
Immunomodulatory Nanoparticles Mitigate Macrophage Inflammation via Inhibition of PAMP Interactions and Lactate-Mediated Functional Reprogramming of NF-κB and p38 MAPK

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. iNP Preparation and Characterization
2.3. Particle-TLR Agonist Association Studies
2.4. Mice
2.5. Isolation and Generation of Bone Marrow-Derived Macrophages (BMMΦs) and Dendritic Cells (BMDCs)
2.6. Flow Cytometry
2.7. Particle-Cell Association Studies
2.8. Cytokine and Chemokine Secretion Analysis
2.9. Immunoblotting for Transcriptional Activity
2.10. Statistical Analyses
3. Results
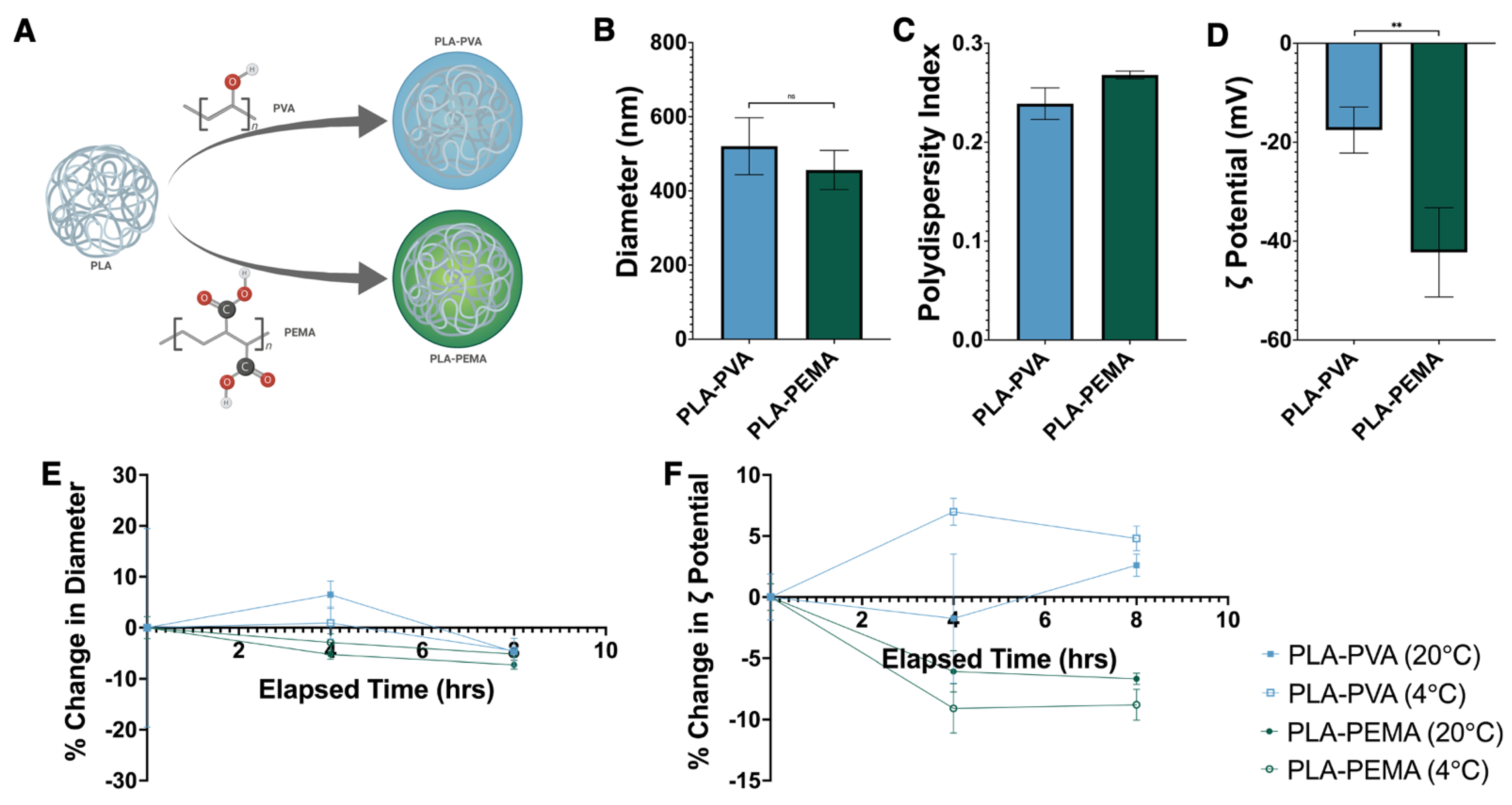
3.1. Fabrication, Characterization, and Stability Assessment of Poly(Lactic Acid) iNPs
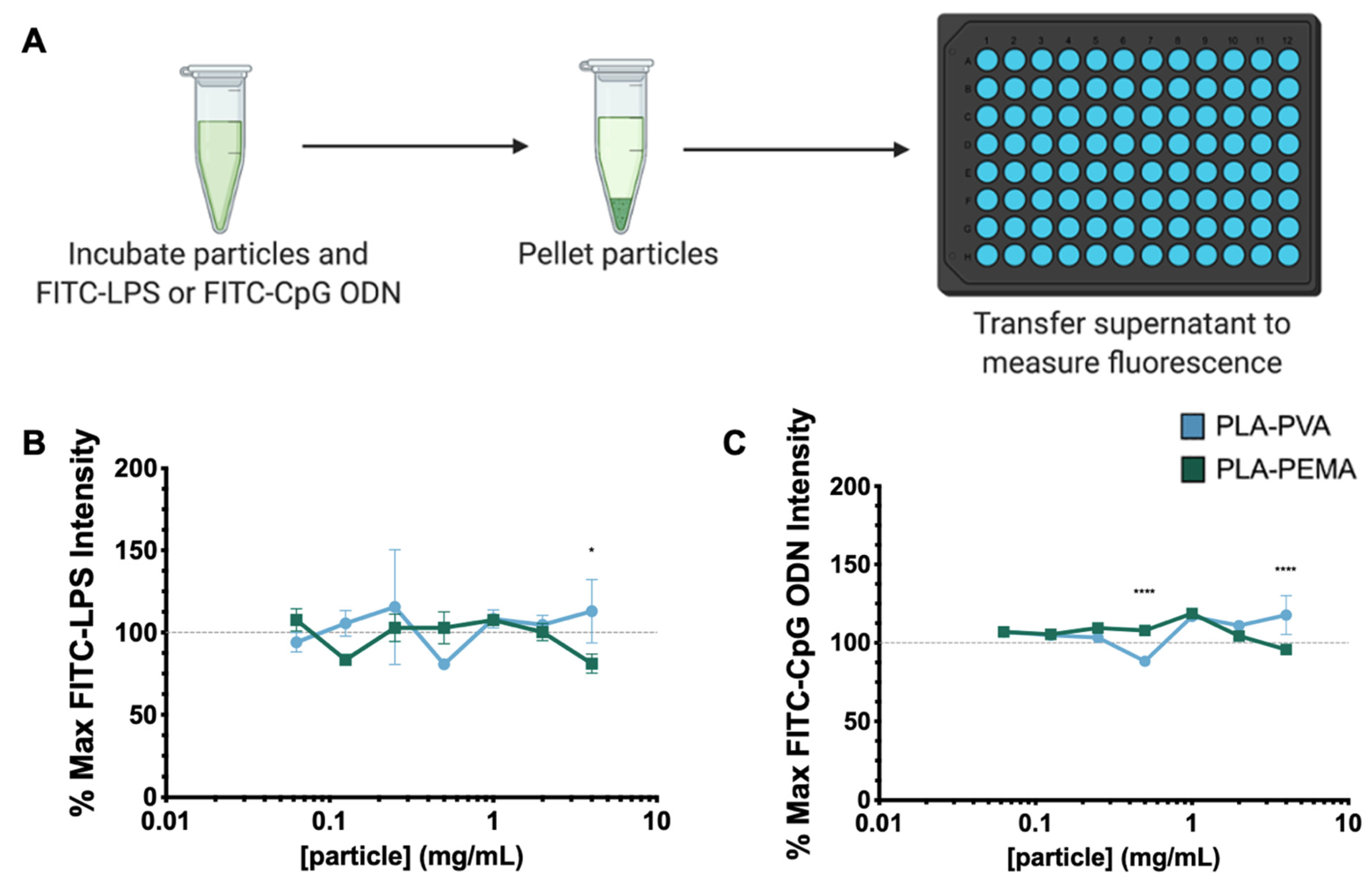
3.2. PLA iNPs Do Not Sequester PAMPs
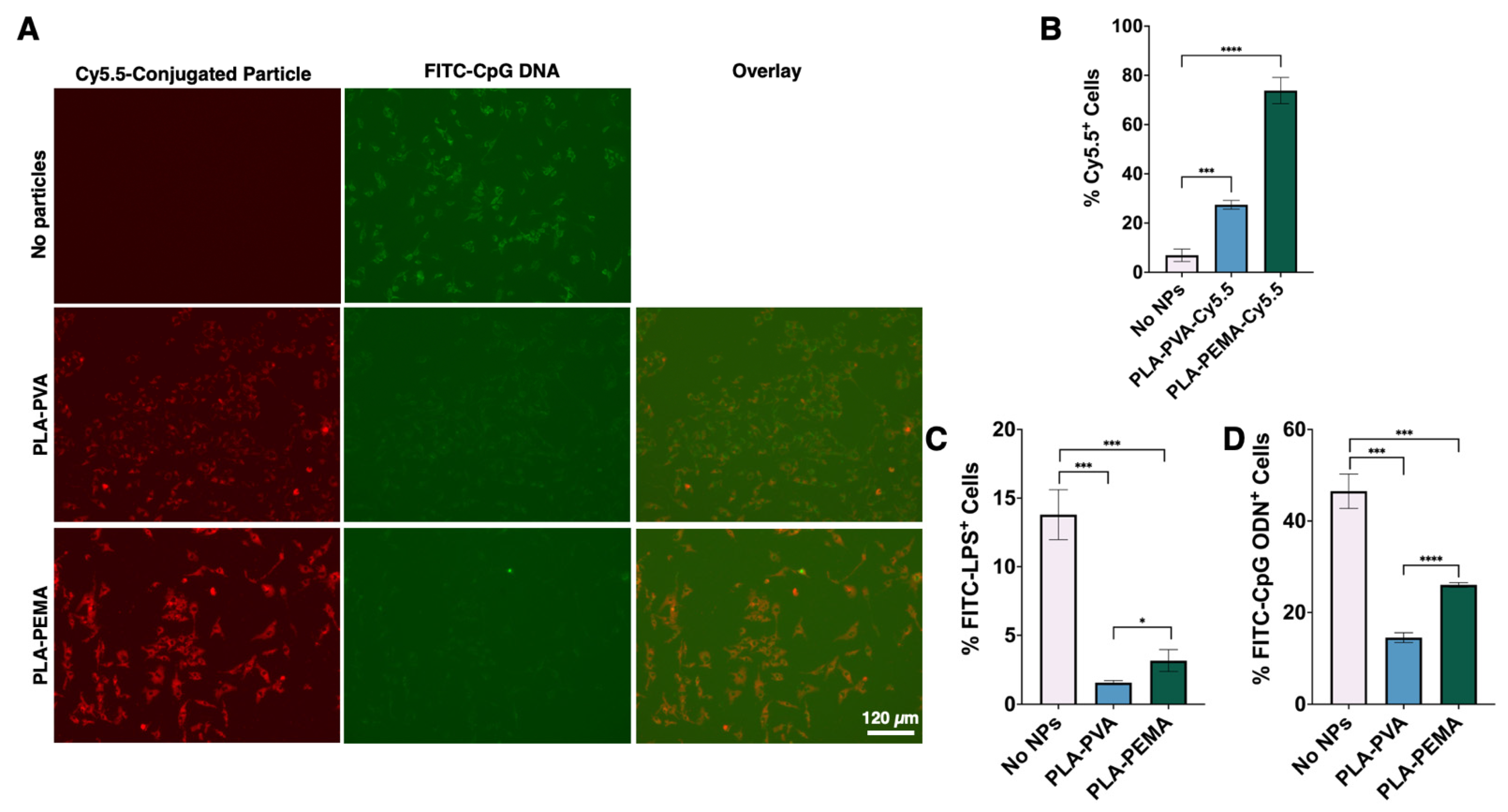
3.3. BMMΦs Associate with and Internalize PLA-PEMA More Extensively Than PLA-PVA
3.4. PLA Particles Hinder LPS and CpG ODN Interaction with BMMΦs
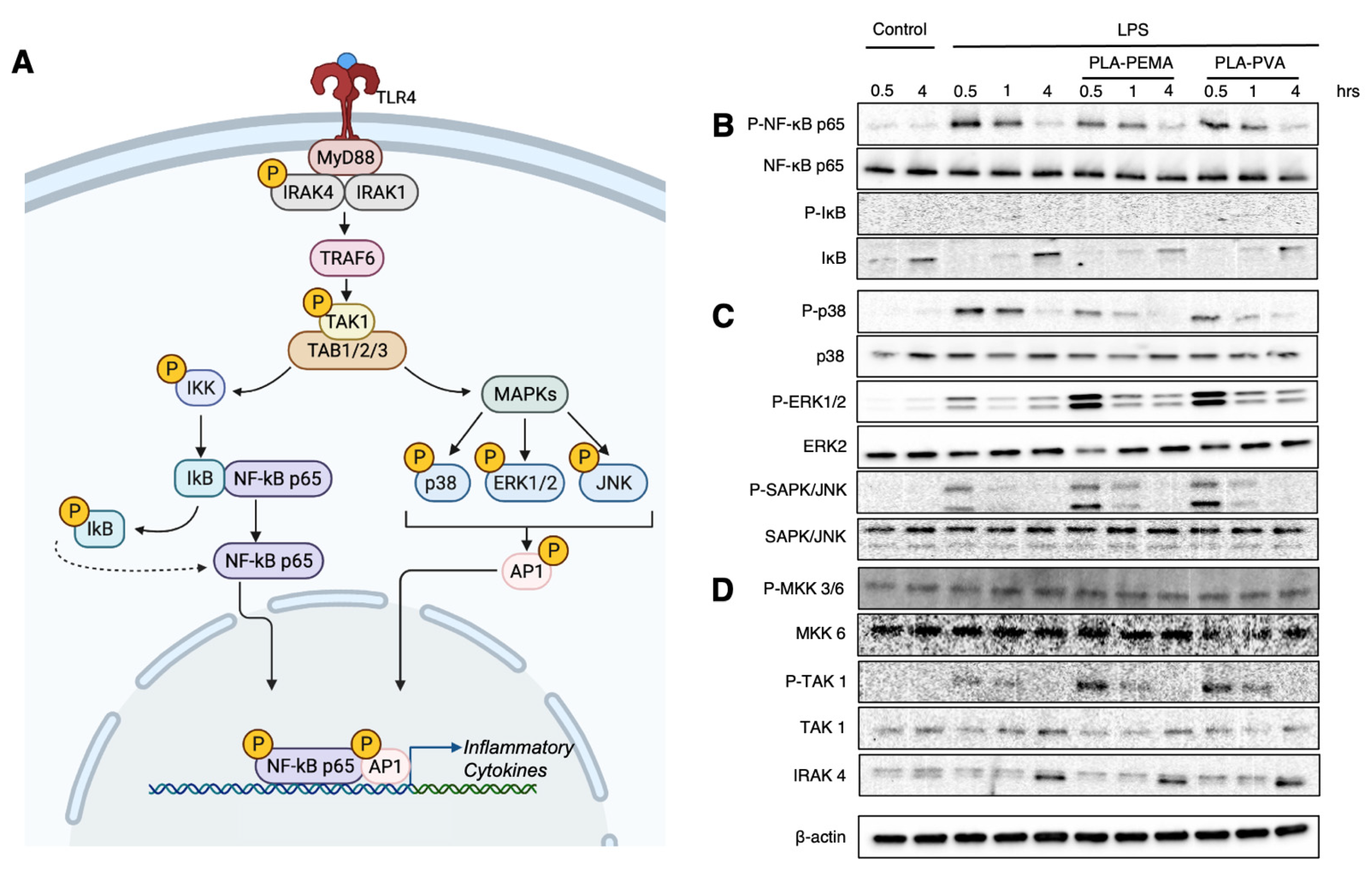
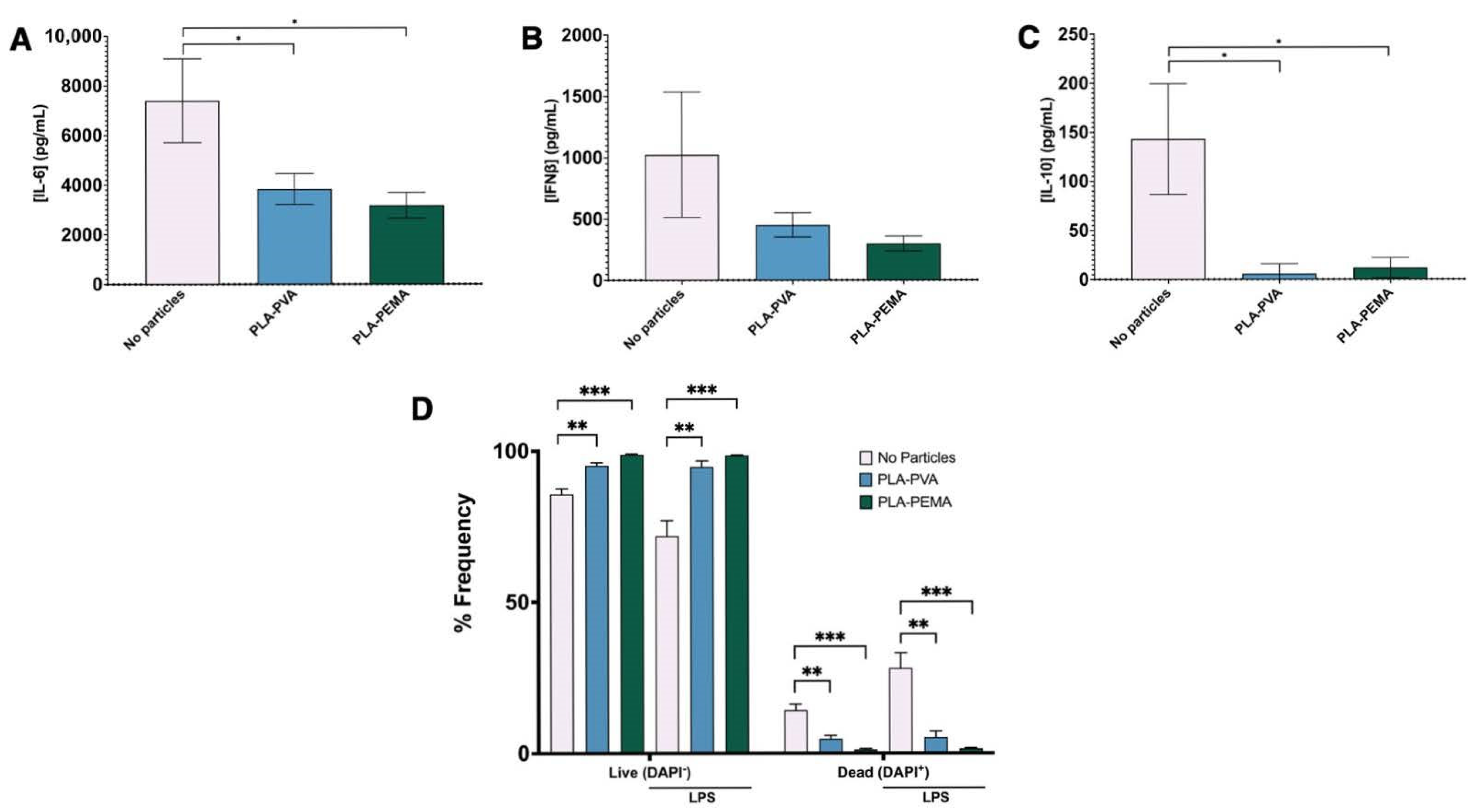
3.5. PLA-PEMA and PLA-PVA Inhibit NF-κB Activation, But Do So at Different Rates
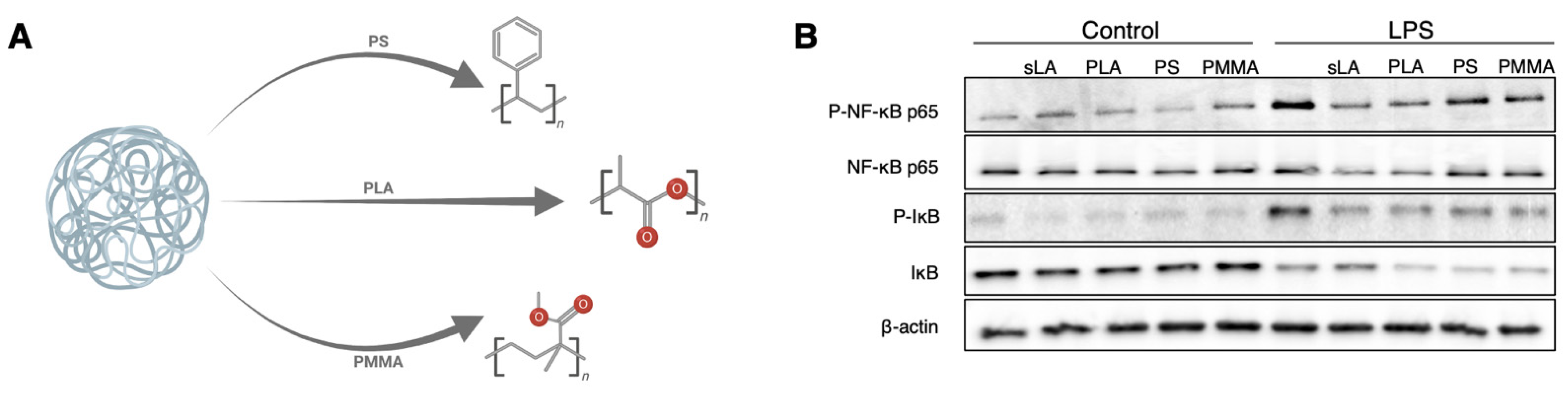
3.6. The PLA Polymer Composition of iNPs Drives the Suppression of NF-κB Signaling
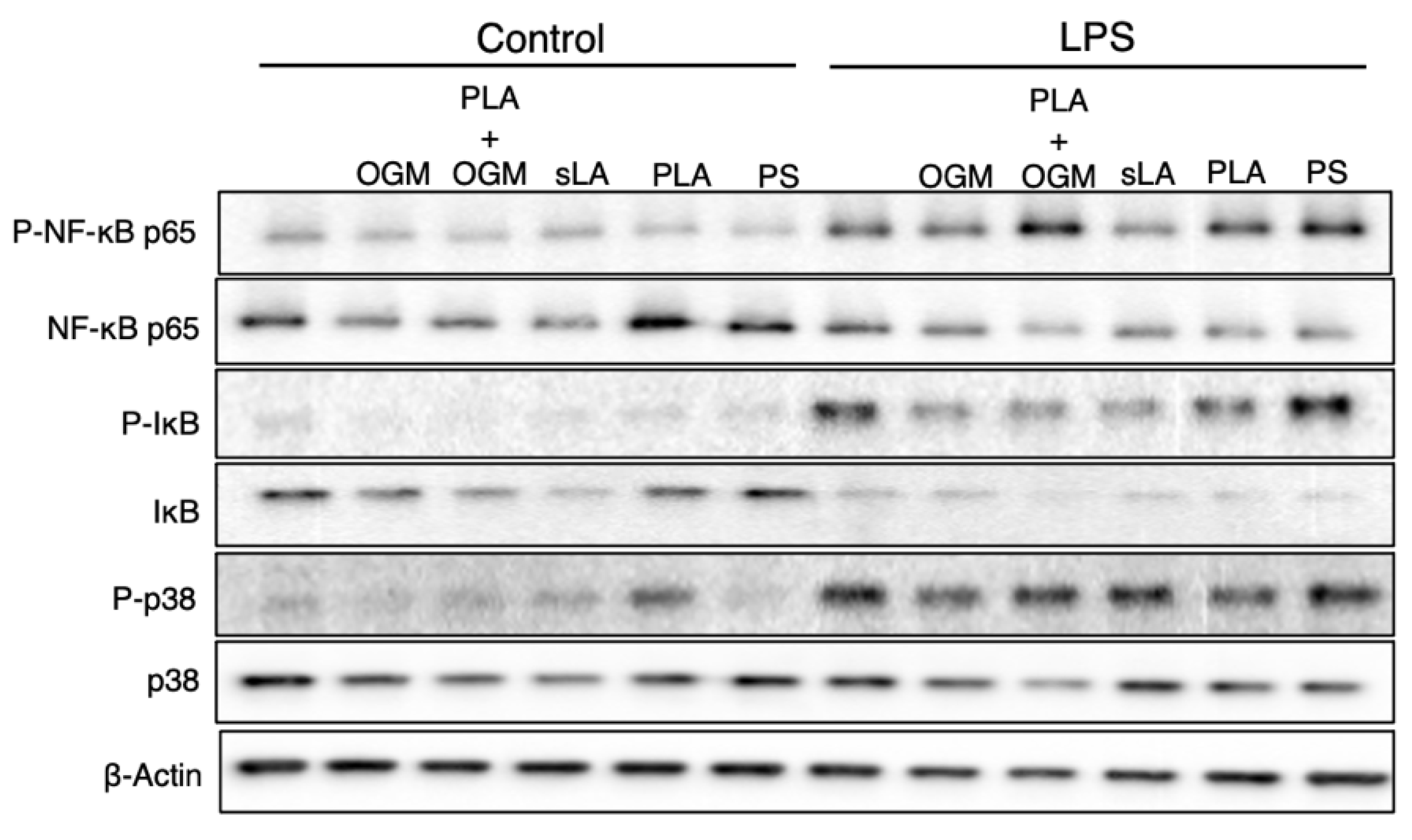
3.7. The Protective Function of the PLA-Based iNPs Depends upon GPR68 Signaling
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rudd, K.E.; Johnson, S.C.; Agesa, K.M.; Shackelford, K.A.; Tsoi, D.; Kievlan, D.R.; Colombara, D.V.; Ikuta, K.S.; Kissoon, N.; Finfer, S.; et al. Global, regional, and national sepsis incidence and mortality, 1990–2017: Analysis for the Global Burden of Disease Study. Lancet 2020, 395, 200–211. [Google Scholar] [CrossRef] [Green Version]
- Delano, M.J.; Ward, P.A. Sepsis-induced immune dysfunction: Can immune therapies reduce mortality? J. Clin. Investig. 2016, 126, 23–31. [Google Scholar] [CrossRef]
- Marshall, J.C. Why have clinical trials in sepsis failed? Trends Mol. Med. 2014, 20, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Fink, M.P. Animal models of sepsis. Virulence 2014, 5, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Remick, D.G. Cytokine therapeutics for the treatment of sepsis: Why has nothing worked? Curr. Pharm. Des. 2003, 9, 75–82. [Google Scholar] [CrossRef]
- Leligdowicz, A.; Matthay, M.A. Heterogeneity in sepsis: New biological evidence with clinical applications. Crit. Care 2019, 23, 80. [Google Scholar] [CrossRef] [Green Version]
- Gomes, A.C.; Mohsen, M.; Bachmann, M.F. Harnessing Nanoparticles for Immunomodulation and Vaccines. Vaccines 2017, 5, 6. [Google Scholar] [CrossRef]
- Feng, X.; Xu, W.; Li, Z.; Song, W.; Ding, J.; Chen, X. Immunomodulatory Nanosystems. Adv. Sci. 2019, 6, 1900101. [Google Scholar] [CrossRef] [PubMed]
- Pearson, R.M.; Casey, L.M.; Hughes, K.R.; Miller, S.D.; Shea, L.D. In vivo reprogramming of immune cells: Technologies for induction of antigen-specific tolerance. Adv. Drug Deliv. Rev. 2017, 114, 240–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lasola, J.J.M.; Kamdem, H.; McDaniel, M.W.; Pearson, R.M. Biomaterial-Driven Immunomodulation: Cell Biology-Based Strategies to Mitigate Severe Inflammation and Sepsis. Front. Immunol. 2020, 11, 1726. [Google Scholar] [CrossRef] [PubMed]
- Casey, L.M.; Kakade, S.; Decker, J.T.; Rose, J.A.; Deans, K.; Shea, L.D.; Pearson, R.M. Cargo-less nanoparticles program innate immune cell responses to toll-like receptor activation. Biomaterials 2019, 218, 119333. [Google Scholar] [CrossRef] [PubMed]
- Getts, D.R.; Terry, R.L.; Getts, M.T.; Deffrasnes, C.; Muller, M.; van Vreden, C.; Ashhurst, T.M.; Chami, B.; McCarthy, D.; Wu, H.; et al. Therapeutic inflammatory monocyte modulation using immune-modifying microparticles. Sci. Transl. Med. 2014, 6, 219ra217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allen, R.P.; Bolandparvaz, A.; Ma, J.A.; Manickam, V.A.; Lewis, J.S. Latent, Immunosuppressive Nature of Poly(lactic-co-glycolic acid) Microparticles. ACS Biomater. Sci. Eng. 2018, 4, 900–918. [Google Scholar] [CrossRef] [PubMed]
- Pearson, R.M.; Podojil, J.R.; Shea, L.D.; King, N.J.C.; Miller, S.D.; Getts, D.R. Overcoming challenges in treating autoimmuntity: Development of tolerogenic immune-modifying nanoparticles. Nanomed. Nanotechnol. Biol. Med. 2018, 18, 282–291. [Google Scholar] [CrossRef] [PubMed]
- Ivashkiv, L.B. The hypoxia-lactate axis tempers inflammation. Nat. Rev. Immunol. 2020, 20, 85–86. [Google Scholar] [CrossRef]
- Zhang, W.; Wang, G.; Xu, Z.G.; Tu, H.; Hu, F.; Dai, J.; Chang, Y.; Chen, Y.; Lu, Y.; Zeng, H.; et al. Lactate Is a Natural Suppressor of RLR Signaling by Targeting MAVS. Cell 2019, 178, 176–189 e115. [Google Scholar] [CrossRef]
- Colegio, O.R.; Chu, N.Q.; Szabo, A.L.; Chu, T.; Rhebergen, A.M.; Jairam, V.; Cyrus, N.; Brokowski, C.E.; Eisenbarth, S.C.; Phillips, G.M.; et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 2014, 513, 559–563. [Google Scholar] [CrossRef]
- Zhang, D.; Tang, Z.; Huang, H.; Zhou, G.; Cui, C.; Weng, Y.; Liu, W.; Kim, S.; Lee, S.; Perez-Neut, M.; et al. Metabolic regulation of gene expression by histone lactylation. Nature 2019, 574, 575–580. [Google Scholar] [CrossRef]
- Certo, M.; Tsai, C.H.; Pucino, V.; Ho, P.C.; Mauro, C. Lactate modulation of immune responses in inflammatory versus tumour microenvironments. Nat. Rev. Immunol. 2021, 21, 151–161. [Google Scholar] [CrossRef]
- Yang, K.; Xu, J.; Fan, M.; Tu, F.; Wang, X.; Ha, T.; Williams, D.L.; Li, C. Lactate Suppresses Macrophage Pro-Inflammatory Response to LPS Stimulation by Inhibition of YAP and NF-kappaB Activation via GPR81-Mediated Signaling. Front. Immunol. 2020, 11, 587913. [Google Scholar] [CrossRef]
- Errea, A.; Cayet, D.; Marchetti, P.; Tang, C.; Kluza, J.; Offermanns, S.; Sirard, J.C.; Rumbo, M. Lactate Inhibits the Pro-Inflammatory Response and Metabolic Reprogramming in Murine Macrophages in a GPR81-Independent Manner. PLoS ONE 2016, 11, e0163694. [Google Scholar] [CrossRef] [Green Version]
- Williams, C.H.; Neitzel, L.R.; Karki, P.; Keyser, B.D.; Thayer, T.E.; Wells, Q.S.; Perry, J.A.; Birukova, A.A.; Birukov, K.G.; Hong, C.C. Coupling Metastasis to pH-Sensing GPR68 Using a Novel Small Molecule Inhibitor. BioRxiv 2021, 612549. [Google Scholar] [CrossRef] [Green Version]
- McCarthy, D.P.; Yap, J.W.; Harp, C.T.; Song, W.K.; Chen, J.; Pearson, R.M.; Miller, S.D.; Shea, L.D. An antigen-encapsulating nanoparticle platform for TH1/17 immune tolerance therapy. Nanomed. Nanotechnol. Biol. Med. 2017, 13, 191–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weischenfeldt, J.; Porse, B. Bone Marrow-Derived Macrophages (BMM): Isolation and Applications. Cold Spring Harb. Protoc. 2008, 2008, 5080. [Google Scholar] [CrossRef] [Green Version]
- Lutz, M.B.; Kukutsch, N.; Ogilvie, A.L.; Rossner, S.; Koch, F.; Romani, N.; Schuler, G. An advanced culture method for generating large quantities of highly pure dendritic cells from mouse bone marrow. J. Immunol. Methods 1999, 223, 77–92. [Google Scholar] [CrossRef]
- Freitag, T.L.; Podojil, J.R.; Pearson, R.M.; Fokta, F.J.; Sahl, C.; Messing, M.; Andersson, L.C.; Leskinen, K.; Saavalainen, P.; Hoover, L.I.; et al. Gliadin Nanoparticles Induce Immune Tolerance to Gliadin in Mouse Models of Celiac Disease. Gastroenterology 2020, 158, 1667–1681.e12. [Google Scholar] [CrossRef]
- Akira, S.; Takeda, K. Toll-like receptor signalling. Nat. Rev. Immunol. 2004, 4, 499–511. [Google Scholar] [CrossRef] [PubMed]
- Hambleton, J.; McMahon, M.; DeFranco, A.L. Activation of Raf-1 and mitogen-activated protein kinase in murine macrophages partially mimics lipopolysaccharide-induced signaling events. J. Exp. Med. 1995, 182, 147–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hambleton, J.; Weinstein, S.L.; Lem, L.; DeFranco, A.L. Activation of c-Jun N-terminal kinase in bacterial lipopolysaccharide-stimulated macrophages. Proc. Natl. Acad. Sci. USA 1996, 93, 2774. [Google Scholar] [CrossRef] [Green Version]
- Platanitis, E.; Decker, T. Regulatory Networks Involving STATs, IRFs, and NFkappaB in Inflammation. Front. Immunol. 2018, 9, 2542. [Google Scholar] [CrossRef] [Green Version]
- Iyer, S.S.; Cheng, G. Role of interleukin 10 transcriptional regulation in inflammation and autoimmune disease. Crit. Rev. Immunol. 2012, 32, 23–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Valliere, C.; Cosin-Roger, J.; Simmen, S.; Atrott, K.; Melhem, H.; Zeitz, J.; Madanchi, M.; Tcymbarevich, I.; Fried, M.; Kullak-Ublick, G.A.; et al. Hypoxia Positively Regulates the Expression of pH-Sensing G-Protein-Coupled Receptor OGR1 (GPR68). Cell. Mol. Gastroenterol. Hepatol. 2016, 2, 796–810. [Google Scholar] [CrossRef] [Green Version]
- de Valliere, C.; Wang, Y.; Eloranta, J.J.; Vidal, S.; Clay, I.; Spalinger, M.R.; Tcymbarevich, I.; Terhalle, A.; Ludwig, M.G.; Suply, T.; et al. G Protein-coupled pH-sensing Receptor OGR1 Is a Regulator of Intestinal Inflammation. Inflamm. Bowel Dis. 2015, 21, 1269–1281. [Google Scholar] [CrossRef] [Green Version]
- Shen, Z.; Jiang, L.; Yuan, Y.; Deng, T.; Zheng, Y.R.; Zhao, Y.Y.; Li, W.L.; Wu, J.Y.; Gao, J.Q.; Hu, W.W.; et al. Inhibition of G protein-coupled receptor 81 (GPR81) protects against ischemic brain injury. CNS Neurosci. Ther. 2015, 21, 271–279. [Google Scholar] [CrossRef] [PubMed]
- Ciesielska, A.; Matyjek, M.; Kwiatkowska, K. TLR4 and CD14 trafficking and its influence on LPS-induced pro-inflammatory signaling. Cell. Mol. Life Sci. 2021, 78, 1233–1261. [Google Scholar] [CrossRef]
- Sangsuwan, R.; Thuamsang, B.; Pacifici, N.; Allen, R.; Han, H.; Miakicheva, S.; Lewis, J.S. Lactate Exposure Promotes Immunosuppressive Phenotypes in Innate Immune Cells. Cell. Mol. Bioeng. 2020, 13, 541–557. [Google Scholar] [CrossRef] [PubMed]
- Husebye, H.; Halaas, O.; Stenmark, H.; Tunheim, G.; Sandanger, O.; Bogen, B.; Brech, A.; Latz, E.; Espevik, T. Endocytic pathways regulate Toll-like receptor 4 signaling and link innate and adaptive immunity. EMBO J. 2006, 25, 683–692. [Google Scholar] [CrossRef] [Green Version]
- Kagan, J.C.; Su, T.; Horng, T.; Chow, A.; Akira, S.; Medzhitov, R. TRAM couples endocytosis of Toll-like receptor 4 to the induction of interferon-beta. Nat. Immunol. 2008, 9, 361–368. [Google Scholar] [CrossRef] [Green Version]
- Leifer, C.A.; Kennedy, M.N.; Mazzoni, A.; Lee, C.; Kruhlak, M.J.; Segal, D.M. TLR9 is localized in the endoplasmic reticulum prior to stimulation. J. Immunol. 2004, 173, 1179–1183. [Google Scholar] [CrossRef] [Green Version]
- Leifer, C.A.; Brooks, J.C.; Hoelzer, K.; Lopez, J.; Kennedy, M.N.; Mazzoni, A.; Segal, D.M. Cytoplasmic targeting motifs control localization of toll-like receptor 9. J. Biol. Chem. 2006, 281, 35585–35592. [Google Scholar] [CrossRef] [Green Version]
- Chockalingam, A.; Brooks, J.C.; Cameron, J.L.; Blum, L.K.; Leifer, C.A. TLR9 traffics through the Golgi complex to localize to endolysosomes and respond to CpG DNA. Immunol. Cell Biol. 2009, 87, 209–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabio, G.; Davis, R.J. TNF and MAP kinase signalling pathways. Semin. Immunol. 2014, 26, 237–245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaestel, M.; Kotlyarov, A.; Kracht, M. Targeting innate immunity protein kinase signalling in inflammation. Nat. Rev. Drug Discov. 2009, 8, 480–499. [Google Scholar] [CrossRef]
- Thamphiwatana, S.; Angsantikul, P.; Escajadillo, T.; Zhang, Q.; Olson, J.; Luk, B.T.; Zhang, S.; Fang, R.H.; Gao, W.; Nizet, V.; et al. Macrophage-like nanoparticles concurrently absorbing endotoxins and proinflammatory cytokines for sepsis management. Proc. Natl. Acad. Sci. USA 2017, 114, 11488–11493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.; Zhang, Y.; Saito, E.; Gurczynski, S.J.; Moore, B.B.; Cummings, B.J.; Anderson, A.J.; Shea, L.D. Intravascular innate immune cells reprogrammed via intravenous nanoparticles to promote functional recovery after spinal cord injury. Proc. Natl. Acad. Sci. USA 2019, 116, 14947–14954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saito, E.; Kuo, R.; Pearson, R.M.; Gohel, N.; Cheung, B.; King, N.J.C.; Miller, S.D.; Shea, L.D. Designing drug-free biodegradable nanoparticles to modulate inflammatory monocytes and neutrophils for ameliorating inflammation. J. Control. Release 2019, 300, 185–196. [Google Scholar] [CrossRef]
- Smarr, C.B.; Yap, W.T.; Neef, T.P.; Pearson, R.M.; Hunter, Z.N.; Ifergan, I.; Getts, D.R.; Bryce, P.J.; Shea, L.D.; Miller, S.D. Biodegradable antigen-associated PLG nanoparticles tolerize Th2-mediated allergic airway inflammation pre- and postsensitization. Proc. Natl. Acad. Sci. USA 2016, 113, 5059–5064. [Google Scholar] [CrossRef] [Green Version]
- Pitt, C.G.; Gratzl, M.M.; Kimmel, G.L.; Surles, J.; Schindler, A. Aliphatic polyesters II. The degradation of poly (DL-lactide), poly (epsilon-caprolactone), and their copolymers in vivo. Biomaterials 1981, 2, 215–220. [Google Scholar] [CrossRef]
- Gopferich, A. Mechanisms of polymer degradation and erosion. Biomaterials 1996, 17, 103–114. [Google Scholar] [CrossRef]
- Williams, D.F. Enzymic Hydrolysis of Polylactic Acid. Eng. Med. 1981, 10, 5–7. [Google Scholar] [CrossRef]
- Nakamura, K.; Tomita, T.; Abe, N.; Kamio, Y. Purification and characterization of an extracellular poly(L-lactic acid) depolymerase from a soil isolate, Amycolatopsis sp. strain K104-1. Appl. Environ. Microbiol. 2001, 67, 345–353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- da Silva, D.; Kaduri, M.; Poley, M.; Adir, O.; Krinsky, N.; Shainsky-Roitman, J.; Schroeder, A. Biocompatibility, biodegradation and excretion of polylactic acid (PLA) in medical implants and theranostic systems. Chem. Eng. J. 2018, 340, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Schakenraad, J.M.; Hardonk, M.J.; Feijen, J.; Molenaar, I.; Nieuwenhuis, P. Enzymatic activity toward poly(L-lactic acid) implants. J. Biomed. Mater. Res. 1990, 24, 529–545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kellum, J.A.; Song, M.; Li, J. Lactic and hydrochloric acids induce different patterns of inflammatory response in LPS-stimulated RAW 264.7 cells. Am. J. Physiol. Integr. Comp. Physiol. 2004, 286, R686–R692. [Google Scholar] [CrossRef] [Green Version]
- Ranganathan, P.; Shanmugam, A.; Swafford, D.; Suryawanshi, A.; Bhattacharjee, P.; Hussein, M.S.; Koni, P.A.; Prasad, P.D.; Kurago, Z.B.; Thangaraju, M.; et al. GPR81, a Cell-Surface Receptor for Lactate, Regulates Intestinal Homeostasis and Protects Mice from Experimental Colitis. J. Immunol. 2018, 200, 1781–1789. [Google Scholar] [CrossRef]
- Hoque, R.; Farooq, A.; Ghani, A.; Gorelick, F.; Mehal, W.Z. Lactate reduces liver and pancreatic injury in Toll-like receptor- and inflammasome-mediated inflammation via GPR81-mediated suppression of innate immunity. Gastroenterology 2014, 146, 1763–1774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiley, S.Z.; Sriram, K.; Salmeron, C.; Insel, P.A. GPR68: An Emerging Drug Target in Cancer. Int. J. Mol. Sci. 2019, 20, 559. [Google Scholar] [CrossRef] [PubMed] [Green Version]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lasola, J.J.M.; Cottingham, A.L.; Scotland, B.L.; Truong, N.; Hong, C.C.; Shapiro, P.; Pearson, R.M. Immunomodulatory Nanoparticles Mitigate Macrophage Inflammation via Inhibition of PAMP Interactions and Lactate-Mediated Functional Reprogramming of NF-κB and p38 MAPK. Pharmaceutics 2021, 13, 1841. https://doi.org/10.3390/pharmaceutics13111841
Lasola JJM, Cottingham AL, Scotland BL, Truong N, Hong CC, Shapiro P, Pearson RM. Immunomodulatory Nanoparticles Mitigate Macrophage Inflammation via Inhibition of PAMP Interactions and Lactate-Mediated Functional Reprogramming of NF-κB and p38 MAPK. Pharmaceutics. 2021; 13(11):1841. https://doi.org/10.3390/pharmaceutics13111841
Chicago/Turabian StyleLasola, Jackline Joy Martín, Andrea L. Cottingham, Brianna L. Scotland, Nhu Truong, Charles C. Hong, Paul Shapiro, and Ryan M. Pearson. 2021. "Immunomodulatory Nanoparticles Mitigate Macrophage Inflammation via Inhibition of PAMP Interactions and Lactate-Mediated Functional Reprogramming of NF-κB and p38 MAPK" Pharmaceutics 13, no. 11: 1841. https://doi.org/10.3390/pharmaceutics13111841