Probing the DNA Reactivity and the Anticancer Properties of a Novel Tubercidin-Pt(II) Complex

 , ,
, ,  , , ,
, , ,  , and
, and
Abstract
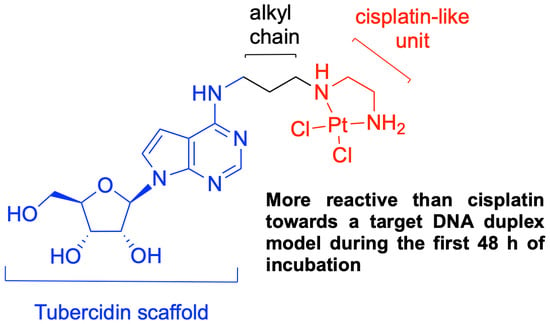
:
1. Introduction
2. Materials and Methods
2.1. General Methods
2.2. Chemistry
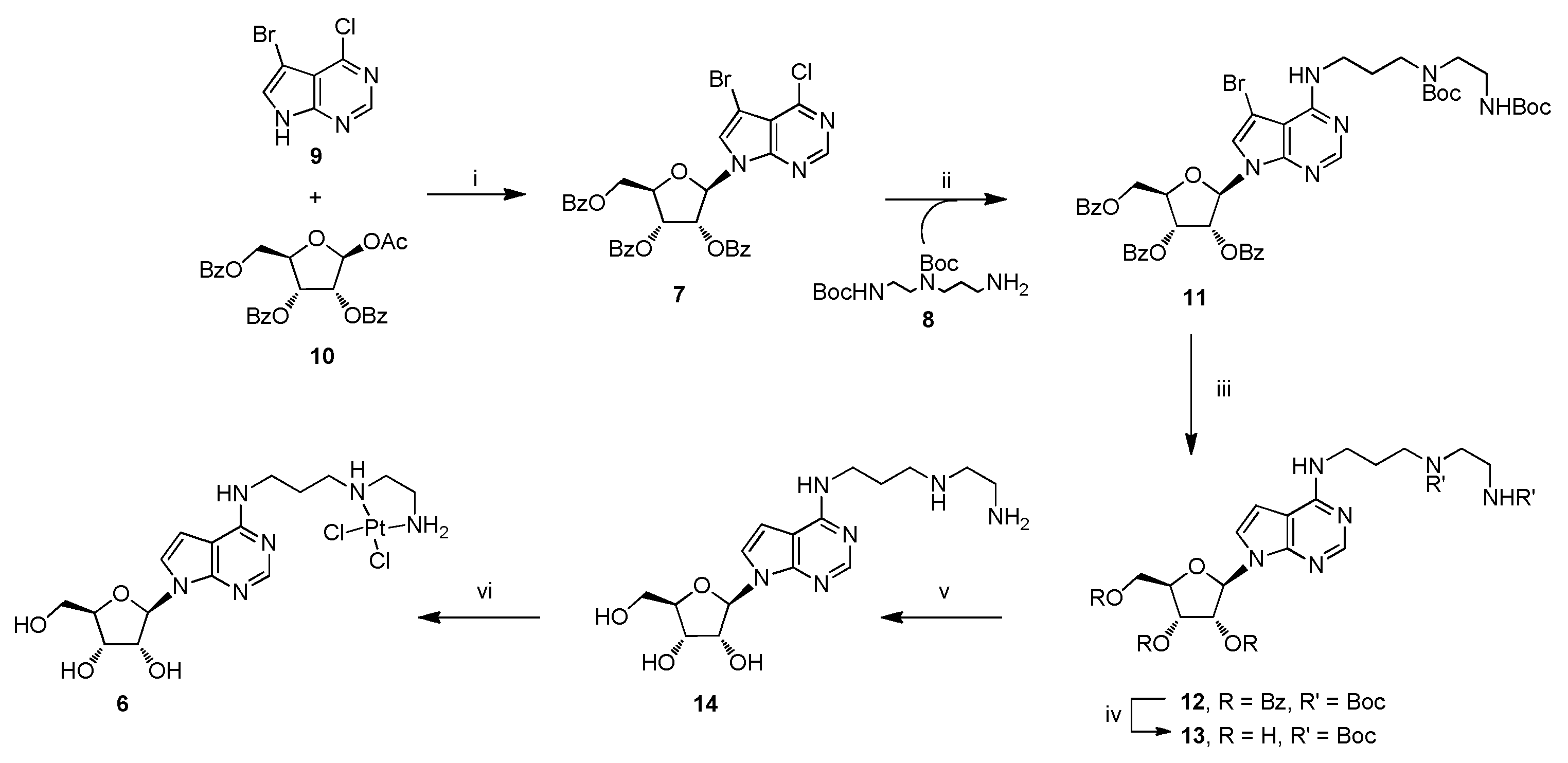
2.2.1. Synthesis of Compound 11
2.2.2. Synthesis of Compound 12
2.2.3. Synthesis of Compound 13
2.2.4. Synthesis of Compound 14
2.2.5. Synthesis of Complex 6
2.2.6. Preparation of ODNs 15, 16
2.2.7. Preparation of Duplex d15/16 and Incubation with Cisplatin, Tubercidin, Complexes 5b and 6 and Diamine 14
2.3. CD Spectroscopy
2.3.1. CD Data Acquisition
2.3.2. CD Melting
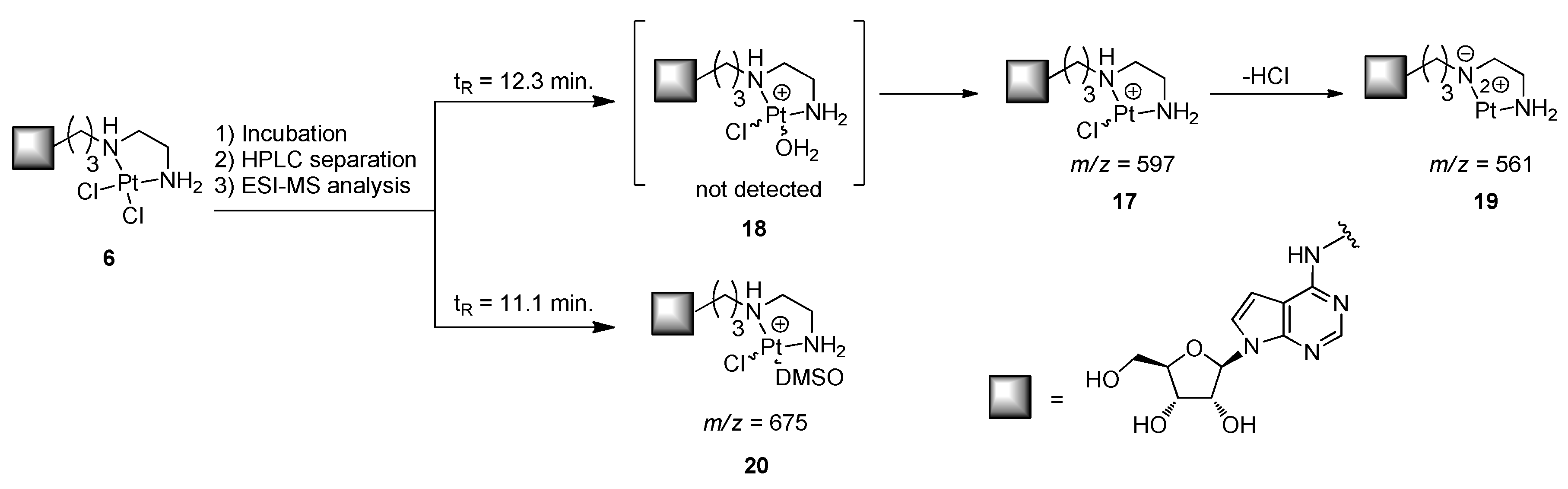
2.4. Studies of Stability of the Complex 6
2.5. Biology
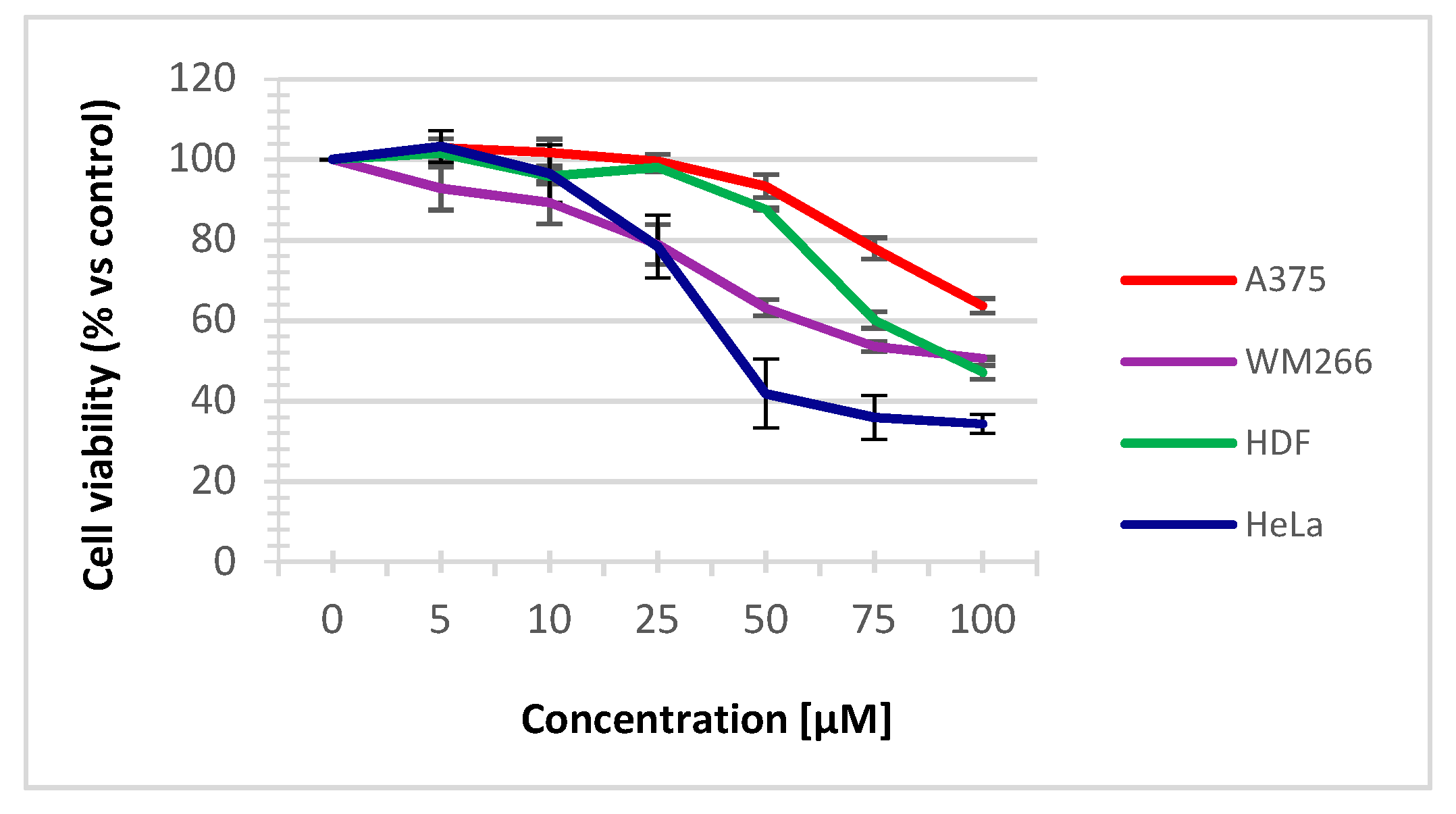
2.5.1. Cell viability assay
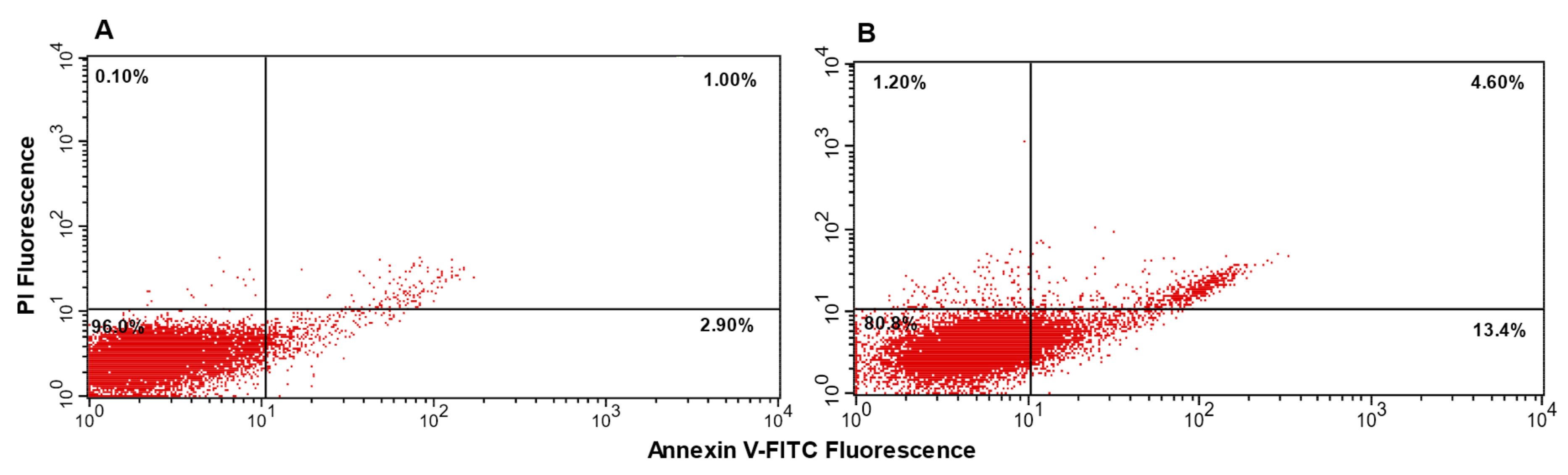
2.5.2. Apoptosis Assay
3. Results and Discussion
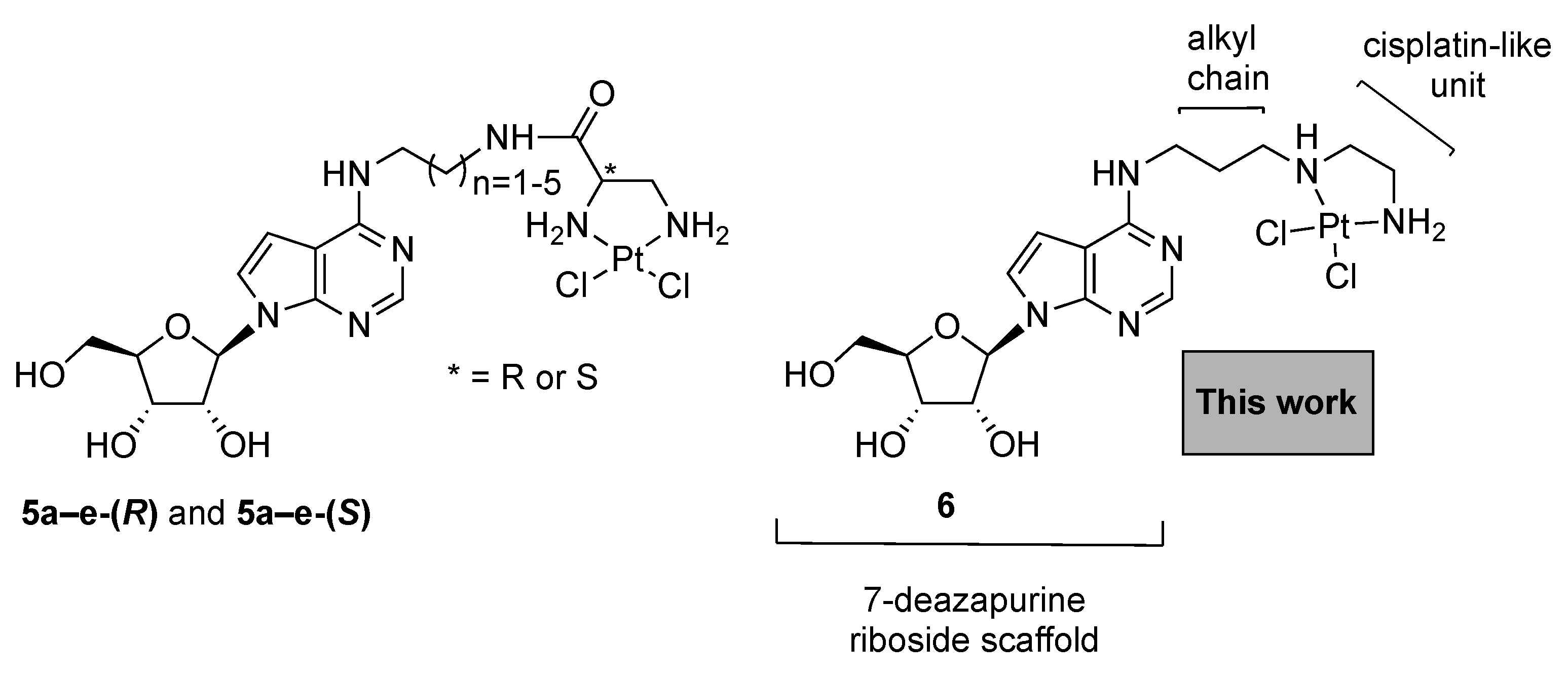
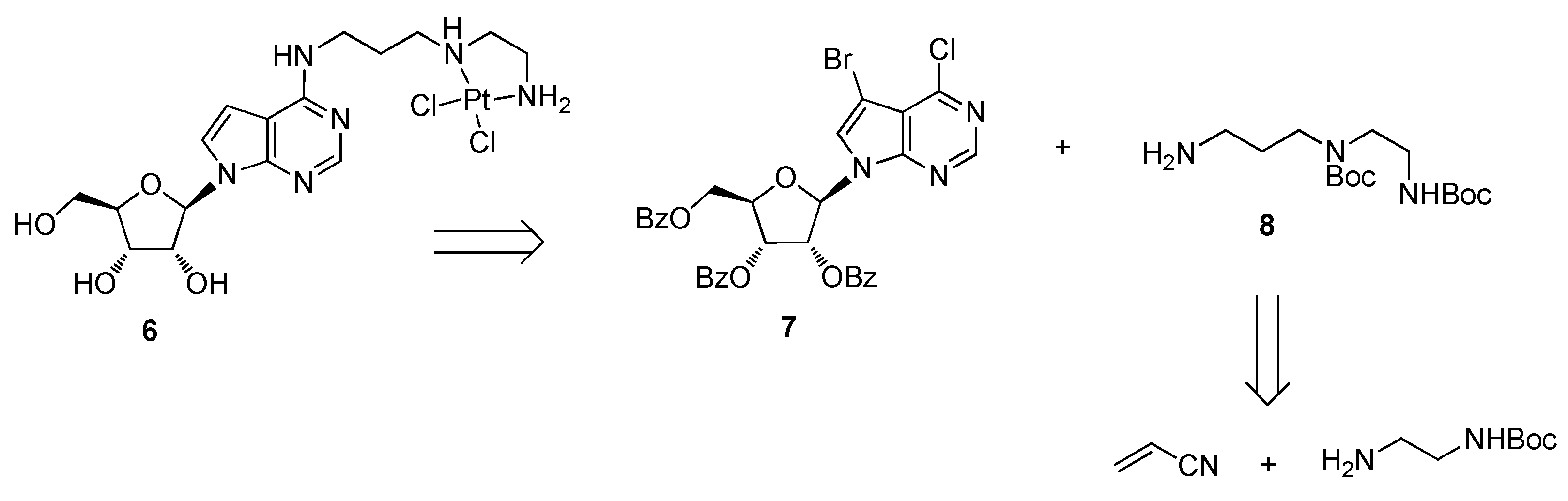
3.1. Chemistry
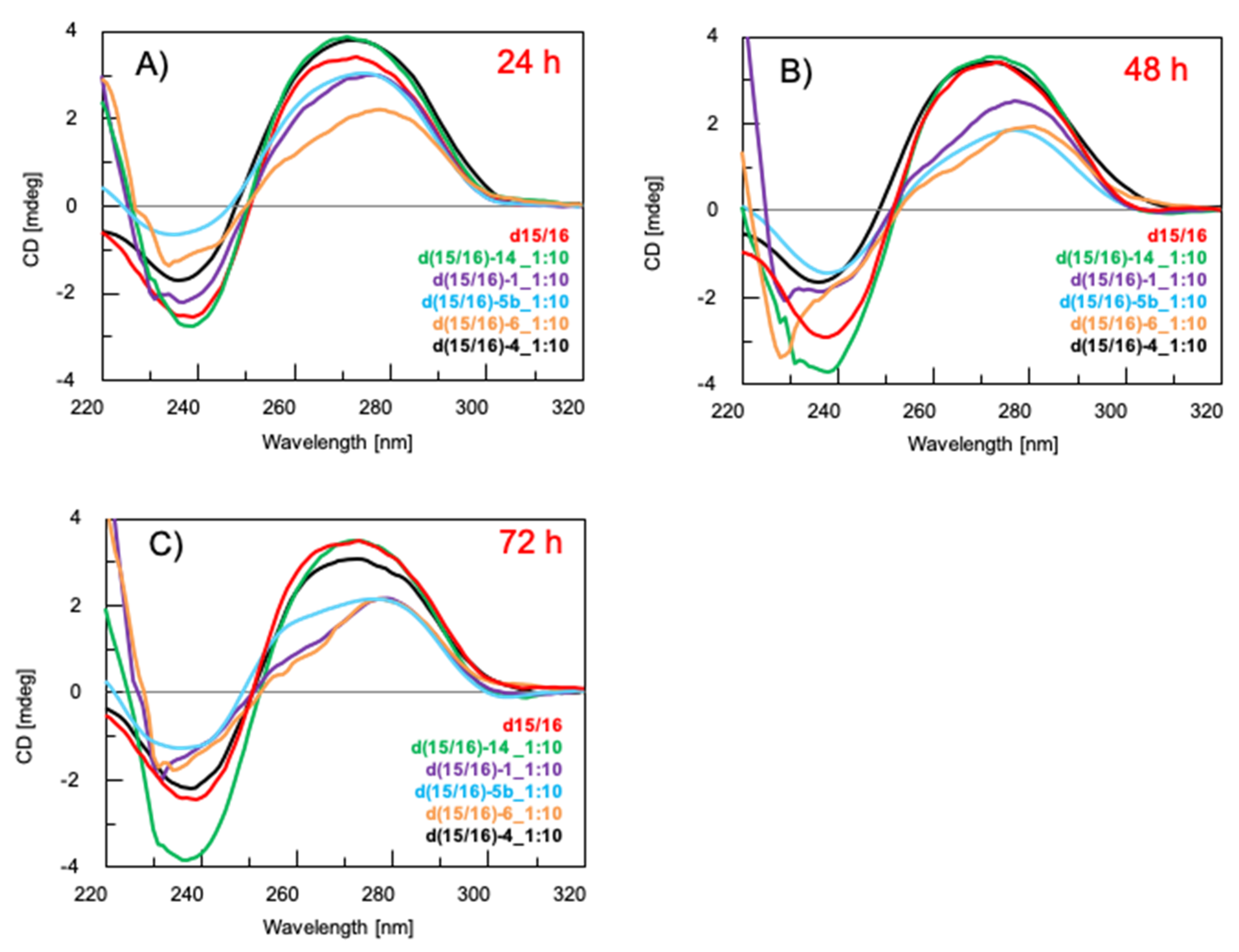
3.2. Study of the Interaction of the Platinum Complex 6 with the Model Duplex DNA d15/16 through CD Spectroscopy
3.3. Study of Stability of the Complex 6 Dissolved in Physiological Solution
3.4. Biology
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Dagenais, G.R.; Leong, D.P.; Rangarajan, S.; Lanas, F.; Lopez-Jaramillo, P.; Gupta, R.; Diaz, R.; Avezum, A.; Oliveira, G.B.F.; Wielgosz, A.; et al. Variations in common diseases, hospital admissions, and deaths in middle-aged adults in 21 countries from five continents (PURE): a prospective cohort study. Lancet 2019, 6736, 1–10. [Google Scholar] [CrossRef]
- Ndagi, U.; Mhlongo, N.; Soliman, M.E. Metal complexes in cancer therapy – An update from drug design perspective. Drug Des. Dev. Ther. 2017, 11, 599–616. [Google Scholar] [CrossRef] [Green Version]
- Rosenberg, B.; VanCapm, L.; Trosko, J.E.; Mansour, V.H. Platinum Compounds: a New Class of Potent Antitumour Agents. Nature 1969, 222, 385–386. [Google Scholar] [CrossRef]
- Ghosh, S. Cisplatin: The First Metal Based Anticancer Drug. Bioorg. Chem. 2019, 88, 102925. [Google Scholar] [CrossRef]
- Dilruba, S.; Kalayda, G.V. Platinum-based drugs: past, present and future. Cancer Chemother. Pharmacol. 2016, 77, 1103–1124. [Google Scholar] [CrossRef]
- Apps, M.G.; Choi, E.H.Y.; Wheate, N.J. The state-of-play and future of platinum drugs. Endocr. Relat. Cancer 2015, 22, R219–R233. [Google Scholar] [CrossRef] [Green Version]
- Mjos, K.D.; Orvig, C. Metallodrugs in medicinal inorganic chemistry. Chem. Rev. 2014, 114, 4540–4563. [Google Scholar] [CrossRef]
- Wang, Z.; Wu, M.; Gou, S. Toward a better understanding of the oxaliplatin mode of action upon the steric hindrance of 1,2-diaminocyclohexane and its analogue. J. Inorg. Biochem. 2016, 157, 1–7. [Google Scholar] [CrossRef]
- Sengupta, P.; Basu, S.; Soni, S.; Pandey, A.; Roy, B.; Oh, M.S.; Chin, K.T.; Paraskar, A.S.; Sarangi, S.; Connor, Y.; et al. Cholesterol-tethered platinum II-based supramolecular nanoparticle increases antitumor efficacy and reduces nephrotoxicity. Proc. Natl. Acad. Sci. USA 2012, 109, 11294–11299. [Google Scholar] [CrossRef] [Green Version]
- Vhora, I.; Khatri, N.; Desai, J.; Thakkar, H.P. Caprylate-conjugated cisplatin for the development of novel liposomal formulation. AAPS PharmSciTech 2014, 15, 845–857. [Google Scholar] [CrossRef] [Green Version]
- Kitteringham, E.; Andriollo, E.; Gandin, V.; Montagner, D.; Griffith, D.M. Synthesis, characterisation and in vitro antitumour potential of novel Pt(II) estrogen linked complexes. Inorg. Chim. Acta 2019, 495, 118944. [Google Scholar] [CrossRef]
- Robillard, M.S.; Bacac, M.; Van Den Elst, H.; Flamigni, A.; Van Der Marel, G.A.; Van Boom, J.H.; Reedijk, J. Automated Parallel Solid-Phase Synthesis and Anticancer Screening of a Library of Peptide-Tethered Platinum(II) Complexes. J. Comb. Chem. 2003, 5, 821–825. [Google Scholar] [CrossRef]
- Robillard, M.S.; Valentijn, A.R.P.M.; Meeuwenoord, N.J.; Van Der Marel, G.A.; Van Boom, J.H.; Reedijk, J. The First Solid-Phase Synthesis of a Peptide- Tethered Platinum (II) Complex. Angew. Chem. Int. Ed. 2000, 39, 3096–3099. [Google Scholar] [CrossRef]
- Cucciolito, M.E.; De Luca Bossa, F.; Esposito, R.; Ferraro, G.; Iadonisi, A.; Petruk, G.; D’Elia, L.; Romanetti, C.; Traboni, S.; Tuzi, A.; et al. C-Glycosylation in platinum-based agents: A viable strategy to improve cytotoxicity and selectivity. Inorg. Chem. Front. 2018, 5, 2921–2933. [Google Scholar] [CrossRef]
- Annunziata, A.; Cucciolito, M.E.; Esposito, R.; Imbimbo, P.; Petruk, G.; Ferraro, G.; Pinto, V.; Tuzi, A.; Monti, D.M.; Merlino, A.; et al. A highly efficient and selective antitumor agent based on a glucoconjugated carbene platinum(ii) complex. Dalton Trans. 2019, 48, 7794–7800. [Google Scholar] [CrossRef]
- Han, J.; Gao, X.; Liu, R.; Yang, J.; Zhang, M.; Mi, Y.; Shi, Y.; Gao, Q. Design, Synthesis of Novel Platinum(II) Glycoconjugates, and Evaluation of Their Antitumor Effects. Chem. Biol. Drug Des. 2016, 87, 867–877. [Google Scholar] [CrossRef]
- Ma, D.-L.; Wu, C.; Cheng, S.-S.; Lee, F.-W.; Han, Q.-B.; Leung, C.-H.; Ma, D.-L.; Wu, C.; Cheng, S.-S.; Lee, F.-W.; et al. Development of Natural Product-Conjugated Metal Complexes as Cancer Therapies. Int. J. Mol. Sci. 2019, 20, 341. [Google Scholar] [CrossRef] [Green Version]
- Burke, M.; Borland, K.; Litosh, V. Base-Modified Nucleosides as Chemotherapeutic Agents: Past and Future. Curr. Top. Med. Chem. 2016, 16, 1231–1241. [Google Scholar] [CrossRef]
- Berdis, A.J. Inhibiting DNA polymerases as a therapeutic intervention against cancer. Front. Mol. Biosci. 2017, 4, 1–12. [Google Scholar] [CrossRef] [Green Version]
- De Clercq, E.; Li, G. Approved antiviral drugs over the past 50 years. Clin. Microbiol. Rev. 2016, 29, 695–747. [Google Scholar] [CrossRef] [Green Version]
- Caso, M.F.; D’Alonzo, D.; D’Errico, S.; Palumbo, G.; Guaragna, A. Highly stereoselective synthesis of lamivudine (3TC) and emtricitabine (FTC) by a novel N -glycosidation procedure. Org. Lett. 2015, 17, 2626–2629. [Google Scholar] [CrossRef] [PubMed]
- Shahabadi, N.; Abbasi, A.R.; Moshtkob, A.; Hadidi, S. Design, synthesis and DNA interaction studies of new fluorescent platinum complex containing anti-HIV drug didanosine. J. Biomol. Struct. Dyn. 2019, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Shahabadi, N.; Fatahi, S.; Maghsudi, M. Synthesis of a new Pt(II) complex containing valganciclovir drug and calf-thymus DNA interaction study using multispectroscopic methods. J. Coord. Chem. 2018, 71, 258–270. [Google Scholar] [CrossRef]
- Chen, J.; Li, K.; Swavey, S.; Church, K.M. Synthesis, characterization and DNA binding activity of PtCl2[DMSO][N4[N-3(4-pyridylmethyl)thymidine]]. Inorg. Chim. Acta 2016, 444, 76. [Google Scholar] [CrossRef]
- Montagner, D.; Gandin, V.; Marzano, C.; Longato, B. Synthesis, characterization and cytotoxic properties of platinum(II) complexes containing the nucleosides adenosine and cytidine. J. Inorg. Biochem. 2011, 105, 919–926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, L.; Lim, K.; Ren, S.; Cadena, R.S.; Beck, W.T. Synthesis and in vitro antitumor activity of oligonucleotide-tethered and related platinum complexes. J. Med. Chem. 2001, 44, 2959–2965. [Google Scholar] [CrossRef] [PubMed]
- Coluccia, M.; Boccarelli, A.; Cermelli, C.; Portolani, M.; Natile, G. Platinum(II)-Acyclovir Complexes: Synthesis, Antiviral and Antitumour Activity. Met. Based Drugs 1995, 2, 249–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Errico, S.; Oliviero, G.; Piccialli, V.; Amato, J.; Borbone, N.; D’Atri, V.; D’Alessio, F.; Di Noto, R.; Ruffo, F.; Salvatore, F.; et al. Solid-phase synthesis and pharmacological evaluation of novel nucleoside-tethered dinuclear platinum(II) complexes. Bioorg. Med. Chem. Lett. 2011, 21, 5835–5838. [Google Scholar] [CrossRef]
- D’Errico, S.; Oliviero, G.; Borbone, N.; Piccialli, V.; Pinto, B.; De Falco, F.; Maiuri, M.C.; Carnuccio, R.; Costantino, V.; Nici, F.; et al. Synthesis and pharmacological evaluation of modified adenosines joined to mono-functional platinum moieties. Molecules 2014, 19, 9339–9353. [Google Scholar] [CrossRef] [Green Version]
- Pastor-Anglada, M.; Pérez-Torras, S. Emerging roles of nucleoside transporters. Front. Pharmacol. 2018, 9, 1–8. [Google Scholar] [CrossRef]
- Mulamoottil, V.A. Tubercidin and Related Analogues: An Inspiration for 50 years in Drug Discovery. Curr. Org. Chem. 2016, 2, 830–838. [Google Scholar] [CrossRef]
- Perlíková, P.; Hocek, M. Pyrrolo[2,3-d]pyrimidine (7-deazapurine) as a privileged scaffold in design of antitumor and antiviral nucleosides. Med. Res. Rev. 2017, 37, 1429–1460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piccolo, M.; Misso, G.; Ferraro, M.G.; Riccardi, C.; Capuozzo, A.; Zarone, M.R.; Maione, F.; Trifuoggi, M.; Stiuso, P.; D’Errico, G.; et al. Exploring cellular uptake, accumulation and mechanism of action of a cationic Ru-based nanosystem in human preclinical models of breast cancer. Sci. Rep. 2019, 9, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Errico, S.; Oliviero, G.; Borbone, N.; Di Gennaro, E.; Zotti, A.I.; Budillon, A.; Cerullo, V.; Nici, F.; Mayol, L.; Piccialli, V.; et al. Synthesis and Evaluation of the Antiproliferative Properties of a Tethered Tubercidin-Platinum(II) Complex. Eur. J. Org. Chem. 2015, 7550–7556. [Google Scholar] [CrossRef]
- D’Errico, S.; Borbone, N.; Piccialli, V.; Di Gennaro, E.; Zotti, A.; Budillon, A.; Vitagliano, C.; Piccialli, I.; Oliviero, G. Synthesis and Evaluation of the Antitumor Properties of a Small Collection of Pt II Complexes with 7-Deazaadenosine as Scaffold. Eur. J. Org. Chem. 2017, 4935–4947. [Google Scholar] [CrossRef]
- Oliviero, G.; Amato, J.; Borbone, N.; D’Errico, S.; Piccialli, G.; Mayol, L. Synthesis of N-1 and ribose modified inosine analogues on solid support. Tetrahedron Lett. 2007, 48, 397–400. [Google Scholar] [CrossRef]
- D’Errico, S.; Piccialli, V.; Oliviero, G.; Borbone, N.; Amato, J.; D’Atri, V.; Piccialli, G. Probing the reactivity of nebularine N1-oxide. A novel approach to C-6 C-substituted purine nucleosides. Tetrahedron 2011, 67, 6138–6144. [Google Scholar] [CrossRef]
- D’Errico, S.; Oliviero, G.; Borbone, N.; Amato, J.; Piccialli, V.; Varra, M.; Mayol, L.; Piccialli, G. Solid-phase synthesis of a new diphosphate 5-aminoimidazole-4-carboxamide riboside (AICAR) derivative and studies toward cyclic AICAR diphosphate ribose. Molecules 2011, 16, 8110–8118. [Google Scholar] [CrossRef] [Green Version]
- Comegna, D.; Zannetti, A.; Del Gatto, A.; De Paola, I.; Russo, L.; Di Gaetano, S.; Liguoro, A.; Capasso, D.; Saviano, M.; Zaccaro, L. Chemical Modification for Proteolytic Stabilization of the Selective αvβ3 Integrin RGDechi Peptide: In Vitro and in Vivo Activities on Malignant Melanoma Cells. J. Med. Chem. 2017, 60, 9874–9884. [Google Scholar] [CrossRef]
- Oliviero, G.; Amato, J.; Borbone, N.; D’Errico, S.; Piccialli, G.; Bucci, E.; Piccialli, V.; Mayol, L. Synthesis of 4-N-alkyl and ribose-modified AICAR analogues on solid support. Tetrahedron 2008, 64, 6475–6481. [Google Scholar] [CrossRef]
- Falanga, A.P.; Cerullo, V.; Marzano, M.; Feola, S.; Oliviero, G.; Piccialli, G.; Borbone, N. Peptide Nucleic Acid-Functionalized Adenoviral Vectors Targeting G-Quadruplexes in the P1 Promoter of Bcl-2 Proto-Oncogene: A New Tool for Gene Modulation in Anticancer Therapy. Bioconjug. Chem. 2019, 30, 572–582. [Google Scholar] [CrossRef] [PubMed]
- Amato, F.; Tomaiuolo, R.; Nici, F.; Borbone, N.; Elce, A.; Catalanotti, B.; D’Errico, S.; Morgillo, C.M.; De Rosa, G.; Mayol, L.; et al. Exploitation of a very small peptide nucleic acid as a new inhibitor of miR-509-3p involved in the regulation of cystic fibrosis disease-gene expression. BioMed Res. Int. 2014, 2014, 68–71. [Google Scholar] [CrossRef] [PubMed]
- Capasso, D.; Di Gaetano, S.; Celentano, V.; Diana, D.; Festa, L.; Di Stasi, R.; De Rosa, L.; Fattorusso, R.; D’Andrea, L.D. Unveiling a VEGF-mimetic peptide sequence in the IQGAP1 protein. Mol. Biosyst. 2017, 13, 1619–1629. [Google Scholar] [CrossRef]
- Seela, F.; Ming, X. 7-Functionalized 7-deazapurine β-D and β-L-ribonucleosides related to tubercidin and 7-deazainosine: glycosylation of pyrrolo[2,3-d]pyrimidines with 1-O-acetyl-2,3,5-tri-O-benzoyl-β-D or β-L-ribofuranose. Tetrahedron 2007, 63, 9850–9861. [Google Scholar] [CrossRef]
- Eisenfu, A.; Arora, P.S.; Sengle, G.; Takaoka, L.R.; Nowick, S.; Famulok, M. A Ribozyme with Michaelase Activity: Synthesis of the Substrate Precursors. Bioorg. Med. Chem. 2003, 11, 235–249. [Google Scholar] [CrossRef]
- D’Errico, S.; Oliviero, G.; Borbone, N.; Amato, J.; D’Alonzo, D.; Piccialli, V.; Mayol, L.; Piccialli, G. A facile synthesis of 5’-Fluoro-5’-deoxyacadesine (5’-F-AICAR): A novel non-phosphorylable AICAR Analogue. Molecules 2012, 17, 13036–13044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, Y.; Zhang, M.-M.; Chen, Z.-F.; Hu, K.; Liu, Y.-C.; Chen, X.; Liang, H. Synthesis, Characterization, and Interaction with Biomolecules of Platinum(II) Complexes with Shikimic Acid-Based Ligands. Bioinorg. Chem. Appl. 2013, 2013, 565032. [Google Scholar] [CrossRef]
- Mügge, C.; Musumeci, D.; Michelucci, E.; Porru, F.; Marzo, T.; Massai, L.; Messori, L.; Weigand, W.; Montesarchio, D. Elucidating the reactivity of Pt(II) complexes with (O, S) bidentate ligands towards DNA model systems. J. Inorg. Biochem. 2016, 160, 198–209. [Google Scholar] [CrossRef]
- Musumeci, D.; Platella, C.; Riccardi, C.; Merlino, A.; Marzo, T.; Massai, L.; Messori, L.; Montesarchio, D. A first-in-class and a fished out anticancer platinum compound:: Cis -[PtCl2(NH3)2] and cis -[PtI2(NH3)2] compared for their reactivity towards DNA model systems. Dalton Trans. 2016, 45, 8587–8600. [Google Scholar] [CrossRef]
- Censi, V.; Caballero, A.B.; Pérez-Hernández, M.; Soto-Cerrato, V.; Korrodi-Gregório, L.; Pérez-Tomás, R.; Dell’Anna, M.M.; Mastrorilli, P.; Gamez, P. DNA-binding and in vitro cytotoxic activity of platinum(II) complexes of curcumin and caffeine. J. Inorg. Biochem. 2019, 198, 110749. [Google Scholar] [CrossRef]
- Sirajuddin, M.; Ali, S.; Badshah, A. Drug-DNA interactions and their study by UV-Visible, fluorescence spectroscopies and cyclic voltametry. J. Photochem. Photobiol. B Biol. 2013, 124, 1–19. [Google Scholar] [CrossRef]
- Roviello, G.N.; Vicidomini, C.; Costanzo, V.; Roviello, V. Nucleic acid binding and other biomedical properties of artificial oligolysines. Int. J. Nanomed. 2016, 11, 5897–5904. [Google Scholar] [CrossRef] [Green Version]
- Varbanov, H.P.; Ortiz, D.; Höfer, D.; Menin, L.; Galanski, M.; Keppler, B.K.; Dyson, P.J. Oxaliplatin reacts with DMSO only in the presence of water. Dalt. Trans. 2017, 46, 8929–8932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Josephsen, J. Diaminehalogenoplatinum(II) complex reactions with DMSO. Inorg. Chim. Acta 2018, 478, 54–58. [Google Scholar] [CrossRef]
- Hall, M.D.; Telma, K.A.; Chang, K.E.; Lee, T.D.; Madigan, J.P.; Lloyd, J.R.; Goldlust, I.S.; Hoeschele, J.D.; Gottesman, M.M. Say no to DMSO: Dimethylsulfoxide inactivates cisplatin, carboplatin, and other platinum complexes. Cancer Res. 2014, 74, 3913–3922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boersma, A.W.M.; Nooter, K.; Oostrum, R.G.; Stoter, G. Quantification of apoptotic cells with fluorescein isothiocyanate-labeled annexin V in Chinese hamster ovary cell cultures treated with cisplatin. Cytometry 1996, 24, 123–130. [Google Scholar] [CrossRef]
- Velàzquez, M.; Maldonado, V.; Melendez-Zajgla, J. Cisplatin-induced apoptosis of HeLa cells. Effect of RNA and protein synthesis inhibitors, Ca2+ chelators and zinc. J. Exp. Clin. Cancer Res. 1998, 17, 277–284. [Google Scholar] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ODN | Sequence |
|---|---|
| 15 | d(5’-GGAGACCAGAGG-3’) |
| 16 | d(5’-CCTCTGGTCTCC-3’) |
| d15/16 | d(5’-GGAGACCAGAGG-3’) d(3’-CCTCTGGTCTCC-5’) |
| Entry | IC50 [μM] | |||
|---|---|---|---|---|
| Cell lines | HeLa | A375 | WM266 | HDF |
| 6 | 55.1 ± 14.6 | >100 | 91.0 ± 11.9 | >100 |
| 14 | >100 | >100 | >100 | >100 |
| Cisplatin | 1.8 ± 0.75 | 1.3 ± 0.28 | 2.4 ± 0.40 | 6.3 ± 1.8 |
| Tubercidin | 0.11 ± 0.3 | 0.085 ± 0.023 | 0.076 ± 0.016 | 0.47 ± 0.2 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
D’Errico, S.; Falanga, A.P.; Capasso, D.; Di Gaetano, S.; Marzano, M.; Terracciano, M.; Roviello, G.N.; Piccialli, G.; Oliviero, G.; Borbone, N. Probing the DNA Reactivity and the Anticancer Properties of a Novel Tubercidin-Pt(II) Complex. Pharmaceutics 2020, 12, 627. https://doi.org/10.3390/pharmaceutics12070627
D’Errico S, Falanga AP, Capasso D, Di Gaetano S, Marzano M, Terracciano M, Roviello GN, Piccialli G, Oliviero G, Borbone N. Probing the DNA Reactivity and the Anticancer Properties of a Novel Tubercidin-Pt(II) Complex. Pharmaceutics. 2020; 12(7):627. https://doi.org/10.3390/pharmaceutics12070627
Chicago/Turabian StyleD’Errico, Stefano, Andrea Patrizia Falanga, Domenica Capasso, Sonia Di Gaetano, Maria Marzano, Monica Terracciano, Giovanni Nicola Roviello, Gennaro Piccialli, Giorgia Oliviero, and Nicola Borbone. 2020. "Probing the DNA Reactivity and the Anticancer Properties of a Novel Tubercidin-Pt(II) Complex" Pharmaceutics 12, no. 7: 627. https://doi.org/10.3390/pharmaceutics12070627