
PEGylated Mesoporous Silica Nanoparticles (MCM-41): A Promising Carrier for the Targeted Delivery of Fenbendazole into Prostrate Cancer Cells

 , , and
, , and
Abstract
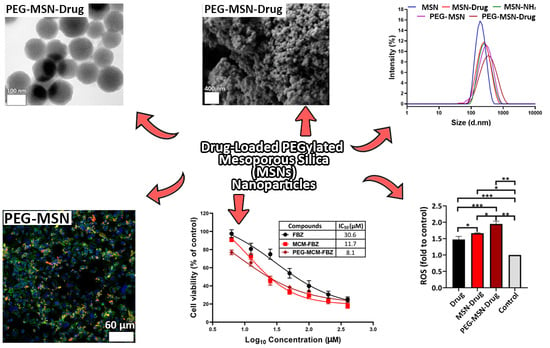
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Synthesis of MCM NPs
2.3. Synthesis of MCM-FBZ NPs
2.4. Surface Functionalization of MCM with Amine Group
2.5. Synthesis of PEG-MCM-FBZ NPs
2.6. Cy-5 Grafting on MCM-NH2 and PEG-MCM NPs
2.7. Physicochemical Characterizations of the Synthesized Drug Nanoformulations
2.7.1. Dynamic Light Scattering (DLS) Measurements
2.7.2. Morphology of the Synthesized Nanoformulations
2.7.3. Thermogravimetric Analysis (TGA) and Differential Scanning Calorimetry (DSC) Measurements
2.7.4. Fourier-Transform Infrared (FTIR) Measurements
2.7.5. Brunauer–Emmett–Teller (BET) Surface Area Analysis
2.8. Release Study
2.9. Qualitative Cellular Uptake Using Confocal Microscopy
2.10. Cell Viability
2.11. Proliferation Assay
2.12. Cell Migration
2.13. Reactive Oxygen Species (ROS) Assay
2.14. Statistical Analysis
3. Results and Discussion
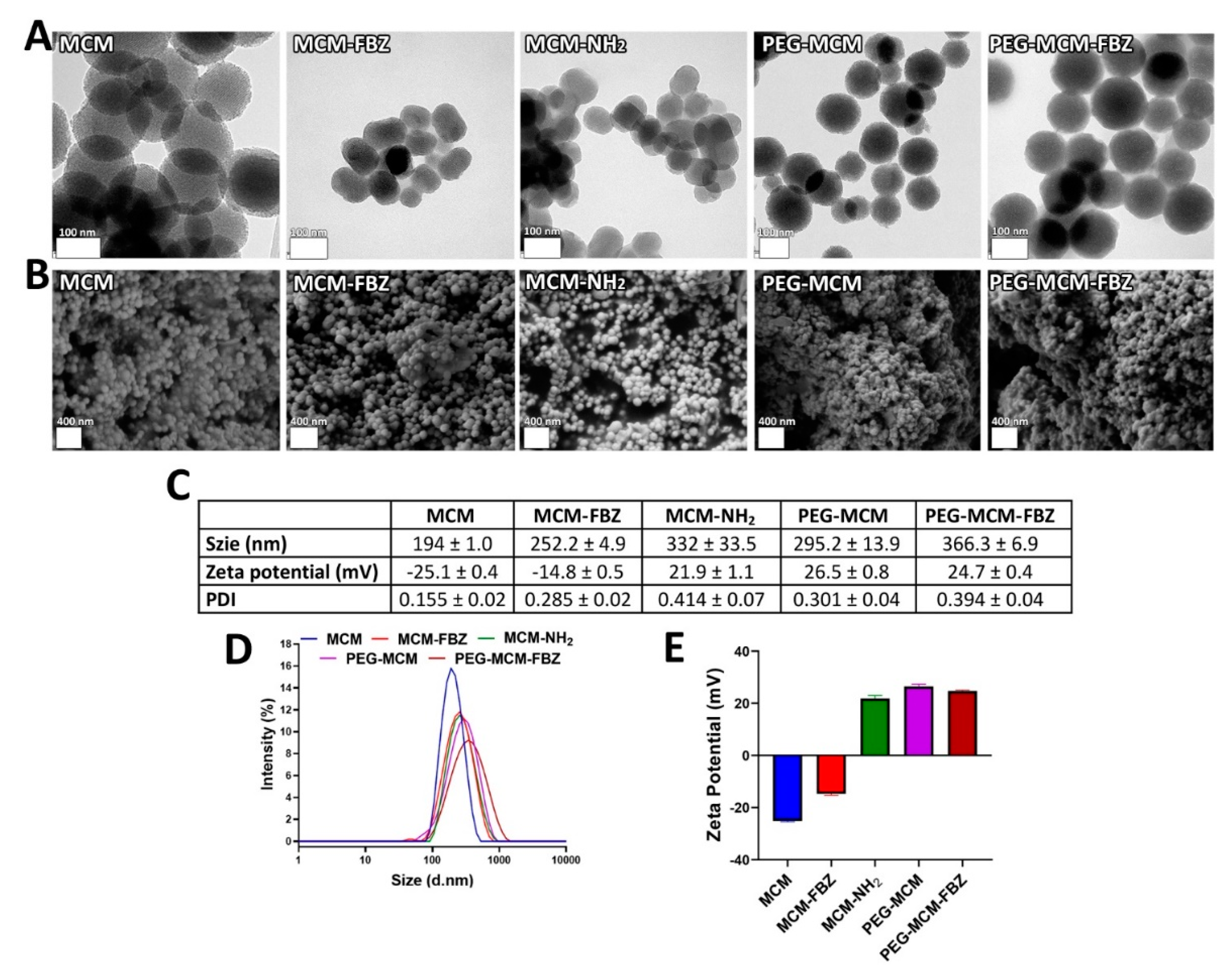
3.1. DLS, TEM, SEM, PDI, and Zeta Potential Characterizations of the Synthesized NPs
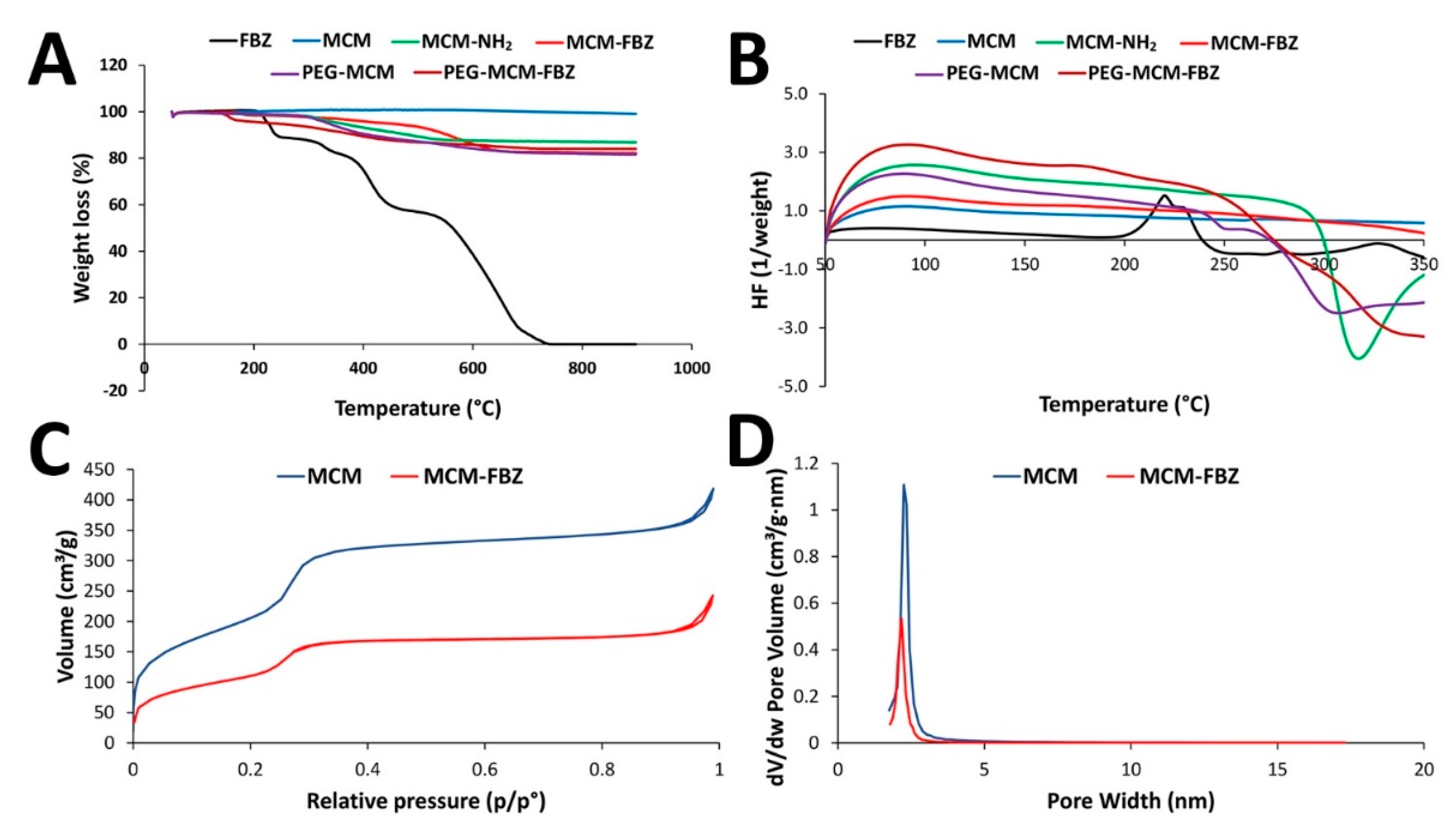
3.2. TGA and DSC Measurements of the Synthesized NPs
3.3. BET Surface Area Analysis
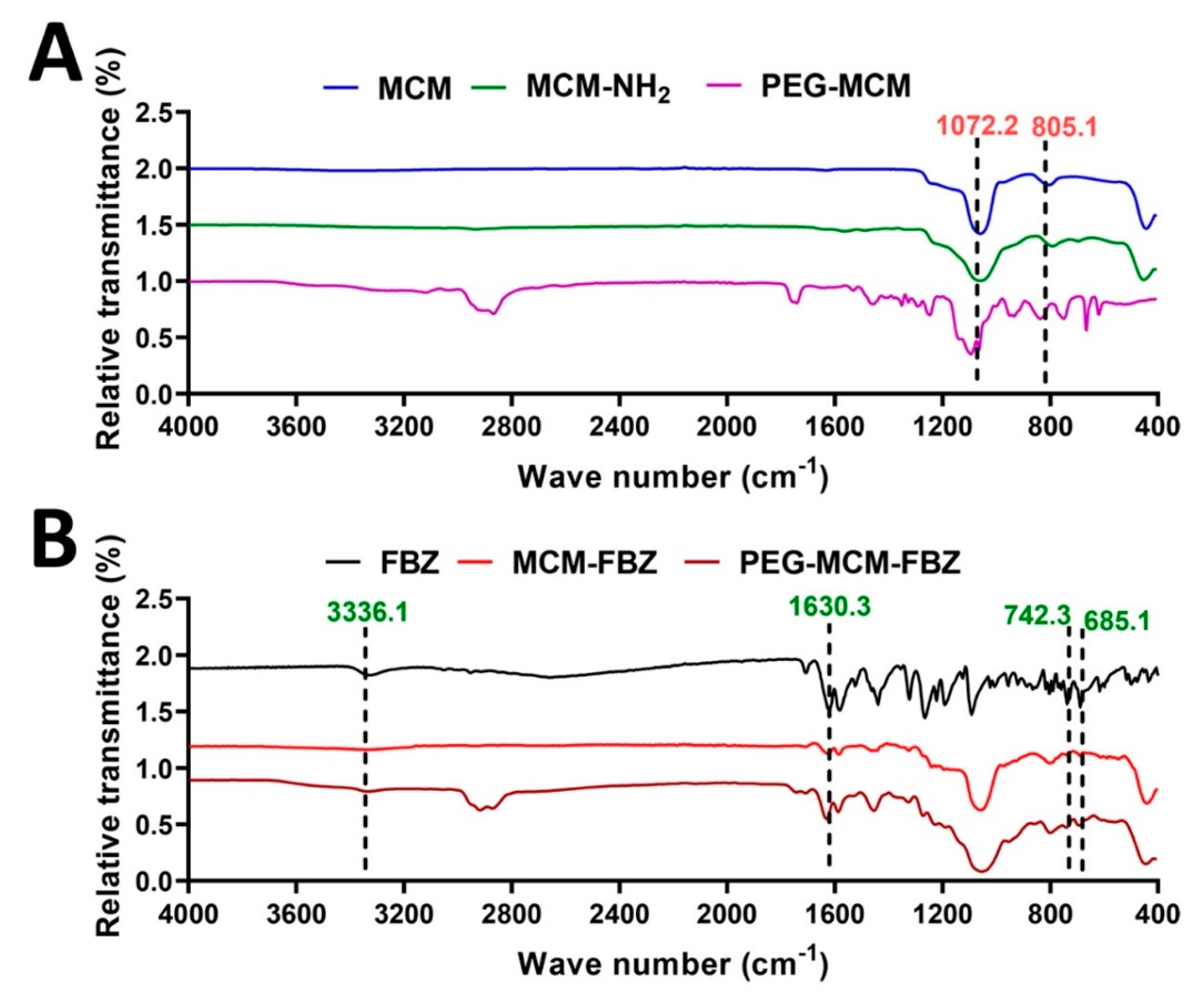
3.4. FTIR
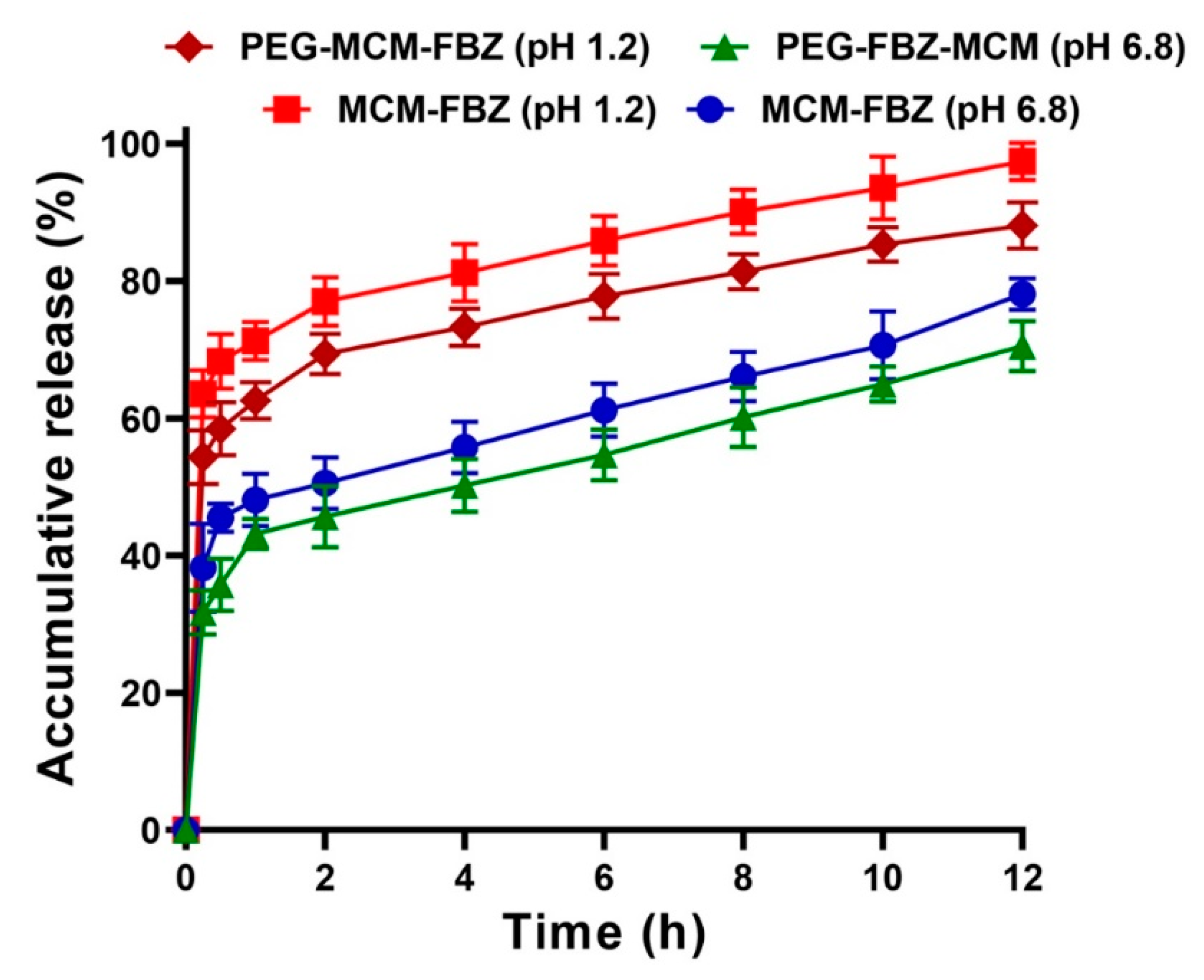
3.5. Release Study
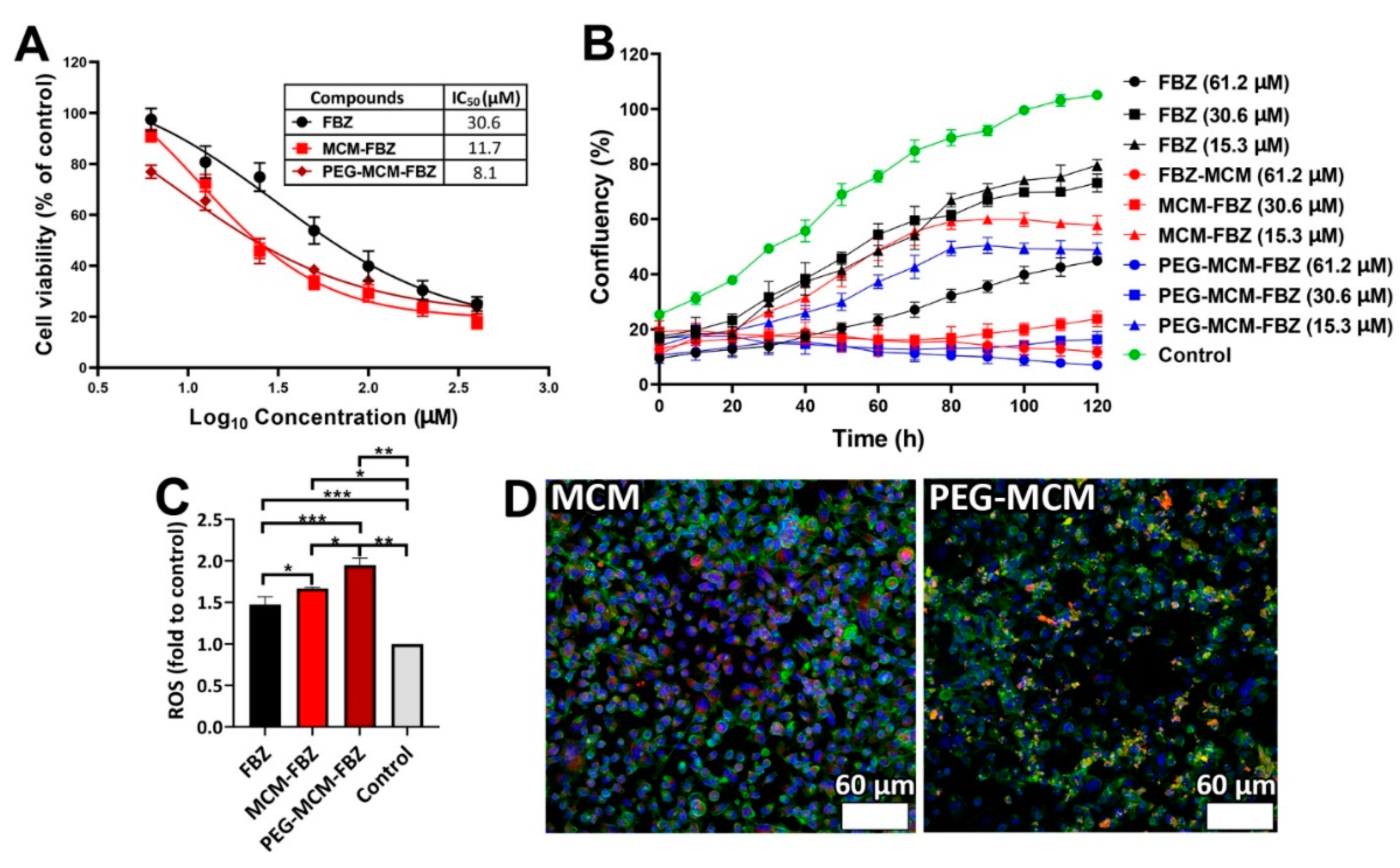
3.6. Cell Viability of the Synthesized Nanoformulations
3.7. Proliferation Assay
3.8. ROS Assay
3.9. Cellular Uptake of FBZ Nanoformulations
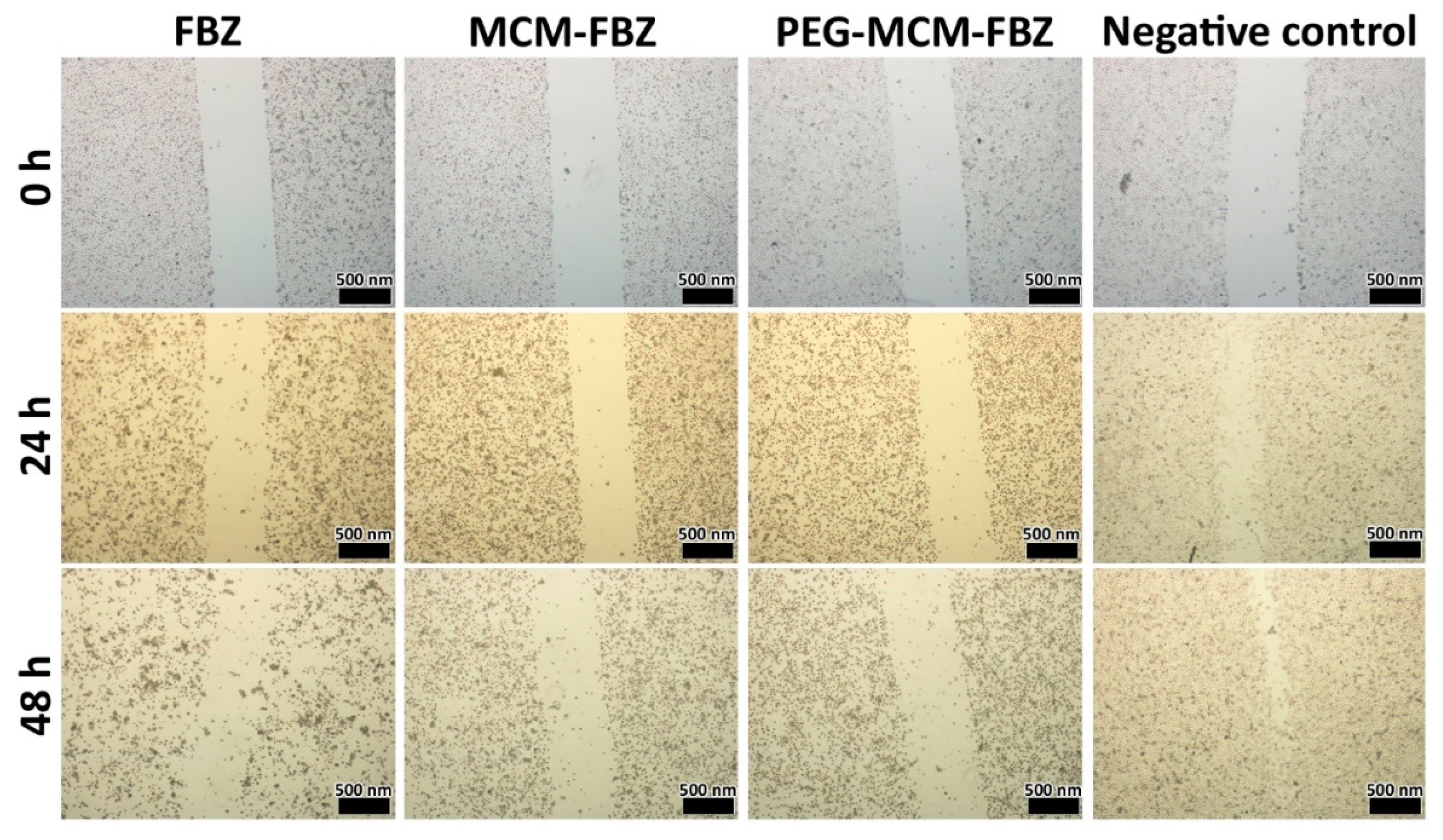
3.10. Cell Migration
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Li, J.; Zheng, S.; Chen, B.; Butte, A.J.; Swamidass, S.J.; Lu, Z. A survey of current trends in computational drug repositioning. Brief. Bioinform. 2016, 17, 2–12. [Google Scholar] [CrossRef] [Green Version]
- Luo, H.; Li, M.; Yang, M.; Wu, F.-X.; Li, Y.; Wang, J. Biomedical data and computational models for drug repositioning: A comprehensive review. Brief. Bioinform. 2020, 22, 1604–1619. [Google Scholar] [CrossRef] [PubMed]
- Jarada, T.N.; Rokne, J.G.; Alhajj, R. A review of computational drug repositioning: Strategies, approaches, opportunities, challenges, and directions. J. Cheminform. 2020, 12, 46. [Google Scholar] [CrossRef] [PubMed]
- Nygren, P.; Fryknäs, M.; Ågerup, B.; Larsson, R. Repositioning of the anthelmintic drug mebendazole for the treatment for colon cancer. J. Cancer Res. Clin. Oncol. 2013, 139, 2133–2140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laudisi, F.; Marônek, M.; Di Grazia, A.; Monteleone, G.; Stolfi, C. Repositioning of Anthelmintic Drugs for the Treatment of Cancers of the Digestive System. Int. J. Mol. Sci. 2020, 21, 4957. [Google Scholar] [CrossRef] [PubMed]
- Arkhipov, I.A.; Khalikov, S.S.; Sadov, K.M.; Dushkin, A.V.; Meteleva, E.S.; Varlamova, A.I.; Odoevskaya, I.M.; Danilevskaya, N.V. Influence of mechanochemical technology on anthelmintic efficacy of the supramolecular complex of fenbendazole with polyvinylpyrrolidone. J. Adv. Vet. Anim. Res. 2019, 6, 133. [Google Scholar] [CrossRef] [PubMed]
- Akkoç, S. Design, synthesis, characterization, and in vitro cytotoxic activity evaluation of 1,2-disubstituted benzimidazole compounds. J. Phys. Org. Chem. 2021, 34, e4125. [Google Scholar] [CrossRef]
- Alavi, S.E.; Shahmabadi, H.E. Anthelmintics for drug repurposing: Opportunities and challenges. Saudi Pharm. J. 2021, 29, 434–445. [Google Scholar] [CrossRef]
- Hernández-Covarrubias, C.; Vilchis-Reyes, M.A.; Yépez-Mulia, L.; Sánchez-Díaz, R.; Navarrete-Vázquez, G.; Hernández-Campos, A.; Castillo, R.; Hernández-Luis, F. Exploring the interplay of physicochemical properties, membrane permeability and giardicidal activity of some benzimidazole derivatives. Eur. J. Med. Chem. 2012, 52, 193–204. [Google Scholar] [CrossRef]
- Dogra, N.; Kumar, A.; Mukhopadhyay, T. Fenbendazole acts as a moderate microtubule destabilizing agent and causes cancer cell death by modulating multiple cellular pathways. Sci. Rep. 2018, 8, 11926. [Google Scholar] [CrossRef]
- Mrkvová, Z.; Uldrijan, S.; Pombinho, A.; Bartůněk, P.; Slaninová, I. Benzimidazoles downregulate Mdm2 and MdmX and activate p53 in MdmX overexpressing tumor cells. Molecules 2019, 24, 2152. [Google Scholar] [CrossRef] [Green Version]
- Lai, S.R.; Castello, S.; Robinson, A.; Koehler, J. In Vitro anti-tubulin effects of mebendazole and fenbendazole on canine glioma cells. Vet. Comp. Oncol. 2017, 15, 1445–1454. [Google Scholar] [CrossRef]
- Shin, H.J.; Jo, M.J.; Jin, I.S.; Park, C.-W.; Kim, J.-S.; Shin, D.H. Optimization and Pharmacokinetic Evaluation of Synergistic Fenbendazole and Rapamycin Co-Encapsulated in Methoxy Poly(Ethylene Glycol)-b-Poly(Caprolactone) Polymeric Micelles. Int. J. Nanomed. 2021, 16, 4873. [Google Scholar] [CrossRef]
- Ghaferi, M.; Koohi Moftakhari Esfahani, M.; Raza, A.; Al Harthi, S.; Ebrahimi Shahmabadi, H.; Alavi, S.E. Mesoporous silica nanoparticles: Synthesis methods and their therapeutic use-recent advances. J. Drug Target. 2020, 29, 131–154. [Google Scholar] [CrossRef]
- Alavi, S.E.; Muflih Al Harthi, S.; Ebrahimi Shahmabadi, H.; Akbarzadeh, A. Cisplatin-loaded polybutylcyanoacrylate nanoparticles with improved properties as an anticancer agent. Int. J. Mol. Sci. 2019, 20, 1531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, Y.; Xie, J.; Chen, H.; Gu, S.; Zhao, R.; Shao, J.; Jia, L. Nanotechnology-based intelligent drug design for cancer metastasis treatment. Biotechnol. Adv. 2014, 32, 761–777. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Li, Z.; Wu, F.; Chen, M.; Wang, R.; Zhu, H.; Li, Q.; Yuan, Y. Improving Solubility and Bioavailability of Breviscapine with Mesoporous Silica Nanoparticles Prepared Using Ultrasound-Assisted Solution-Enhanced Dispersion by Supercritical Fluids Method. Int. J. Nanomed. 2020, 15, 1661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almomen, A.; El-Toni, A.M.; Badran, M.; Alhowyan, A.; Abul Kalam, M.; Alshamsan, A.; Alkholief, M. The design of anionic surfactant-based amino-functionalized mesoporous silica nanoparticles and their application in transdermal drug delivery. Pharmaceutics 2020, 12, 1035. [Google Scholar] [CrossRef]
- Wang, X.; Li, C.; Fan, N.; Li, J.; Zhang, H.; Shang, L.; He, Z.; Sun, J. Amino functionalized chiral mesoporous silica nanoparticles for improved loading and release of poorly water-soluble drug. Asian J. Pharm. Sci. 2019, 14, 405–412. [Google Scholar] [CrossRef]
- Tawfeek, G.M.; Baki, M.H.A.; Ibrahim, A.N.; Mostafa, M.A.H.; Fathy, M.M.; Diab, M.S.E.D.M. Enhancement of the therapeutic efficacy of praziquantel in murine Schistosomiasis mansoni using silica nanocarrier. Parasitol. Res. 2019, 118, 3519–3533. [Google Scholar] [CrossRef] [PubMed]
- Talavera-Pech, W.A.; Ávila-Ortega, A.; Pacheco-Catalán, D.; Quintana-Owen, P.; Barrón-Zambrano, J.A. Effect of Functionalization Synthesis Type of Amino-MCM-41 Mesoporous Silica Nanoparticles on Its RB5 Adsorption Capacity and Kinetics. Silicon 2019, 11, 1547–1555. [Google Scholar] [CrossRef]
- Cheng, W.; Liang, C.; Xu, L.; Liu, G.; Gao, N.; Tao, W.; Luo, L.; Zuo, Y.; Wang, X.; Zhang, X.; et al. TPGS-Functionalized Polydopamine-Modified Mesoporous Silica as Drug Nanocarriers for Enhanced Lung Cancer Chemotherapy against Multidrug Resistance. Small 2017, 13, 1700623. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.X.; Wen, X.; Bell, C.; Appiah, S. Liposome-delivered baicalein induction of myeloid leukemia K562 cell death via reactive oxygen species generation. Mol. Med. Rep. 2018, 17, 4524–4530. [Google Scholar] [CrossRef] [Green Version]
- Kang, B.-S.; Choi, J.-S.; Lee, S.-E.; Lee, J.-K.; Kim, T.-H.; Jang, W.S.; Tunsirikongkon, A.; Kim, J.-K.; Park, J.-S. Enhancing the in vitro anticancer activity of albendazole incorporated into chitosan-coated PLGA nanoparticles. Carbohydr. Polym. 2017, 159, 39–47. [Google Scholar] [CrossRef]
- Patsula, V.; Moskvin, M.; Dutz, S.; Horák, D. Size-dependent magnetic properties of iron oxide nanoparticles. J. Phys. Chem. Solids 2016, 88, 24–30. [Google Scholar] [CrossRef]
- Jiao, Y.; Li, D.; Liu, C.; Chang, Y.; Song, J.; Xiao, Y. Polypeptide–decorated nanoliposomes as novel delivery systems for lutein. RSC Adv. 2018, 8, 31372–31381. [Google Scholar] [CrossRef] [Green Version]
- Dave, V.; Tak, K.; Sohgaura, A.; Gupta, A.; Sadhu, V.; Reddy, K.R. Lipid-polymer hybrid nanoparticles: Synthesis strategies and biomedical applications. J. Microbiol. Methods 2019, 160, 130–142. [Google Scholar] [CrossRef]
- Shah, P.; Rajput, S.J. Investigation of in vitro permeability and in vivo pharmacokinetic behavior of bare and functionalized MCM-41 and MCM-48 mesoporous silica nanoparticles: A burst and controlled drug release system for raloxifene. Drug Dev. Ind. Pharm. 2019, 45, 587–602. [Google Scholar] [CrossRef]
- Khezri, K.; Haddadi-Asl, V.; Roghani-Mamaqani, H. Introduction of a double bond containing modifier on the surface of MCM-41 nanoparticles: Application for SR&NI ATRP of styrene. Nano 2014, 9, 1450023. [Google Scholar]
- Arias, J.L.; López-Viota, M.; Clares, B.; Ruiz, M.A. Stability of fenbendazole suspensions for veterinary use: Correlation between zeta potential and sedimentation. Eur. J. Pharm. Sci. 2008, 34, 257–262. [Google Scholar] [CrossRef] [PubMed]
- Melian, M.E.; Munguía, A.B.; Faccio, R.; Palma, S.; Domínguez, L. The impact of solid dispersion on formulation, using confocal micro Raman spectroscopy as tool to probe distribution of components. J. Pharm. Innov. 2018, 13, 58–68. [Google Scholar] [CrossRef]
- Shah, B.A.; Patel, A.V.; Bagia, M.I.; Oluyinka, O.A. Removal of Cr (VI) from aqueous solutions using EDCC-MCM-41: Isotherm, kinetics and thermodynamic evaluation. J. Dispers. Sci. Technol. 2019, 40, 1827–1841. [Google Scholar] [CrossRef]
- Costa, J.A.; Sarmento, V.H.; Romão, L.P.; Paranhos, C.M. Performance of the MCM-41-NH 2 Functionalized Mesoporous Material Synthetized from the Rice Husk Ash on the Removal of the Polycyclic Aromatic Hydrocarbons. Silicon 2019, 12, 1913–1923. [Google Scholar] [CrossRef]
- Melian, M.E.; Paredes, A.; Munguía, B.; Colobbio, M.; Ramos, J.C.; Teixeira, R.; Manta, E.; Palma, S.; Faccio, R.; Domínguez, L. Nanocrystals of Novel Valerolactam-Fenbendazole Hybrid with Improved in vitro Dissolution Performance. AAPS PharmSciTech 2020, 21, 237. [Google Scholar] [CrossRef]
- Zhang, H.-x.; Liu, E. Spectroscopic and molecular modeling investigation on the binding of a synthesized steroidal amide to protein. J. Lumin. 2014, 153, 182–187. [Google Scholar] [CrossRef]
- Vasvári, G.; Kalmár, J.; Veres, P.; Vecsernyés, M.; Bácskay, I.; Fehér, P.; Ujhelyi, Z.; Haimhoffer, Á.; Rusznyák, Á.; Fenyvesi, F. Matrix systems for oral drug delivery: Formulations and drug release. Drug Discov. Today Technol. 2018, 27, 71–80. [Google Scholar] [CrossRef]
- Caraballo, I. Critical points in the formulation of pharmaceutical swellable controlled release dosage forms—Influence of particle size. Particuology 2009, 7, 421–425. [Google Scholar] [CrossRef]
- Koohi Moftakhari Esfahani, M.; Islam, N.; Cabot, P.J.; Izake, E.L. Development of Thiabendazole-Loaded Mesoporous Silica Nanoparticles for Cancer Therapy. ACS Biomater. Sci. Eng. 2021. [Google Scholar] [CrossRef]
- Ghaferi, M.; Amari, S.; Mohrir, B.V.; Raza, A.; Shahmabadi, H.E.; Alavi, S.E. Preparation, characterization, and evaluation of cisplatin-loaded polybutylcyanoacrylate nanoparticles with improved in vitro and in vivo anticancer activities. Pharmaceuticals 2020, 13, 44. [Google Scholar] [CrossRef] [Green Version]
- Lombardi, B.M.; Sanchez, R.M.T.; Eloy, P.; Genet, M. Interaction of thiabendazole and benzimidazole with montmorillonite. Appl. Clay Sci. 2006, 33, 59–65. [Google Scholar] [CrossRef]
- Muto, S.; Imai, H. Relationship between mesostructures and pH conditions for the formation of silica–cationic surfactant complexes. Microporous Mesoporous Mater. 2006, 95, 200–205. [Google Scholar] [CrossRef]
- Franco, M.S.; Gomes, E.R.; Roque, M.C.; Oliveira, M.C. Triggered Drug Release from Liposomes: Exploiting the Outer and Inner Tumor Environment. Front. Oncol. 2021, 11, 470. [Google Scholar] [CrossRef]
- Alavi, S.E.; Cabot, P.J.; Moyle, P.M. Glucagon-Like Peptide-1 Receptor Agonists and Strategies to Improve Their Efficiency. Mol. Pharm. 2019, 16, 2278–2295. [Google Scholar] [CrossRef] [PubMed]
- Alavi, S.E.; Cabot, P.J.; Yap, G.Y.; Moyle, P.M. Optimized Methods for the Production and Bioconjugation of Site-Specific, Alkyne-Modified Glucagon-like Peptide-1 (GLP-1) Analogs to Azide-Modified Delivery Platforms Using Copper-Catalyzed Alkyne–Azide Cycloaddition. Bioconjugate Chem. 2020, 31, 1820–1834. [Google Scholar] [CrossRef] [PubMed]
- Alavi, S.E.; Cabot, P.J.; Raza, A.; Moyle, P.M. Developing GLP-1 Conjugated Self-Assembling Nanofibers Using Copper-Catalyzed Alkyne–Azide Cycloaddition and Evaluation of Their Biological Activity. Bioconjugate Chem. 2021, 32, 810–820. [Google Scholar] [CrossRef] [PubMed]
- Ghaferi, M.; Asadollahzadeh, M.J.; Akbarzadeh, A.; Ebrahimi Shahmabadi, H.; Alavi, S.E. Enhanced efficacy of pegylated liposomal cisplatin: In vitro and in vivo evaluation. Int. J. Mol. Sci. 2020, 21, 559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaminskas, L.M.; McLeod, V.M.; Kelly, B.D.; Sberna, G.; Boyd, B.J.; Williamson, M.; Owen, D.J.; Porter, C.J. A comparison of changes to doxorubicin pharmacokinetics, antitumor activity, and toxicity mediated by PEGylated dendrimer and PEGylated liposome drug delivery systems. Nanomed. Nanotechnol. Biol. Med. 2012, 8, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Chen, X.; Li, M.; Tan, J.; Zhang, Y.; Yuan, W.; Zhou, J.; Wang, G. 2-D08 as a SUMOylation inhibitor induced ROS accumulation mediates apoptosis of acute myeloid leukemia cells possibly through the deSUMOylation of NOX2. Biochem. Biophys. Res. Commun. 2019, 513, 1063–1069. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Villani, R.M.; Wang, H.; Simpson, M.J.; Roberts, M.S.; Tang, M.; Liang, X. The role of cellular reactive oxygen species in cancer chemotherapy. J. Exp. Clin. Cancer Res. 2018, 37, 266. [Google Scholar] [CrossRef]
- Movahedi, F.; Wu, Y.; Gu, W.; Xu, Z.P. Nanostructuring a Widely Used Antiworm Drug into the Lipid-Coated Calcium Phosphate Matrix for Enhanced Skin Tumor Treatment. ACS Appl. Bio. Mater. 2020, 3, 4230–4238. [Google Scholar] [CrossRef]
- Cui, Q.; Wang, J.-Q.; Assaraf, Y.G.; Ren, L.; Gupta, P.; Wei, L.; Ashby, C.R., Jr.; Yang, D.-H.; Chen, Z.-S. Modulating ROS to overcome multidrug resistance in cancer. Drug Resist. Updates 2018, 41, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Pelaz, B.; del Pino, P.; Maffre, P.; Hartmann, R.; Gallego, M.; Rivera-Fernandez, S.; de la Fuente, J.M.; Nienhaus, G.U.; Parak, W.J. Surface functionalization of nanoparticles with polyethylene glycol: Effects on protein adsorption and cellular uptake. ACS Nano 2015, 9, 6996–7008. [Google Scholar] [CrossRef] [PubMed]
- Sanz-Moreno, V.; Marshall, C.J. The plasticity of cytoskeletal dynamics underlying neoplastic cell migration. Curr. Opin. Cell Biol. 2010, 22, 690–696. [Google Scholar] [CrossRef] [PubMed]
- Canales, J.; Morales, D.; Blanco, C.; Rivas, J.; Díaz, N.; Angelopoulos, I.; Cerda, O. A TR (i) P to cell migration: New roles of TRP channels in mechanotransduction and cancer. Front. Physiol. 2019, 10, 757. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Esfahani, M.K.M.; Alavi, S.E.; Cabot, P.J.; Islam, N.; Izake, E.L. PEGylated Mesoporous Silica Nanoparticles (MCM-41): A Promising Carrier for the Targeted Delivery of Fenbendazole into Prostrate Cancer Cells. Pharmaceutics 2021, 13, 1605. https://doi.org/10.3390/pharmaceutics13101605
Esfahani MKM, Alavi SE, Cabot PJ, Islam N, Izake EL. PEGylated Mesoporous Silica Nanoparticles (MCM-41): A Promising Carrier for the Targeted Delivery of Fenbendazole into Prostrate Cancer Cells. Pharmaceutics. 2021; 13(10):1605. https://doi.org/10.3390/pharmaceutics13101605
Chicago/Turabian StyleEsfahani, Maedeh Koohi Moftakhari, Seyed Ebrahim Alavi, Peter J. Cabot, Nazrul Islam, and Emad L. Izake. 2021. "PEGylated Mesoporous Silica Nanoparticles (MCM-41): A Promising Carrier for the Targeted Delivery of Fenbendazole into Prostrate Cancer Cells" Pharmaceutics 13, no. 10: 1605. https://doi.org/10.3390/pharmaceutics13101605