Pharmacokinetic Interaction between Sorafenib and Atorvastatin, and Sorafenib and Metformin in Rats

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animal Experiments
2.2. Reagents
2.3. HPLC-UV Assays
2.4. UPLC-MS/MS Assay
2.5. Pharmacokinetic Evaluation
- The elimination rate constant (ke);
- The absorption rate constant (ka);
- The elimination half-life (t1/2);
- The maximum plasma concentration (Cmax);
- The time to reach the Cmax (tmax);
- The total area under the concentration–time curve (AUC0-t and AUC0--∞);
- The apparent plasma drug clearance (Cl/F);
- The apparent volume of distribution (Vd/F).
2.6. Statistical Analysis
3. Results
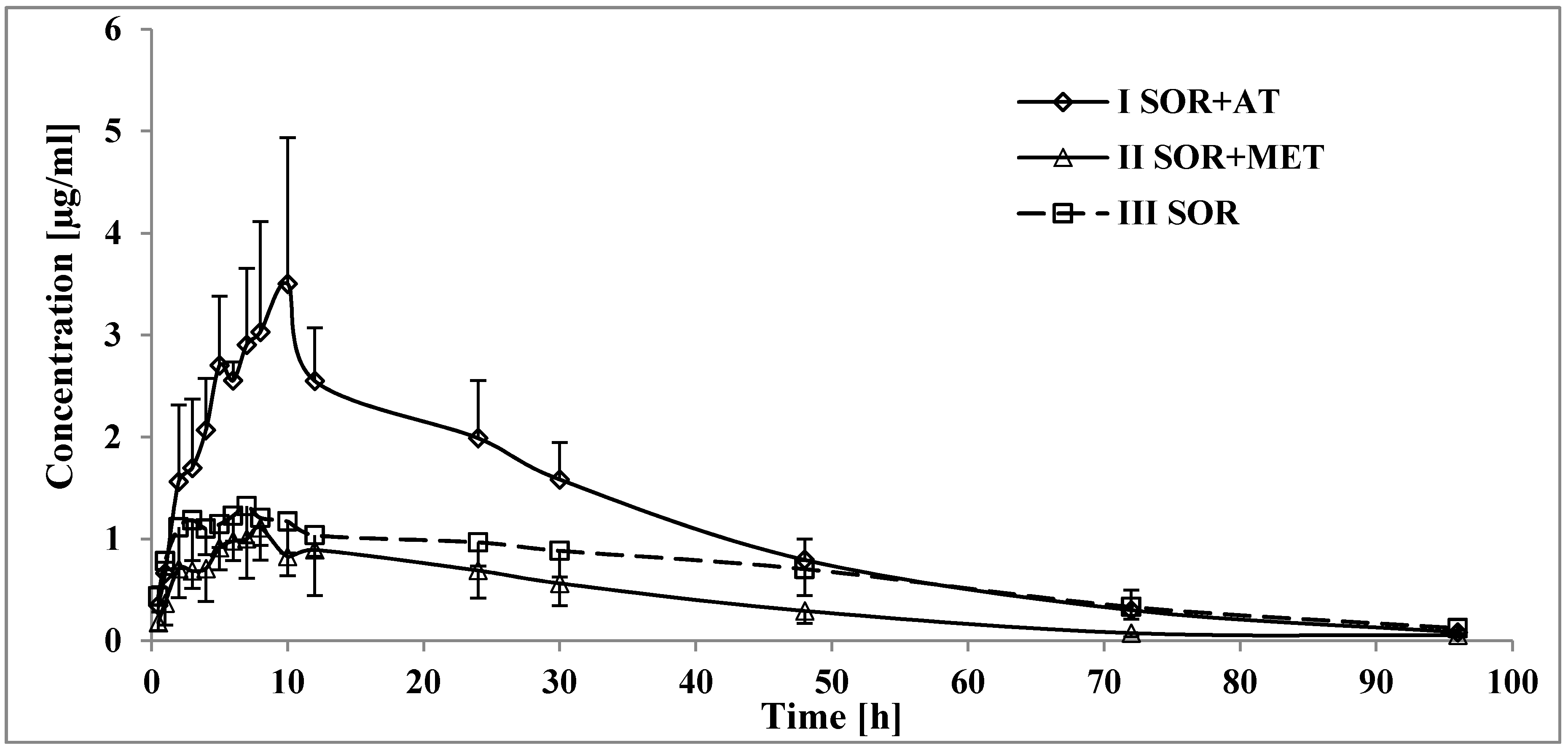
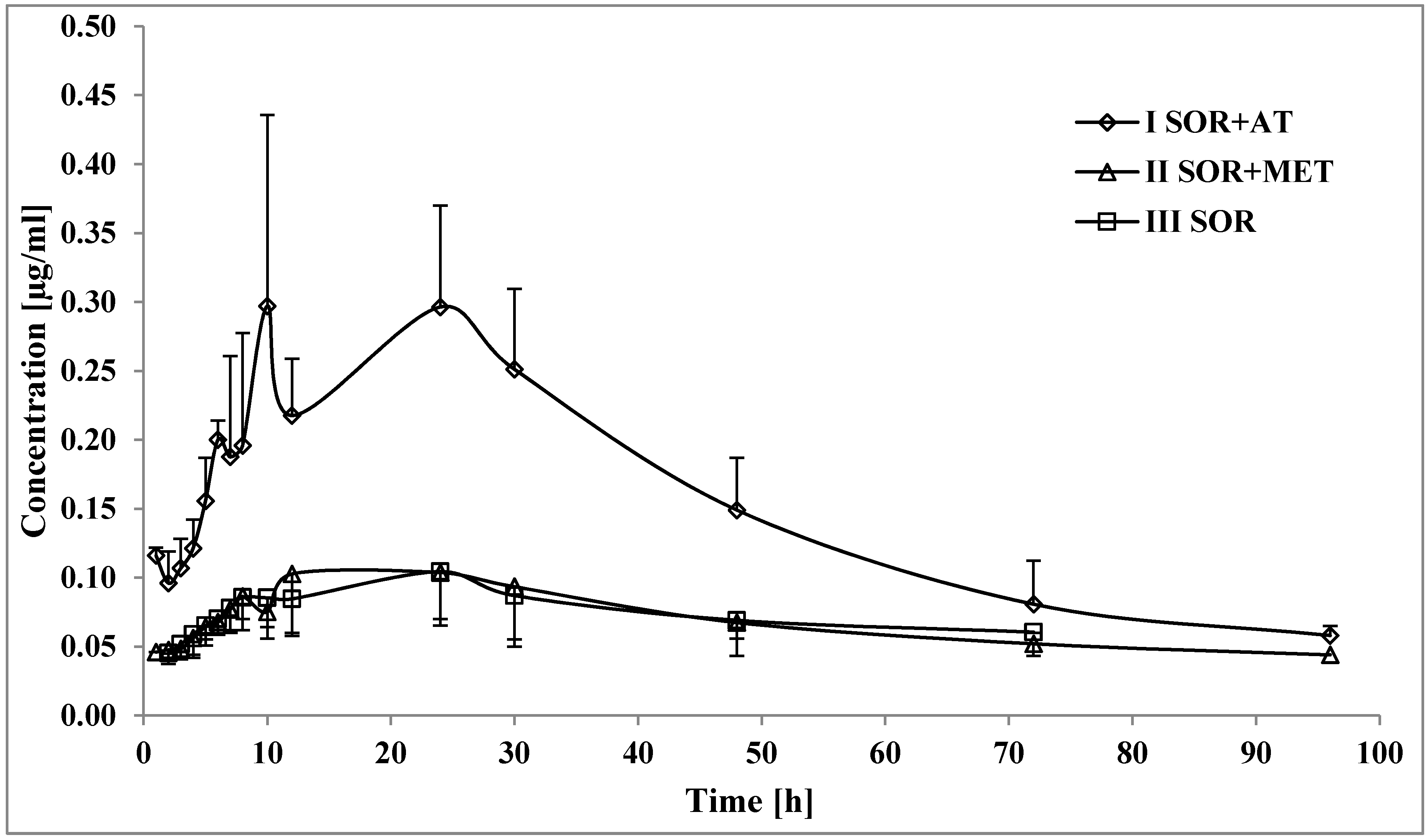
3.1. The Influence of Atorvastatin on the Pharmacokinetics of Sorafenib and SR_NO
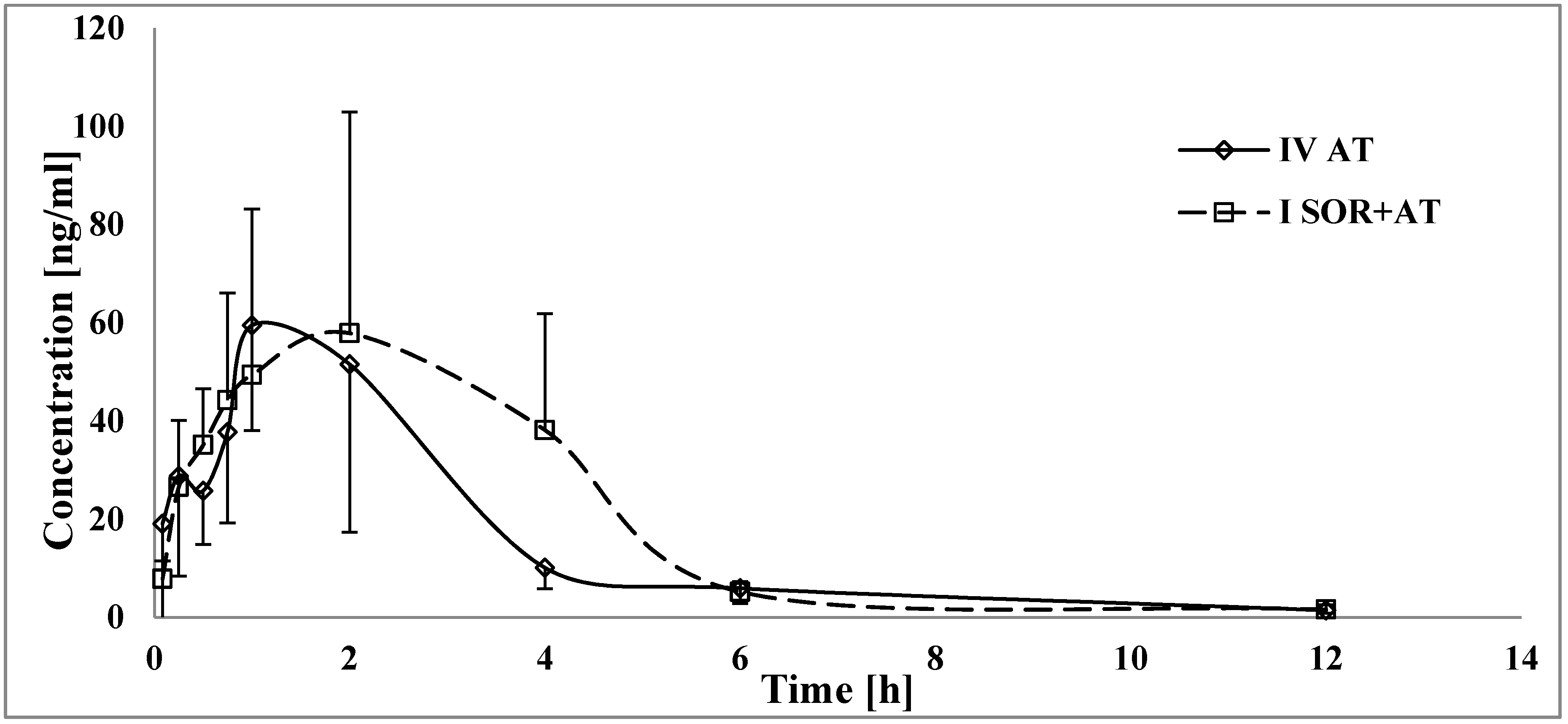
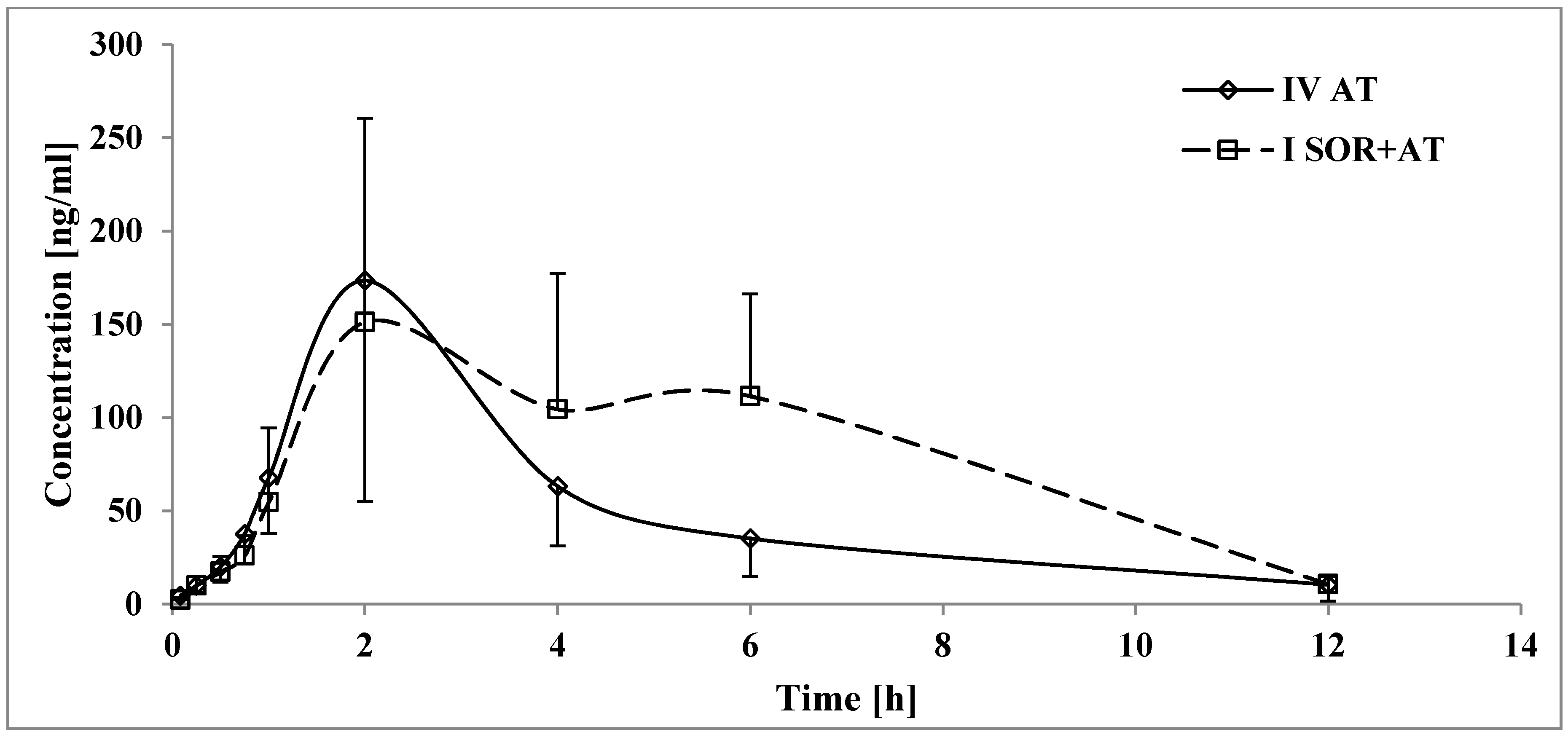
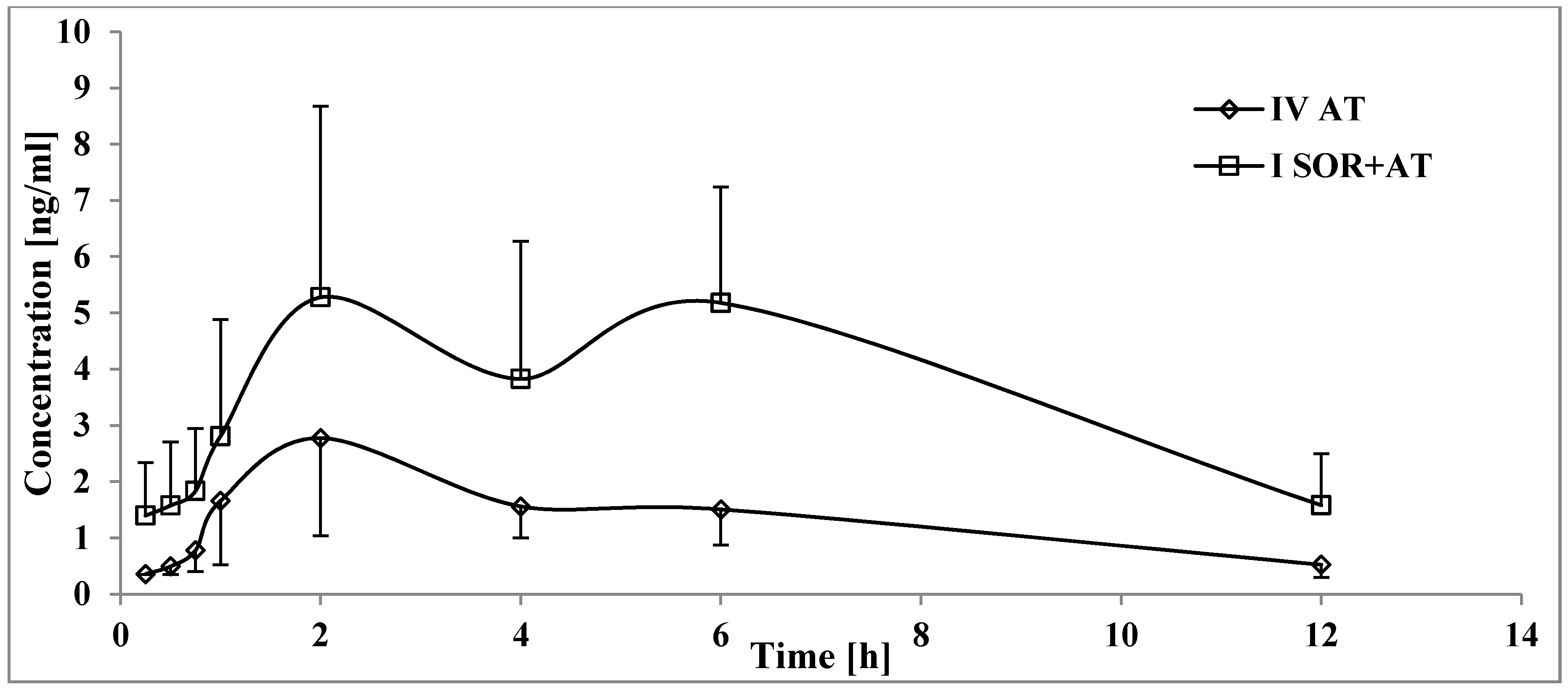
3.2. The Influence of Sorafenib on the Pharmacokinetics of Atorvastatin, 2-OH AT, and 4-OH AT
3.3. The Influence of Metformin on the Pharmacokinetics of Sorafenib and SR_NO
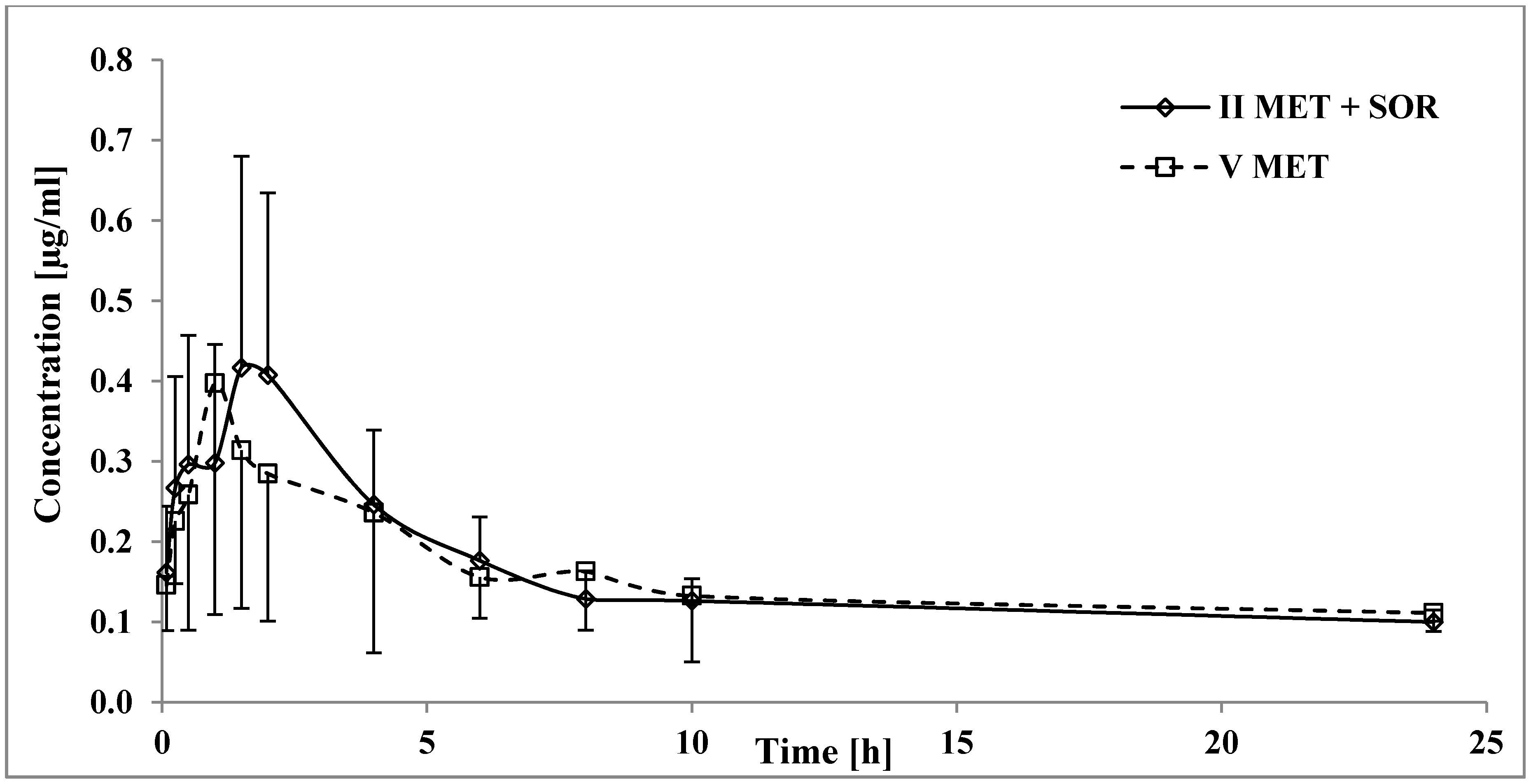
3.4. The Influence of Sorafenib on the Pharmacokinetics of Metformin
4. Discussion
4.1. The Influence of Atorvastatin on the Pharmacokinetics of Sorafenib and SR_NO
4.2. The Influence of Sorafenib on the Pharmacokinetics of Atorvastatin, 2-OH AT, and 4-OH AT
4.3. The Influence of Metformin on the Pharmacokinetics of Sorafenib and SR_NO
4.4. The Influence of Sorafenib on the Pharmacokinetics of Metformin
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Bertot, L.C.; Adams, L.A. Trends in hepatocellular carcinoma due to non-alcoholic fatty liver disease. Expert Rev. Gastroenterol. Hepatol. 2019, 13, 179–187. [Google Scholar] [CrossRef] [PubMed]
- Mao, M.; Wang, X.; Sheng, H.; Yijun, L.; Zhang, L.; Dai, S.; Chi, P.D. A novel score based on serum apolipoprotein A-1 and C-reactive protein is a prognostic biomarker in hepatocellular carcinoma patients. BMC Cancer 2018, 18, 1178. [Google Scholar] [CrossRef] [PubMed]
- Di Costanzo, G.G.; Tortora, R.; Morisco, F.; Addario, L.; Guarino, M.; Cordone, G.; Falco, L.; Caporaso, N. Impact of diabetes on outcomes of sorafenib therapy for hepatocellular carcinoma. Target Oncol. 2017, 12, 61–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hassan, M.M.; Curley, S.A.; Li, D.; Kaseb, A.; Davila, M.; Abdalla, E.K.; Javle, M.; Moghazy, D.M.; Lozano, R.D.; Abbruzzese, J.L.; et al. Association of diabetes duration and diabetes treatment with the risk of hepatocellular carcinoma. Cancer 2010, 116, 1938–1946. [Google Scholar] [CrossRef] [Green Version]
- Gong, L.; Giacomini, M.M.; Giacomini, C.; Maitland, M.L.; Altman, R.B.; Klein, T.E. PharmGKB summary: Sorafenib pathways. Pharm. Genom. 2017, 27, 240–246. [Google Scholar] [CrossRef]
- Chen, M.; Neul, C.; Schaeffeler, E.; Frisch, F.; Winter, S.; Schwab, M.; Koepsell, H.; Hu, S.; Laufer, S.; Baker, S.D.; et al. Sorafenib activity and disposition in liver cancer does not depend on organic cation transporter 1. Clin. Pharm. Ther. 2020, 107, 227–237. [Google Scholar] [CrossRef]
- Vasilyeva, A.; Durmus, S.; Li, L.; Wagenaar, E.; Hu, S.; Gibson, A.A.; Panetta, J.C.; Mani, S.; Sparreboom, A.; Baker, S.D.; et al. Hepatocellular shuttling and recirculation of sorafenib-glucuronide is dependent on Abcc2, Abcc3, and Oatp1a/1b. Cancer Res. 2015, 75, 2729–2736. [Google Scholar] [CrossRef] [Green Version]
- International Transporter Consortium; Giacomini, K.M.; Huang, S.M.; Tweedie, D.J.; Benet, L.Z.; Brouwer, K.L.R.; Chu, X.; Dahlin, A.; Evers, R.; Fischer, V.; et al. Membrane transporters in drug development. Nat. Rev. Drug. Discov. 2010, 9, 215–236. [Google Scholar] [CrossRef]
- Wang, Z.; Yang, H.; Xu, J.; Zhao, K.; Chen, Y.; Liang, L.; Li, P.; Chen, N.; Geng, D.; Zhang, X.; et al. Prediction of atorvastatin pharmacokinetics in high-fat diet and low-dose streptozotocin-induced diabetic rats using a semiphysiologically based pharmacokinetic model involving both enzymes and transporters. Drug Metab. Dispos. 2019, 47, 1066–1079. [Google Scholar] [CrossRef]
- Bil, J.; Zapala, L.; Nowis, D.; Jakobisiak, M.; Golab, J. Statins potentiate cytostatic/cytotoxic activity of sorafenib but not sunitinib against tumor cell lines in vitro. Cancer Lett. 2010, 288, 57–67. [Google Scholar] [CrossRef]
- Mohammadalipour, A.; Hashemnia, M.; Goudarzi, F.; Ravan, A.P. Increasing the effectiveness of tyrosine kinase inhibitor (TKI) in combination with a statin in reducing liver fibrosis. Clin. Exp. Pharm. Physiol. 2019, 46, 1183–1193. [Google Scholar] [CrossRef]
- Hwang, K.E.; Kwon, S.J.; Kim, Y.S.; Park, D.S.; Kim, B.R.; Yoon, K.H.; Jeong, E.K.; Kim, H.R. Effect of simvastatin on the resistance to EGFR tyrosine kinase inhibitors in a non-small cell lung cancer with the T790M mutation of EGFR. Exp. Cell Res. 2014, 323, 288–296. [Google Scholar] [CrossRef]
- Hung, M.S.; Chen, I.C.; Lee, C.P.; Huang, R.J.; Chen, P.C.; Tsai, Y.H.; Yang, Y.H. Statin improves survival in patients with EGFR-TKI lung cancer: A nationwide population-based study. PLoS ONE 2017, 12, e0171137. [Google Scholar] [CrossRef] [Green Version]
- Scheen, A.J. Clinical Pharmacokinetics of Metformin. Clin. Pharm. 1996, 30, 359–371. [Google Scholar] [CrossRef] [PubMed]
- Gong, L.; Goswami, S.; Giacomini, K.M.; Altman, R.B.; Klein, T.E. Metformin pathways: Pharmacokinetics and pharmacodynamics. Pharm. Genom. 2012, 22, 820–827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stage, T.B.; Brøsen, K.; Christensen, M.M. A comprehensive review of drug–drug interactions with metformin. Clin. Pharm. 2015, 54, 811–824. [Google Scholar] [CrossRef] [PubMed]
- Chung, Y.G.; Tak, E.; Hwang, S.; Lee, J.Y.; Kim, J.Y.; Kim, Y.Y.; Song, G.W.; Lee, K.J.; Kim, N. Synergistic effect of metformin on sorafenib in in vitro study using hepatocellular carcinoma cell lines. Ann. Hepatobiliary Pancreat Surg. 2018, 22, 179–184. [Google Scholar] [CrossRef]
- Orsi, G.; Casadei-Gardini, A. Sorafenib and metformin: To be, or not to be, that is the question. Hepatobiliary Surg Nutr. 2019, 8, 411–413. [Google Scholar] [CrossRef]
- Wang, Q.Q.; Guo, X.C.; Li, L.; Gao, Z.H.; Ji, M. Treatment with metformin and sorafenib alleviates endometrial hyperplasia in polycystic ovary syndrome by promoting apoptosis via synergically regulating autophagy. J. Cell Physiol. 2020, 235, 1339–1348. [Google Scholar] [CrossRef]
- Ling, S.; Song, L.; Fan, N.; Feng, T.; Liu, L.; Yang, X.; Wang, M.; Li, Y.; Tian, Y.; Zhao, F. Combination of metformin and sorafenib suppresses proliferation and induces autophagy of hepatocellular carcinoma via targeting the mTOR pathway. Int. J. Oncol. 2017, 50, 297–309. [Google Scholar] [CrossRef] [Green Version]
- Chen, G.; Nicula, D.; Renko, K.; Derwahl, M. Synergistic anti-proliferative effect of metformin and sorafenib on growth of anaplastic thyroid cancer cells and their stem cells. Oncol. Rep. 2015, 33, 1994–2000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schulte, L.; Scheiner, B.; Voigtländer, T.; Koch, S.; Schweitzer, N.; Marhenke, S.; Ivanyi, R.; Manns, M.P.; Rodt, T.; Hinrichs, J.B. Treatment with metformin is associated with a prolonged survival in patients with hepatocellular carcinoma. Liver Int. 2019, 39, 714–726. [Google Scholar] [CrossRef] [PubMed]
- Gardini, A.C.; Faloppi, L.; De Matteis, S.; Foschi, F.G.; Silvestris, N.; Tovoli, F.; Palmieri, V.; Marisi, G.; Brunetti, O.; Vespasiani-Gentilucci, U.; et al. Metformin and insulin impact on clinical outcome in patients with advanced hepatocellular carcinoma receiving sorafenib: Validation study and biological rationale. Eur. J. Cancer 2017, 86, 106–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gardini, A.C.; Marisi, G.; Scarpi, E.; Scartozzi, M.; Faloppi, L.; Silvestris, N.; Masi, C.; Vivaldi, C.; Brunetti, O.; Tamberi, S.; et al. Effects of metformin on clinical outcome in diabetic patients with advanced HCC receiving sorafenib. Expert Opin. Pharm. 2015, 16, 2719–2725. [Google Scholar] [CrossRef]
- Minematsu, T.; Giacomini, K.M. Interactions of tyrosine kinase inhibitors with organic cation transporters and multidrug and toxic compound extrusion proteins. Mol. Cancer Ther. 2011, 10, 531–539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Zhang, X.; Liu, F.; Wang, M.; Qin, S. The effects of triptolide on the pharmacokinetics of sorafenib in rats and its potential mechanism. Pharm. Biol. 2017, 55, 1863–1867. [Google Scholar] [CrossRef] [Green Version]
- Shu, N.; Hu, M.; Ling, Z.; Liu, P.; Wang, F.; Xu, P.; Zhong, Z.; Sun, B.; Zhang, M.; Li, F.; et al. The enhanced atorvastatin hepatotoxicity in diabetic rats was partly attributed to the upregulated hepatic Cyp3a and SLCO1B1. Sci. Rep. 2016, 6, 33072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, R.; Xu, Z.; Xu, X.; Jin, J.; Zhao, Y.; Wang, T.; Li, Y.; Ma, Y. Organic cation transporter and multidrug and toxin extrusion 1 co-mediated interaction between metformin and berberine. Eur. J. Pharm. Sci. 2019, 127, 282–290. [Google Scholar] [CrossRef]
- Afify, S.; Rapp, U.R.; Högger, P. Validation of a liquid chromatography assay for the quantification of the Raf kinase inhibitor BAY 43-9006 in small volumes of mouse serum. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2004, 809, 99–103. [Google Scholar] [CrossRef]
- Gabr, R.Q.; Padwal, R.S.; Brocks, D.R. Determination of metformin in human plasma and urine by high-performance liquid chromatography using small sample volume and conventional octadecyl silane column. J. Pharm. Pharm. Sci. 2010, 13, 486–494. [Google Scholar] [CrossRef] [Green Version]
- Karbownik, A.; Stachowiak, A.; Urjasz, H.; Sobańska, K.; Szczecińska, A.; Grabowski, T.; Stanisławiak-Rudowicz, J.; Wolc, A.; Grześkowiak, E.; Szałek, E. The oxidation and hypoglycaemic effect of sorafenib in streptozotocin-induced diabetic rats. Pharm. Rep. 2020, 72, 254–259. [Google Scholar] [CrossRef] [PubMed]
- Summary of Product Characteristics for Atorvastatin 10 mg film coated tablets. Datapharm Ltd., Electronic Medicines Compendium. Available online: https://www.medicines.org.uk/emc/product/4109/smpc (accessed on 10 February 2020).
- Drug interaction studies-study design, data analysis, implications for dosing, and labeling recommendations. U.S. Food and Drug Administration. Available online: http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm292362.pdf (accessed on 29 February 2020).
- Summary of Product Characteristic for Nexavar 200 mg, European Medicines Agency. Available online: https://www.ema.europa.eu/en/documents/product-information/nexavar-epar-product-information_en.pdf (accessed on 10 February 2020).
- Kalliokoski, A.; Backman, J.; Kurkinen, K.J.; Neuvonen, P.J.; Niemi, M. Effects of gemfibrozil and atorvastatin on the pharmacokinetics of repaglinide in relation to SLCO1B1 polymorphism. Clin. Pharmacol. Ther. 2008, 84, 488–496. [Google Scholar] [CrossRef] [PubMed]
- Bogman, K.; Peyer, A.K.; Török, M.; Küsters, E.; Drewe, J. HMG-CoA reductase inhibitors and P-glycoprotein modulation. Br. J. Pharmacol. 2001, 132, 1183–1192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghassabian, S.; Gillani, T.B.; Rawling, T.; Crettol, S.; Nair, P.C.; Murray, M. Sorafenib N-Oxide is an inhibitor of human hepatic CYP3A4. AAPS J. 2019, 21, 15. [Google Scholar] [CrossRef]
- Holtzman, C.W.; Wiggins, B.S.; Spinler, S.A. Role of P-glycoprotein in statin drug interactions. Pharmacotherapy 2016, 26, 1601–1607. [Google Scholar] [CrossRef]
- Summary of Product Characteristics for Glucophage 500 mg film coated tablets. Datapharm Ltd., Electronic Medicines Compendium. Available online: https://www.medicines.org.uk/emc/product/987/smpc (accessed on 10 February 2020).
- Sugiyama, M.; Fujita, K.; Murayama, N.; Akiyama, Y.; Yamazaki, H.; Sasaki, Y. Sorafenib and sunitinib, two anticancer drugs, inhibit CYP3A4-mediated and activate CY3A5-mediated midazolam 1’-hydroxylation. Drug. Metab. Dispos. 2011, 39, 757–762. [Google Scholar] [CrossRef]
- Krausova, L.; Stejskalova, L.; Wang, H.; Vrzal, R.; Dvorak, Z.; Mani, S.; Pavek, P. Metformin suppresses pregnane X receptor (PXR)-regulated transactivation of CYP3A4 gene. Biochem. Pharmacol. 2011, 82, 1771–1780. [Google Scholar] [CrossRef] [Green Version]
- Tsuda, M.; Terada, T.; Ueba, M.; Sato, T.; Masuda, S.; Katsura, T.; Inui, K.I. Involvement of human multidrug and toxin extrusion 1 in the drug interaction between cimetidine and metformin in renal epithelial cells. J. Pharmacol. Exp. Ther. 2009, 329, 185–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jayasagar, G.; Krishna Kumar, M.; Chandrasekhar, K.; Madhusudan Rao, C.; Madhusudan Rao, Y. Effect of cephalexin on the pharmacokinetics of metformin in healthy human volunteers. Drug. Metabol. Drug. Interact. 2002, 19, 41–48. [Google Scholar] [CrossRef]
- Manitpisitkul, P.; Curtin, C.R.; Shalayda, K.; Wang, S.S.; Ford, L.; Heald, D. Pharmacokinetic interactions between topiramate and pioglitazone and metformin. Epilepsy Res. 2014, 108, 1519–1532. [Google Scholar] [CrossRef]
- Grün, B.; Kiessling, M.K.; Burhenne, J.; Riedel, K.D.; Weiss, J.; Rauch, G.; Haefeli, W.E.; Czock, D. Trimethoprim-metformin interaction and its genetic modulation by OCT2 and MATE1 transporters. Br. J. Clin. Pharm. 2013, 76, 787–796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johansson, S.; Read, J.; Oliver, S.; Steinberg, M.; Li, Y.; Lisbon, E.; Mathews, D.; Leese, P.T.; Martin, P. Pharmacokinetic evaluations of the co-administrations of vandetanib and metformin, digoxin, midazolam, omeprazole or ranitidine. Clin. Pharm. 2014, 53, 837–847. [Google Scholar] [CrossRef] [PubMed]
- Kusuhara, H.; Ito, S.; Kumagai, Y.; Jiang, M.; Shiroshita, T.; Moriyama, Y.; Inoue, K.; Yuasa, H.; Sugiyama, Y. Effects of a MATE protein inhibitor, pyrimethamine, on the renal elimination of metformin at oral microdose and at therapeutic dose in healthy subjects. Clin. Pharm. Ther. 2011, 89, 837–844. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reagent | CAS Number |
|---|---|
| 1-butanol 1 | 71-36-3 |
| 2-hydroxy atorvastatin dihydrate monosodium salt 2 | 214217-86-4 |
| 4-hydroxy atorvastatin disodium salt 2 | 1276537-18-8 |
| acetaminophen 3 | 103-90-2 |
| acetonitrile 3 | 75-05-8 |
| ammonium acetate 3 | 631-61-8 |
| ammonium formate 3 | 540-69-2 |
| atorvastatin calcium salt trihydrate 2 | 344423-98-9 |
| dimethyl sulfoxide 3 | 67-68-5 |
| ethyl acetate 3 | 141-78-6 |
| formic acid 3 | 64-18-6 |
| lapatinib 3 | 231277-92-9 |
| metformin hydrochloride 3 | 1115-70-4 |
| methanol 3 | 67-56-1 |
| n-heptane 4 | 142-82-5 |
| sodium hydroxide 3 | 67-56-1 |
| sodium hydroxide microprills 5 | 1310-73-2 |
| sorafenib 6 | 284461-73-0 |
| sorafenib N-oxide 6 | 583840-03 |
| rosuvastatin calcium 3 | 147098-20-2 |
| ultrapure water (deionized, distilled, and filtered through Direct Q3 system) 4 | 7732-18-5 |
| Pharmacokinetic Parameters 1 | IIISOR (n = 8) | ISOR+AT (n = 8) | IISOR+MET (n = 7) | p-Value ISOR+AT vs. IIISOR | p-Value IISOR+MET vs. IIISOR |
|---|---|---|---|---|---|
| Sorafenib | |||||
| Cmax (µg/mL) | 1.56 ± 0.35 (22.6) | 3.66 ± 1.21 (33.1) | 1.27 ± 0.38 (30.2) | <0.0001 | 0.7499 |
| AUC0-t (µg × h/mL) | 62.83 ± 16.14 (25.7) | 104.67 ± 18.90 (18.1) | 35.16 ± 8.36 (23.8) | <0.0001 | 0.0065 |
| AUC0--∞ (µg × h/mL) | 67.05 ± 16.70 (24.9) | 106.41 ± 19.33 (18.2) | 36.79 ± 8.96 (24.4) | 0.0002 | 0.0041 |
| tmax (h) | 5.13 ± 2.17 (42.3) | 11.13 ± 5.54 (49.8) | 7.43 ± 2.29 (30.9) | 0.0117 | 0.4722 |
| ka (h−1) | 0.74 ± 0.31 (42.5) | 0.20 ± 0.11 (53.2) | 0.35 ± 0.23 (66.8) | 0.0005 | 0.0106 |
| kel (h−1) | 0.035 ± 0.01 (30.3) | 0.05 ± 0.02 (30.6) | 0.05 ± 0.01 (24.4) | 0.0637 | 0.3306 |
| t1/2 (h) | 21.89 ± 7.79 (35.6) | 14.70 ± 3.17 (21.5) | 16.33 ± 3.72 (22.8) | 0.0372 | 0.1386 |
| Cl/F (L/h × kg) | 0.80 ± 0.22 (27.1) | 0.48 ± 0.09 (18.3) | 1.41 ± 0.50 (35.3) | 0.1118 | 0.0026 |
| Vd/F (L) | 25.30 ± 11.59 (45.8) | 9.98 ± 2.48 (24.9) | 34.15 ± 18.74 (54.9) | 0.0570 | 0.3716 |
| Sorafenib N-oxide | |||||
| Cmax (µg/mL) | 0.11 ± 0.02 (21.8) | 0.38 ± 0.09 (24.0) | 0.13 ± 0.04 (31.5) | <0.0001 | 0.8981 |
| AUC0-t (µg × h/mL) | 4.10 ± 1.56 (38.1) | 14.04 ± 2.11 (15.1) | 5.49 ± 2.39 (43.6) | <0.0001 | 0.4029 |
| AUC0-∞ (µg × h/mL) | 8.61 ± 2.19 (25.4) | 16.32 ± 2.50 (15.3) | 8.55 ± 2.36 (27.5) | <0.0001 | 0.9988 |
| tmax (h) | 16.38 ± 8.21 (50.1) | 19.50 ± 8.12 (41.7) | 21.43 ± 6.80 (31.7) | 0.7055 | 0.4363 |
| kel (h−1) | 0.016 ± 0.010 (60.9) | 0.028 ± 0.009 (31.4) | 0.018 ± 0.009 (51.9) | 0.0638 | 0.9473 |
| t1/2 (h) | 53.31 ± 25.23 (47.3) | 27.04 ± 8.49 (31.4) | 45.98 ± 20.71 (45.0) | 0.0347 | 0.7487 |
| Sorafenib N-oxide/Sorafenib | |||||
| Cmax (µg/mL) | 0.07 ± 0.02 (26.8) | 0.11 ± 0.03 (26.9) | 0.10 ± 0.03 (26.0) | 0.0410 | 0.1231 |
| AUC0-t (µg × h/mL) | 0.07 ± 0.02 (37.1) | 0.14 ± 0.02 (12.7) | 0.16 ± 0.06 (35.6) | 0.0026 | 0.0002 |
| AUC0--∞ (µg × h/mL) | 0.14 ± 0.05 (38.1) | 0.16 ± 0.03 (16.7) | 0.25 ± 0.11 (42.2) | 0.8226 | 0.0108 |
| Pharmacokinetic Parameters 1 | IVAT (n = 8) | ISOR+AT (n = 8) | p-Value ISOR+AT vs. IVAT |
|---|---|---|---|
| Atorvastatin | |||
| Cmax (ng/mL) | 72.76 ± 27.21 (37.4) | 81.77 ± 38.24 (46.8) | 0.5957 1 |
| AUC0-t (ng × h/mL) | 186.70 ± 74.03 (39.7) | 362.21 ± 90.82 (25.1) | 0.0038 2 |
| AUC0-∞ (ng × h/mL) | 194.01 ± 72.41 (37.3) | 374.12 ± 87.23 (23.3) | 0.0038 2 |
| tmax (h) | 1.26 ± 0.69 (54.5) | 2.75 ± 1.75 (63.7) | 0.0419 1 |
| ka (h−1) | 1.95 ± 3.03 (155.4) | 0.79 ± 0.62 (78.7) | 0.1893 2 |
| kel (h−1) | 0.24 ± 0.08 (33.7) | 0.34 ± 0.11 (31.6) | 0.0704 1 |
| t1/2 (h) | 3.22 ± 1.33 (41.4) | 2.27 ± 0.80 (35.1) | 0.1071 1 |
| Cl/F (L/h × kg) | 54.31 ± 13.87 (25.5) | 27.71 ± 7.46 (29.9) | 0.0003 1 |
| Vd/F (L) | 257.67 ± 132.63 (51.5) | 93.91 ± 50.93 (54.2) | 0.0028 2 |
| 2-OH AT | |||
| Cmax (ng/mL) | 175.20 ± 116.62 (66.6) | 185.82 ± 90.16 (48.5) | 0.4948 2 |
| AUC0-t (ng × h/mL) | 617.32 ± 277.54 (45.0) | 958.91 ± 342.91 (35.8) | 0.0239 2 |
| AUC0--∞ (ng × h/mL) | 695.68 ± 297.16 (42.7) | 993.97 ± 343.87 (34.6) | 0.0846 1 |
| 2-OH AT/atorvastatin | |||
| Cmax (ng/mL) | 2.35 ± 1.12 (47.6) | 2.43 ± 1.06 (43.6) | 1.0000 2 |
| AUC0-t (ng × h/mL) | 3.30 ± 0.70 (21.3) | 2.66 ± 0.74 (27.8) | 0.0520 2 |
| AUC0-∞ (ng × h/mL) | 3.61 ± 1.12 (31.0) | 2.67 ± 0.76 (28.3) | 0.0312 2 |
| 4-OH AT | |||
| Cmax (ng/mL) | 3.18 ± 1.78 (55.9) | 7.22 ± 2.42 (33.5) | 0.0019 1 |
| AUC0-t (ng × h/mL) | 14.63 ± 5.57 (38.1) | 45.26 ± 16.72 (34.9) | 0.0002 1 |
| AUC0-∞ (ng × h/mL) | 19.22 ± 7.06 (36.7) | 61.59 ± 21.50 (34.9) | 0.0001 1 |
| 4-OH AT/atorvastatin | |||
| Cmax (ng/mL) | 0.04 ± 0.02 (40.7) | 0.10 ± 0.05 (50.3) | 0.0084 1 |
| AUC0-t (ng × h/mL) | 0.08 ± 0.02 (28.3) | 0.13 ± 0.06 (44.0) | 0.0239 2 |
| AUC0-∞ (ng × h/mL) | 0.10 ± 0.04 (38.1) | 0.17 ± 0.08 (47.2) | 0.0239 2 |
| Pharmacokinetic Parameters 1 | VMET (n = 8) | IISOR+MET (n = 8) | p-Value IISOR+MET vs. VMET |
|---|---|---|---|
| Cmax (µg/mL) | 0.50 ± 0.29 (58.0) | 0.46 ± 0.26 (56.5) | 0.9581 1 |
| AUC0-t (µg × h/mL) | 3.83 ± 1.23 (32.1) | 3.64 ± 1.15 (31.6) | 0.5635 1 |
| AUC0-∞ (µg × h/mL) | 7.90 ± 1.93 (24.4) | 10.88 ± 4.50 (41.4) | 0.1035 1 |
| tmax (h) | 1.09 ± 0.57 (52.3) | 1.88 ± 0.23 (12.2) | 0.0069 2 |
| ka (h−1) | 5.20 ± 5.22 (100.4) | 5.06 ± 4.36 (86.2) | 0.9536 2 |
| kel (h−1) | 0.03 ± 0.01 (33.3) | 0.03 ± 0.04 (133.3) | 0.9039 2 |
| t1/2 (h) | 26.54 ± 14.52 (54.7) | 51.51 ± 36.59 (71.0) | 0.0944 2 |
| Cl/F (L/h) | 6.31 ± 1.48 (23.5) | 5.50 ± 3.08 (56.0) | 0.1893 1 |
| Vd/F (L) | 227.66 ± 90.97 (39.9) | 297.89 ± 133.23 (44.7) | 0.2271 1 |
| MRT0-t (h) | 9.81 ± 1.27 (12.9) | 8.65 ± 2.22 (25.7) | 0.2202 2 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karbownik, A.; Szkutnik-Fiedler, D.; Czyrski, A.; Kostewicz, N.; Kaczmarska, P.; Bekier, M.; Stanisławiak-Rudowicz, J.; Karaźniewicz-Łada, M.; Wolc, A.; Główka, F.; et al. Pharmacokinetic Interaction between Sorafenib and Atorvastatin, and Sorafenib and Metformin in Rats. Pharmaceutics 2020, 12, 600. https://doi.org/10.3390/pharmaceutics12070600
Karbownik A, Szkutnik-Fiedler D, Czyrski A, Kostewicz N, Kaczmarska P, Bekier M, Stanisławiak-Rudowicz J, Karaźniewicz-Łada M, Wolc A, Główka F, et al. Pharmacokinetic Interaction between Sorafenib and Atorvastatin, and Sorafenib and Metformin in Rats. Pharmaceutics. 2020; 12(7):600. https://doi.org/10.3390/pharmaceutics12070600
Chicago/Turabian StyleKarbownik, Agnieszka, Danuta Szkutnik-Fiedler, Andrzej Czyrski, Natalia Kostewicz, Paulina Kaczmarska, Małgorzata Bekier, Joanna Stanisławiak-Rudowicz, Marta Karaźniewicz-Łada, Anna Wolc, Franciszek Główka, and et al. 2020. "Pharmacokinetic Interaction between Sorafenib and Atorvastatin, and Sorafenib and Metformin in Rats" Pharmaceutics 12, no. 7: 600. https://doi.org/10.3390/pharmaceutics12070600