, H.; Riaz, H.; Raza, H.; Rashid, Z.; Raza, S.A.; Abrar, M.A.; Abbas, K.; et al. Application of various polymers and polymers based techniques used to improve solubility of poorly water soluble drugs: A review. Acta Pol. Pharm. 2017, 74, 347–356. [Google Scholar]
- Lu, Y.; Park, K. Polymeric micelles and alternative nanonized delivery vehicles for poorly soluble drugs. Int. J. Pharm. 2013, 453, 198–214. [Google Scholar] [CrossRef] [Green Version]
- Carneiro, S.; Duarte, F.I.C.; Heimfarth, L.; Quintans, S.; Quintans-Júnior, L.J.; Veiga Júnior, V.; de Neves Lima, Á.A. Cy-clodextrin–Drug Inclusion Complexes: In Vivo and In Vitro Approaches. Int. J. Mol. Sci. 2019, 20, 642–665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Irie, T.; Uekama, K. Pharmaceutical Applications of Cyclodextrins. III. Toxicological Issues and Safety Evaluation. J. Pharm. Sci. 1997, 86, 147–162. [Google Scholar] [CrossRef] [PubMed]
- Ono, N.; Rima, H.A.; Irayama, F.H.; Uekama, K. A moderate interaction of maltosyl-α-cyclodextrin with caco-2 cells in comparison with the parent cyclodextrin. Biol. Pharm. Bull. 2001, 24, 395–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yasmin, N.; Ishitsuka, Y.; Fukaura, M.; Yamada, Y.; Nakahara, S.; Ishii, A.; Kondo, Y.; Takeo, T.; Nakagata, N.; Motoyama, K.; et al. In Vitro and In Vivo Evaluation of 6-O-α-Maltosyl-β-Cyclodextrin as a Potential Therapeutic Agent Against Niemann-Pick Disease Type C. Int. J. Mol. Sci. 2019, 20, 1152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Barghouthi, M.I.; Masoud, N.A.; Al-Kafawein, J.K.; Abdoh, A.A. Inclusion complexation of Itraconazole with β-and 2-hydroxypropyl-β-cyclodextrins in aqueous solutions. Russ. J. Phys. Chem. 2006, 80, 1050–1055. [Google Scholar] [CrossRef]
- Alsarra, I.A.; Alanazi, F.K.; Ahmed, S.M.; Bosela, A.A.; Alhamed, S.S.; Mowafy, H.A.; Neau, S.H. Comparative study of itraconazole-cyclodextrin inclusion complex and its commercial product. Arch. Pharmacal Res. 2010, 33, 1009–1017. [Google Scholar] [CrossRef]
- Cevher, E.; Açma, A.; Sinani, G.; Aksu, B.; Zloh, M.; Mülazımoğlu, L. Bioadhesive tablets containing cyclodextrin complex of itraconazole for the treatment of vaginal candidiasis. Int. J. Biol. Macromol. 2014, 69, 124–136. [Google Scholar] [CrossRef] [PubMed]
- Siafaka, P.; Okur, N.Ü.; Mone, M.; Giannakopoulou, S.; Er, S.; Pavlidou, E.; Karavas, E.; Bikiaris, D.N. Two different approaches for oral administration of voriconazole laded formulations: Electrospun fibers versus β-cyclodextrin complexes. Int. J. Mol. Sci. 2016, 17, 282. [Google Scholar] [CrossRef]
- Fifere, A.; Marangoci, N.; Maier, S.S.; Coroaba, A.; Maftei, D.; Pinteala, M. Theoretical study on β-cyclodextrin inclusion complexes with propiconazole and protonated propiconazole. Beilstein J. Org. Chem. 2012, 8, 2191–2201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneiderman, E.; Stalcup, A.M. Cyclodextrins: A versatile tool in separation science. J. Chromatogr. 2000, 745, 83–102. [Google Scholar]
- Tang, P.; Wang, L.; Ma, X.; Xu, K.; Xiong, X.; Liao, X.; Li, H. Characterization and In Vitro Evaluation of the Complexes of Posaconazole with β- and 2,6-di-O-methyl-β-cyclodextrin. AAPS PharmSciTech 2017, 18, 104–114. [Google Scholar] [CrossRef] [Green Version]
- Tang, P.; Ma, X.; Wu, D.; Li, S.; Xu, K.; Tang, B.; Li, H. Posaconazole/hydroxypropyl-β-cyclodextrin host–guest system: Improving dissolution while maintaining antifungal activity. Carbohydr. Polym. 2016, 142, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Miyake, K.; Irie, T.; Arima, H.; Hirayama, F.; Uekama, K.; Hirano, M.; Okamoto, Y. Characterization of itraconazole/2-hydroxypropyl-β-cyclodextrin inclusion complex in aqueous propylene glycol solution. Int. J. Pharm. 1999, 179, 237–245. [Google Scholar] [CrossRef]
- Sayed, S.; Elsayed, I.; Ismail, M.M. Optimization of β-cyclodextrin consolidated micellar dispersion for promoting the transcorneal permeation of a practically insoluble drug. Int. J. Pharm. 2018, 549, 249–260. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhang, S.; Zhou, Y.; Guan, S.; Zhang, L. Inclusion complexes of fluconazole with β-cyclodextrin and 2-hydroxypropyl-β-cyclodextrin in aqueous solution: Preparation, characterization and a structural insight. J. Incl. Phenom. Macrocycl. Chem. 2016, 84, 209–217. [Google Scholar] [CrossRef]
- Kırımlıoğlu, G.Y.; Müzeyyen, D.; Lütfi, G. Inclusion complexes of fluconazole with β-cyclodextrin: Physicochemical charac-terization and in vitro evaluation of its formulation. J. Incl. Phenom. Macrocycl. Chem. 2011, 70, 429–435. [Google Scholar]
- Fernández-Ferreiro, A.; Bargiela, N.F.; Varela, M.S.; Martínez, M.G.; Pardo, M.; Ces, A.P.; Méndez, J.B.; Barcia, M.G.; Lamas, M.J.; Otero-Espinar, F. Cyclodextrin–polysaccharide-based, in situ-gelled system for ocular antifungal delivery. Beilstein J. Org. Chem. 2014, 10, 2903–2911. [Google Scholar] [CrossRef] [Green Version]
- Sayeed, F.; Ahmed, A.; Sayeed, A. Formulation and in vitro evaluation of solid dispersion of fluconazole. Int. J. Pharm. Sci. Res. 2016, 7, 4170–4179. [Google Scholar]
- Volkova, T.V.; Simonova, O.R.; Levshin, I.B.; Perlovich, G.L. Physicochemical profile of new antifungal compound: pH-dependent solubility, distribution, permeability and ionization assay. J. Mol. Liq. 2021, 336, 116535. [Google Scholar] [CrossRef]
- Miller, J.M.; Beig, A.; Carr, R.A.; Webster, G.K.; Dahan, A. The Solubility–Permeability Interplay When Using Cosolvents for Solubilization: Revising the Way We Use Solubility-Enabling Formulations. Mol. Pharm. 2012, 9, 581–590. [Google Scholar] [CrossRef]
- Miller, J.M.; Dahan, A. Predicting the solubility-permeability interplay when using cyclodextrins in solubility-enabling for-mulations: Model validation. Int. J. Pharm. 2012, 430, 388–391. [Google Scholar] [CrossRef] [PubMed]
- Higuchi, T.; Connors, K.A. Phase Solubility Techniques. In Advanced Analytical Chemistry of Instrumentation; Wiley-Interscience Publication: Hoboken, NJ, USA, 1965. [Google Scholar]
- Di Cagno, M.; Bibi, H.A.; Bauer-Brandl, A. New biomimetic barrier Permeapad™ for efficient investigation of passive per-meability of drugs. Eur. J. Pharm. Sci. 2015, 73, 29–34. [Google Scholar] [CrossRef]
- Brewster, M.E.; Loftsson, T. Cyclodextrins as pharmaceutical solubilizers. Adv. Drug Deliv. Rev. 2007, 59, 645–666. [Google Scholar] [CrossRef] [PubMed]
- Al Omari, M.M.; El-Barghouthi, M.I.; Zughul, M.B.; Davies, J.E.D.; Badwan, A.A. The role of drug hydrophobicity in β-cyclodextrin complexes. J. Mol. Liq. 2010, 155, 103–108. [Google Scholar] [CrossRef]
- Szejtli, J. Cyclodextrin Technology; Kluwer Academic: Dordrecht, The Netherlands, 1988. [Google Scholar]
- Orgován, G.; Kelemen, H.; Noszál, B. Protonation and β-cyclodextrin complex formation equilibria of fluconazole. J. Incl. Phenom. Macrocycl. Chem. 2016, 84, 189–196. [Google Scholar] [CrossRef]
- Jezequel, S.G. Fluconazole: Interspecies Scaling and Allometric Relationships of Pharmacokinetic Properties. J. Pharm. Pharmacol. 1994, 46, 196–199. [Google Scholar] [CrossRef] [PubMed]
- Perlovich, G.L.; Skara, M.; Bauer-Brandl, A. Driving forces and the influence of the buffer composition on the complexation reaction between ibuprofen and HPβ-CD. Eur. J. Pharm. Sci. 2003, 20, 197–200. [Google Scholar] [CrossRef]
- Rekharsky, M.V.; Inoue, Y. Complexation Thermodynamics of Cyclodextrins. Chem. Rev. 1998, 98, 1875. [Google Scholar] [CrossRef]
- Kopecký, F.; Kopecká, B.; Kaclík, P. Solubility Study of Nimodipine Inclusion Complexation with α- and β-Cyclodextrin and some Substituted Cyclodextrins. J. Incl. Phenom. Macrocycl. Chem. 2021, 39, 215–217. [Google Scholar] [CrossRef]
- Buvári-Barcza, Á.; Barcza, L. Influence of the guests, the type and degree of substitution on inclusion complex formation of substituted beta-cyclodextrins. Talanta 1999, 49, 577–585. [Google Scholar] [CrossRef]
- Tomoshige, N.; Takoshi, K. Thermodinamics of solubilities in mixed solvents. J. Chem. Eng. Jap. 1975, 8, 175–180. [Google Scholar]
- Ueberreiter, K. Change of water structure by solvents and polymers. Colloid Polym. Sci. 1982, 260, 37–45. [Google Scholar] [CrossRef]
- Sanghvi, R.; Narazaki, R.; Machatha, S.G.; Yalkowsky, S.H. Solubility Improvement of Drugs using N-Methyl Pyrrolidone. AAPS PharmSciTech 2008, 9, 366–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, J.; Fu, H.; Peng, G.; Cao, H.; Zhang, Y.; Liu, M.; Wu, W.; Qing, X.; Zhou, J. Solubility and solution thermodynamics of flofenicol in binary PEG 400+ water systems. Fluid Phase Equilibria 2014, 376, 159–164. [Google Scholar] [CrossRef]
- Balata, G.; Mahdi, M.; Bakera, R.A. Improvement of Solubility and Dissolution Properties of Clotrimazole by Solid Disper-sions and Inclusion Complexes. Indian J. Pharm. Sci. 2011, 73, 517–526. [Google Scholar] [CrossRef] [Green Version]
- Vinarov, Z.; Katev, V.; Burdzhiev, N.; Tcholakova, S.; Denkov, N. Effect of Surfactant–Bile Interactions on the Solubility of Hydrophobic Drugs in Biorelevant Dissolution Media. Mol. Pharm. 2018, 15, 5741–5753. [Google Scholar] [CrossRef]
- Florence, A.T.; Attwood, D. Physicochemical Principles of Pharmacy; Pharmaceutical Press: London, UK, 2006; Chapter 6; pp. 177–227. [Google Scholar]
- Stephenson, B.C.; Rangel-Yagui, C.O.; Junior, A.P.; Tavares, L.C.; Beers, K.; Blankschtein, D. Experimental and Theoretical Investigation of the Micellar-Assisted Solubilization of Ibuprofen in Aqueous Media. Langmuir 2006, 22, 1514–1525. [Google Scholar] [CrossRef]
- Obradović, S.; Poša, M. The influence of the structure of selected Brij and Tween homologues on the thermodynamic stability of their binary mixed micelles. J. Chem. Thermodyn. 2017, 110, 41–50. [Google Scholar] [CrossRef]
- Sezgin, Z.; Yüksel, N.; Baykara, T. Preparation and characterization of polymeric micelles for solubilization of poorly soluble anticancer drugs. Eur. J. Pharm. Biopharm. 2006, 64, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Alexandridis, P.; Holzwarth, J.F.; Hatton, T.A. Micellization of Poly(ethylene oxide)-Poly(propylene oxide)-Poly(ethylene oxide) Triblock Copolymers in Aqueous Solutions: Thermodynamics of Copolymer Association. Macromolecules 1994, 27, 2414–2425. [Google Scholar] [CrossRef]
- Croy, S.R.; Kwon, G.S. The effects of Pluronic block copolymers on the aggregation state of nystatin. J. Control. Release 2004, 95, 161–171. [Google Scholar] [CrossRef]
- Wilhelm, M.; Le Zhao, C.; Wang, Y.; Xu, R.; Winnik, M.A.; Mura, J.L.; Riess, G.; Croucher, M.D. Poly(styrene-ethylene oxide) block copolymer micelle formation in water: A fluorescence probe study. Macromolecules 1991, 24, 1033–1040. [Google Scholar] [CrossRef]
- Ibrahim, M.A. Ketoconazole binary and ternary solid dispersions in different macromolecular matrices. Macromol. Indian J. 2009, 5, 1–8. [Google Scholar]
- Másson, M.; Loftsson, T.; Masson, G.; Stefansson, E. Cyclodextrins as permeation enhancers: Some theoretical evaluations and in vitro testing. J. Control. Release 1999, 59, 107–118. [Google Scholar] [CrossRef]
- Loftsson, T.; Masson, M.; Sigurdsson, H. Cyclodextrins and drug permeability through semi-permeable cellophane membranes. Int. J. Pharm. 2002, 232, 35–43. [Google Scholar] [CrossRef]
- Avdeef, A.; Nielsen, P.E.; Tsinman, O. PAMPA-a drug absorption in vitro model 11. Matching the in vivo unstirred water layer thickness by individual-well stirring in microtitre plates. Eur. J. Pharm. Sci. 2004, 22, 365–374. [Google Scholar] [PubMed]
- Loftsson, T.; Vogensen, S.B.; Brewster, M.E.; Konráðsdóttir, F. Effects of cyclodextrins on drug delivery through biological membranes. J. Pharm. Sci. 2007, 96, 2532–2546. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| System | PermeaPad (PP) | Cellulose Membrane (RC) | ||||
|---|---|---|---|---|---|---|
| C (M) | J (µM∙cm−2∙s−1) | Papp (cm∙s−1) | C (M) | J (µM∙cm−2∙s−1) | Papp (cm∙s−1) | |
| S-119 | ||||||
| Buffer | 1.50 × 10−3 | 2.20 × 10−5 | (1.48 ± 0.04) × 10−5 | 1.29 × 10−3 | 3.49 × 10−5 | (2.71 ± 0.08) × 10−5 |
| β-CD | 1.63 × 10−3 | 9.93 × 10−6 | (6.12 ± 0.31) × 10−6 | 1.49 × 10−3 | 2.90 × 10−5 | (1.96 ± 0.04) × 10−5 |
| HP-β-CD | 1.28 × 10−3 | 1.00 × 10−5 | (7.81 ± 0.24) × 10−6 | 1.53 × 10−3 | 2.21 × 10−5 | (1.45 ± 0.03) × 10−5 |
| 6-O-M-β-CD | 1.81 × 10−3 | 1.01 × 10−5 | (5.62 ± 0.14) × 10−6 | 1.16 × 10−3 | 2.68 × 10−5 | (2.31 ± 0.07) × 10−5 |
| FCZ | ||||||
| Buffer | 2.01 × 10−3 | 3.50 × 10−5 | (1.74 ± 0.03) × 10−5 * | 2.67 × 10−3 | 1.31 × 10−4 | (4.90 ± 0.19) × 10−5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Volkova, T.V.; Simonova, O.R.; Perlovich, G.L. New Antifungal Compound: Impact of Cosolvency, Micellization and Complexation on Solubility and Permeability Processes. Pharmaceutics 2021, 13, 1865. https://doi.org/10.3390/pharmaceutics13111865
Volkova TV, Simonova OR, Perlovich GL. New Antifungal Compound: Impact of Cosolvency, Micellization and Complexation on Solubility and Permeability Processes. Pharmaceutics. 2021; 13(11):1865. https://doi.org/10.3390/pharmaceutics13111865
Chicago/Turabian StyleVolkova, Tatyana V., Olga R. Simonova, and German L. Perlovich. 2021. "New Antifungal Compound: Impact of Cosolvency, Micellization and Complexation on Solubility and Permeability Processes" Pharmaceutics 13, no. 11: 1865. https://doi.org/10.3390/pharmaceutics13111865