Fabrication of Core Crosslinked Polymeric Micelles as Nanocarriers for Doxorubicin Delivery: Self-Assembly, In Situ Diselenide Metathesis and Redox-Responsive Drug Release

 ,
,  ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
2.2.1. Synthesis of β-Benzyl-l-Aspartate N-Carboxy Anhydride (BLA-NCA)
2.2.2. Synthesis of Poly(ethylene oxide)-b-Poly(β-Benzyl-l-Aspartate) (mPEG-PBLA)
2.2.3. Synthesis of Poly(ethylene oxide)-b-Poly(β-Benzyl-l-Aspartate)-b-Polycaprolactone (mPEG-PBLA-PCL)
2.2.4. Synthesis of Poly(ethylene oxide)-b-Poly(l-Aspartic Acid)-b-Polycaprolactone (mPEG-PLA-PCL)
2.2.5. Synthesis of mPEG-P(LA-DSeDEA)-PCL
2.2.6. Characterization
2.2.7. Preparation of mPEG-P(LA-DSeDEA)-PCL Micelles
2.2.8. Determination of Critical Micelle Concentration (CMC)
2.2.9. Preparation of DOX-loaded mPEG-P(LA-DSeDEA)-PCL Micelles
2.2.10. Redox Sensitivity and DOX-Releasing Behavior of Micelles
2.2.11. In Vitro Cytotoxicity Study
2.2.12. Cellular Uptake Study
2.2.13. Flow Cytometry Analysis
2.2.14. Statistical Analysis
3. Results and Discussion
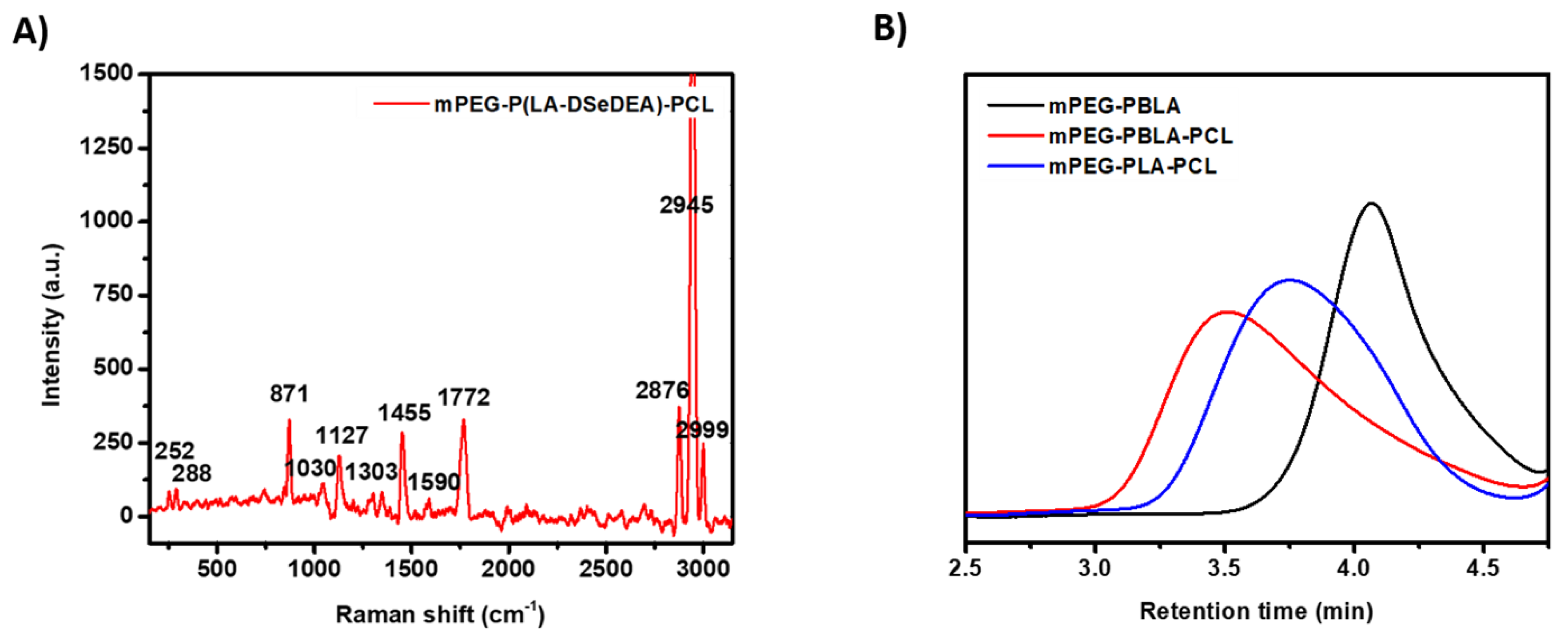
3.1. Synthesis and Characterization of Copolymers
3.2. Self-Assembly and Preparation of Core Crosslinked Polymeric Micelles
3.3. Critical Micelle Concentration of mPEG-P(LA-DSeDEA)-PCL
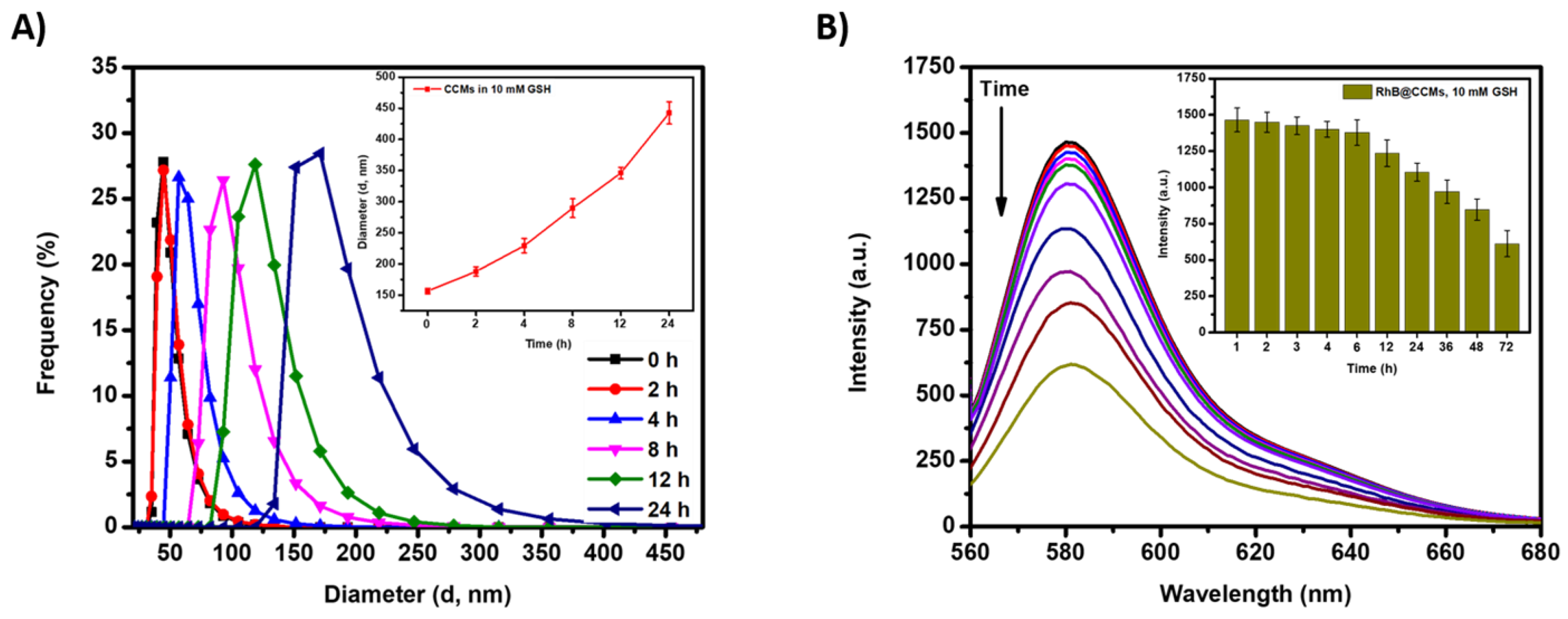
3.4. Colloidal Stability of Blank and DOX-Loaded Micelles
3.5. Redox Sensitivity of mPEG-P(LA-DSeDEA)-PCL Micelles
3.6. Drug-Loading and Releasing Behavior of Micelles
3.7. In Vitro Cytotoxicity and Anticancer Effect
3.8. Cellular Uptake and Localization Study
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Yang, J.; Pan, S.; Gao, S.; Dai, Y.; Xu, H. Anti-recurrence/metastasis and chemosensitization therapy with thioredoxin reductase-interfering drug delivery system. Biomaterials 2020, 249, 120054. [Google Scholar] [CrossRef]
- Hailemeskel, B.Z.; Hsu, W.-H.; Addisu, K.D.; Andrgie, A.T.; Chou, H.-Y.; Lai, J.-Y.; Tsai, H.-C. Diselenide linkage containing triblock copolymer nanoparticles based on Bi(methoxyl poly(ethylene glycol))-poly(ε-carprolactone): Selective intracellular drug delivery in cancer cells. Mater. Sci. Eng. C 2019, 103, 109803. [Google Scholar] [CrossRef]
- Birhan, Y.S.; Hailemeskel, B.Z.; Mekonnen, T.W.; Hanurry, E.Y.; Darge, H.F.; Andrgie, A.T.; Chou, H.-Y.; Lai, J.-Y.; Hsiue, G.-H.; Tsai, H.-C. Fabrication of redox-responsive Bi(mPEG-PLGA)-Se2 micelles for doxorubicin delivery. Int. J. Pharm. 2019, 567, 118486. [Google Scholar] [CrossRef]
- Mekonnen, T.W.; Birhan, Y.S.; Andrgie, A.T.; Hanurry, E.Y.; Darge, H.F.; Chou, H.-Y.; Lai, J.-Y.; Tsai, H.-C.; Yang, J.M.; Chang, Y.-H. Encapsulation of gadolinium ferrite nanoparticle in generation 4.5 poly(amidoamine) dendrimer for cancer theranostics applications using low frequency alternating magnetic field. Colloid Surf. B 2019, 184, 110531. [Google Scholar] [CrossRef]
- Hu, X.; Zhai, S.; Liu, G.; Xing, D.; Liang, H.; Liu, S. Concurrent Drug Unplugging and Permeabilization of Polyprodrug-Gated Crosslinked Vesicles for Cancer Combination Chemotherapy. Adv. Mater. 2018, 30, 1706307. [Google Scholar] [CrossRef]
- Qiu, L.; Zhao, L.; Xing, C.; Zhan, Y. Redox-responsive polymer prodrug/AgNPs hybrid nanoparticles for drug delivery. Chin. Chem. Lett. 2018, 29, 301–304. [Google Scholar] [CrossRef]
- Zhao, S.-Q.; Hu, G.; Xu, X.-H.; Kang, S.-M.; Liu, N.; Wu, Z.-Q. Synthesis of Redox-Responsive Core Cross-Linked Micelles Carrying Optically Active Helical Poly(phenyl isocyanide) Arms and Their Applications in Drug Delivery. ACS Macro Lett. 2018, 7, 1073–1079. [Google Scholar] [CrossRef]
- Zhang, K.; Liu, J.; Ma, X.; Lei, L.; Li, Y.; Yang, H.; Lei, Z. Temperature, pH, and reduction triple-stimuli-responsive inner-layer crosslinked micelles as nanocarriers for controlled release. J. Appl. Polym. Sci. 2018, 135, 46714. [Google Scholar] [CrossRef]
- Ling, L.; Ismail, M.; Du, Y.; Xia, Q.; He, W.; Yao, C.; Li, X. High Drug Loading, Reversible Disulfide Core-Cross-Linked Multifunctional Micelles for Triggered Release of Camptothecin. Mol. Pharm. 2018, 15, 5479–5492. [Google Scholar] [CrossRef]
- Xiong, D.; Zhang, R.; Luo, W.; Gu, H.; Peng, S.; Zhang, L. Hydrazone cross-linked micelles based on redox degradable block copolymer for enhanced stability and controlled drug release. React. Funct. Polym. 2017, 119, 64–74. [Google Scholar] [CrossRef]
- Yi, X.-Q.; Zhang, Q.; Zhao, D.; Xu, J.-Q.; Zhong, Z.-L.; Zhuo, R.-X.; Li, F. Preparation of pH and redox dual-sensitive core crosslinked micelles for overcoming drug resistance of DOX. Polym. Chem. 2016, 7, 1719–1729. [Google Scholar] [CrossRef]
- Xia, Y.; He, H.; Liu, X.; Hu, D.; Yin, L.; Lu, Y.; Xu, W. Redox-responsive, core-crosslinked degradable micelles for controlled drug release. Polym. Chem. 2016, 7, 6330–6339. [Google Scholar] [CrossRef]
- Hu, J.; He, J.; Cao, D.; Zhang, M.; Ni, P. Core cross-linked polyphosphoester micelles with folate-targeted and acid-cleavable features for pH-triggered drug delivery. Polym. Chem. 2015, 6, 3205–3216. [Google Scholar] [CrossRef]
- He, J.; Xia, Y.; Niu, Y.; Hu, D.; Xia, X.; Lu, Y.; Xu, W. pH-responsive core crosslinked polycarbonate micelles via thiol-acrylate Michael addition reaction. J. Appl. Polym. Sci. 2017, 134. [Google Scholar] [CrossRef]
- Biswas, D.; An, S.Y.; Li, Y.; Wang, X.; Oh, J.K. Intracellular Delivery of Colloidally Stable Core-Cross-Linked Triblock Copolymer Micelles with Glutathione-Responsive Enhanced Drug Release for Cancer Therapy. Mol. Pharm. 2017, 14, 2518–2528. [Google Scholar] [CrossRef]
- Zhou, Y.; Yu, J.; Feng, X.; Li, W.; Wang, Y.; Jin, H.; Huang, H.; Liu, Y.; Fan, D. Reduction-responsive core-crosslinked micelles based on a glycol chitosan–lipoic acid conjugate for triggered release of doxorubicin. RSC Adv. 2016, 6, 31391–31400. [Google Scholar] [CrossRef]
- Cao, Y.; He, J.; Liu, J.; Zhang, M.; Ni, P. Folate-Conjugated Polyphosphoester with Reversible Cross-Linkage and Reduction Sensitivity for Drug Delivery. ACS Appl. Mater. Interfaces 2018, 10, 7811–7820. [Google Scholar] [CrossRef]
- Lili, Y.; Ruihua, M.; Li, L.; Fei, L.; Lin, Y.; Li, S. Intracellular Doxorubicin Delivery of a Core Cross-linked, Redox-responsive Polymeric Micelles. Int. J. Pharm. 2016, 498, 195–204. [Google Scholar] [CrossRef]
- Cao, X.T.; Kim, Y.H.; Park, J.M.; Lim, K.T. One-pot syntheses of dual-responsive core cross-linked polymeric micelles and covalently entrapped drug by click chemistry. Eur. Polym. J. 2016, 78, 264–273. [Google Scholar] [CrossRef]
- Zhang, Z.; Yin, L.; Tu, C.; Song, Z.; Zhang, Y.; Xu, Y.; Tong, R.; Zhou, Q.; Ren, J.; Cheng, J. Redox-Responsive, Core Cross-Linked Polyester Micelles. ACS Macro Lett. 2013, 2, 40–44. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Tang, L.; Tu, C.; Song, Z.; Yin, Q.; Yin, L.; Zhang, Z.; Cheng, J. Redox-Responsive, Core-Cross-Linked Micelles Capable of On-Demand, Concurrent Drug Release and Structure Disassembly. Biomacromolecules 2013, 14, 3706–3712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, N.; An, S.Y.; Oh, J.K. Dual location disulfide degradable interlayer-crosslinked micelles with extended sheddable coronas exhibiting enhanced colloidal stability and rapid release. Polym. Chem. 2014, 5, 1637–1649. [Google Scholar] [CrossRef]
- Zhai, S.; Hu, X.; Hu, Y.; Wu, B.; Xing, D. Visible light-induced crosslinking and physiological stabilization of diselenide-rich nanoparticles for redox-responsive drug release and combination chemotherapy. Biomaterials 2017, 121, 41–54. [Google Scholar] [CrossRef] [PubMed]
- Najafi, M.; Kordalivand, N.; Moradi, M.-A.; van den Dikkenberg, J.; Fokkink, R.; Friedrich, H.; Sommerdijk, N.A.J.M.; Hembury, M.; Vermonden, T. Native Chemical Ligation for Cross-Linking of Flower-Like Micelles. Biomacromolecules 2018, 19, 3766–3775. [Google Scholar] [CrossRef] [Green Version]
- Xia, J.; Zhao, P.; Pan, S.; Xu, H. Diselenide-Containing Polymeric Vesicles with Osmotic Pressure Response. ACS Macro Lett. 2019, 8, 629–633. [Google Scholar] [CrossRef]
- Tian, K.; Jia, X.; Zhao, X.; Liu, P. pH/Reductant Dual-Responsive Core-Cross-Linked Micelles via Facile in Situ ATRP for Tumor-Targeted Delivery of Anticancer Drug with Enhanced Anticancer Efficiency. Mol. Pharm. 2016, 13, 2683–2690. [Google Scholar] [CrossRef]
- Cheng, X.; Jin, Y.; Qi, R.; Fan, W.; Li, H.; Sun, X.; Lai, S. Dual pH and oxidation-responsive nanogels crosslinked by diselenide bonds for controlled drug delivery. Polymer 2016, 101, 370–378. [Google Scholar] [CrossRef]
- Deepagan, V.G.; Kwon, S.; You, D.G.; Nguyen, V.Q.; Um, W.; Ko, H.; Lee, H.; Jo, D.-G.; Kang, Y.M.; Park, J.H. In situ diselenide-crosslinked polymeric micelles for ROS-mediated anticancer drug delivery. Biomaterials 2016, 103, 56–66. [Google Scholar] [CrossRef]
- Deng, Z.; Qian, Y.; Yu, Y.; Liu, G.; Hu, J.; Zhang, G.; Liu, S. Engineering Intracellular Delivery Nanocarriers and Nanoreactors from Oxidation-Responsive Polymersomes via Synchronized Bilayer Cross-Linking and Permeabilizing Inside Live Cells. J. Am. Chem. Soc. 2016, 138, 10452–10466. [Google Scholar] [CrossRef]
- Zhang, L.; Liu, Y.; Zhang, K.; Chen, Y.; Luo, X. Redox-responsive comparison of diselenide micelles with disulfide micelles. Colloid Polym. Sci. 2019, 297, 225–238. [Google Scholar] [CrossRef]
- Hailemeskel, B.Z.; Addisu, K.D.; Prasannan, A.; Mekuria, S.L.; Kao, C.-Y.; Tsai, H.-C. Synthesis and characterization of diselenide linked poly(ethylene glycol) nanogel as multi-responsive drug carrier. Appl. Surf. Sci. 2018, 449, 15–22. [Google Scholar] [CrossRef]
- Suzuki, N.; Takahashi, A.; Ohishi, T.; Goseki, R.; Otsuka, H. Enhancement of the stimuli-responsiveness and photo-stability of dynamic diselenide bonds and diselenide-containing polymers by neighboring aromatic groups. Polymer 2018, 154, 281–290. [Google Scholar] [CrossRef]
- Salma, S.A.; Patil, M.P.; Kim, D.W.; Le, C.M.Q.; Ahn, B.-H.; Kim, G.-D.; Lim, K.T. Near-infrared light-responsive, diselenide containing core-cross-linked micelles prepared by the Diels–Alder click reaction for photocontrollable drug release application. Polym. Chem. 2018, 9, 4813–4823. [Google Scholar] [CrossRef]
- Waliczek, M.; Pehlivan, Ö.; Stefanowicz, P. Light-Driven Diselenide Metathesis in Peptides. Chem. Open 2019, 8, 1199–1203. [Google Scholar] [CrossRef] [Green Version]
- Buwalda, S.; Nottelet, B.; Bethry, A.; Kok, R.J.; Sijbrandi, N.; Coudane, J. Reversibly core-crosslinked PEG-P(HPMA) micelles: Platinum coordination chemistry for competitive-ligand-regulated drug delivery. J. Colloid Interface Sci. 2019, 535, 505–515. [Google Scholar] [CrossRef]
- Maiti, C.; Parida, S.; Kayal, S.; Maiti, S.; Mandal, M.; Dhara, D. Redox-Responsive Core-Cross-Linked Block Copolymer Micelles for Overcoming Multidrug Resistance in Cancer Cells. ACS Appl. Mater. Interfaces 2018, 10, 5318–5330. [Google Scholar] [CrossRef]
- Kim, J.H.; Oh, Y.T.; Lee, K.S.; Yun, J.M.; Park, B.T.; Oh, K.T. Development of a pH-sensitive polymer using poly(aspartic acid-graft-imidazole)-block-poly(ethylene glycol) for acidic pH targeting systems. Macromol. Res. 2011, 19, 453–460. [Google Scholar] [CrossRef]
- Lv, S.; Li, M.; Tang, Z.; Song, W.; Sun, H.; Liu, H.; Chen, X. Doxorubicin-loaded amphiphilic polypeptide-based nanoparticles as an efficient drug delivery system for cancer therapy. Acta Biomater. 2013, 9, 9330–9342. [Google Scholar] [CrossRef]
- Ponta, A.; Bae, Y. PEG-poly(amino acid) Block Copolymer Micelles for Tunable Drug Release. Pharm. Res. 2010, 27, 2330–2342. [Google Scholar] [CrossRef]
- Veeren, A.; Bhaw-Luximon, A. Polymer-Drug Encapsulation using Various PEG-and Polypeptide-Based Block Copolymer Micelles. Macromol. Symp. 2012, 313–314, 59–68. [Google Scholar] [CrossRef]
- Yang, L.; Hu, X.; Wang, W.; Liu, S.; Sun, T.; Huang, Y.; Jing, X.; Xie, Z. Y-shaped block copolymer (methoxy-poly(ethylene glycol))2-b-poly(l-glutamic acid): Preparation, self-assembly, and use as drug carriers. RSC Adv. 2014, 4, 41588–41596. [Google Scholar] [CrossRef]
- Ji, S.; Cao, W.; Yu, Y.; Xu, H. Dynamic Diselenide Bonds: Exchange Reaction Induced by Visible Light without Catalysis. Angew. Chem. Int. Ed. 2014, 53, 6781–6785. [Google Scholar] [CrossRef] [PubMed]
- Sill, K.N.; Sullivan, B.; Carie, A.; Semple, J.E. Synthesis and Characterization of Micelle-Forming PEG-Poly(Amino Acid) Copolymers with Iron-Hydroxamate Cross-Linkable Blocks for Encapsulation and Release of Hydrophobic Drugs. Biomacromolecules 2017, 18, 1874–1884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, Z.; Feng, R.; Sun, M.; Guo, C.; Gao, Y.; Li, L.; Zhai, G. Curcumin-loaded PLGA-PEG-PLGA triblock copolymeric micelles: Preparation, pharmacokinetics and distribution in vivo. J. Colloid Interface Sci. 2011, 354, 116–123. [Google Scholar] [CrossRef]
- Debele, T.A.; Mekuria, S.L.; Tsai, H.-C. A pH-sensitive micelle composed of heparin, phospholipids, and histidine as the carrier of photosensitizers: Application to enhance photodynamic therapy of cancer. Int. J. Biol. Macromol. 2017, 98, 125–138. [Google Scholar] [CrossRef]
- Andrgie, A.T.; Birhan, Y.S.; Mekonnen, T.W.; Hanurry, E.Y.; Darge, H.F.; Lee, R.-H.; Chou, H.-Y.; Tsai, H.-C. Redox-Responsive Heparin–Chlorambucil Conjugate Polymeric Prodrug for Improved Anti-Tumor Activity. Polymers 2020, 12, 43. [Google Scholar] [CrossRef] [Green Version]
- Addisu, K.D.; Hailemeskel, B.Z.; Mekuria, S.L.; Andrgie, A.T.; Lin, Y.-C.; Tsai, H.-C. Bioinspired, Manganese-Chelated Alginate–Polydopamine Nanomaterials for Efficient in Vivo T1-Weighted Magnetic Resonance Imaging. ACS Appl. Mater. Interfaces 2018, 10, 5147–5160. [Google Scholar] [CrossRef]
- Darge, H.F.; Andrgie, A.T.; Hanurry, E.Y.; Birhan, Y.S.; Mekonnen, T.W.; Chou, H.-Y.; Hsu, W.-H.; Lai, J.-Y.; Lin, S.-Y.; Tsai, H.-C. Localized controlled release of bevacizumab and doxorubicin by thermo-sensitive hydrogel for normalization of tumor vasculature and to enhance the efficacy of chemotherapy. Int. J. Pharm. 2019, 572, 118799. [Google Scholar] [CrossRef]
- Hanurry, E.Y.; Mekonnen, T.W.; Andrgie, A.T.; Darge, H.F.; Birhan, Y.S.; Hsu, W.-H.; Chou, H.-Y.; Cheng, C.-C.; Lai, J.-Y.; Tsai, H.-C. Biotin-Decorated PAMAM G4.5 Dendrimer Nanoparticles to Enhance the Delivery, Anti-Proliferative, and Apoptotic Effects of Chemotherapeutic Drug in Cancer Cells. Pharmaceutics 2020, 12, 443. [Google Scholar] [CrossRef]
- Gradišar, Š.; Žagar, E.; Pahovnik, D. Ring-Opening Polymerization of N-Carboxyanhydrides Initiated by a Hydroxyl Group. ACS Macro Lett. 2017, 6, 637–640. [Google Scholar] [CrossRef]
- Chan, B.A.; Xuan, S.; Horton, M.; Zhang, D. 1,1,3,3-Tetramethylguanidine-Promoted Ring-Opening Polymerization of N-Butyl N-Carboxyanhydride Using Alcohol Initiators. Macromolecules 2016, 49, 2002–2012. [Google Scholar] [CrossRef]
- Pandey, B.; Patil, N.G.; Bhosle, G.S.; Ambade, A.V.; Gupta, S.S. Amphiphilic Glycopolypeptide Star Copolymer-Based Cross-Linked Nanocarriers for Targeted and Dual-Stimuli-Responsive Drug Delivery. Bioconjug. Chem. 2019, 30, 633–646. [Google Scholar] [CrossRef]
- Li, L.; Li, D.; Zhang, M.; He, J.; Liu, J.; Ni, P. One-Pot Synthesis of pH/Redox Responsive Polymeric Prodrug and Fabrication of Shell Cross-Linked Prodrug Micelles for Antitumor Drug Transportation. Bioconjug. Chem. 2018, 29, 2806–2817. [Google Scholar] [CrossRef] [PubMed]
- Laskar, P.; Saha, B.; Ghosh, S.K.; Dey, J. PEG based random copolymer micelles as drug carriers: The effect of hydrophobe content on drug solubilization and cytotoxicity. RSC Adv. 2015, 5, 16265–16276. [Google Scholar] [CrossRef]
- Fluksman, A.; Benny, O. A robust method for critical micelle concentration determination using coumarin-6 as a fluorescent probe. Anal. Methods 2019, 11, 3810–3818. [Google Scholar] [CrossRef]
- He, Y.; Guo, S.; Wu, L.; Chen, P.; Wang, L.; Liu, Y.; Ju, H. Near-infrared boosted ROS responsive siRNA delivery and cancer therapy with sequentially peeled upconversion nano-onions. Biomaterials 2019, 225, 119501. [Google Scholar] [CrossRef] [PubMed]
- Deng, H.; Zhang, Y.; Wang, X.; Cao, Y.; Liu, J.; Liu, J.; Deng, L.; Dong, A. Balancing the stability and drug release of polymer micelles by the coordination of dual-sensitive cleavable bonds in cross-linked core. Acta Biomater. 2015, 11, 126–136. [Google Scholar] [CrossRef]
- Du, A.W.; Lu, H.; Stenzel, M.H. Core-Cross-Linking Accelerates Antitumor Activities of Paclitaxel–Conjugate Micelles to Prostate Multicellular Tumor Spheroids: A Comparison of 2D and 3D Models. Biomacromolecules 2015, 16, 1470–1479. [Google Scholar] [CrossRef]
- Ji, S.; Xia, J.; Xu, H. Dynamic Chemistry of Selenium: Se–N and Se–Se Dynamic Covalent Bonds in Polymeric Systems. ACS Macro Lett. 2016, 5, 78–82. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Xia, J.; Ji, S.; Fan, Z.; Xu, H. Visible-light-induced metathesis reaction between diselenide and ditelluride. Chem. Commun. 2019, 55, 2813–2816. [Google Scholar] [CrossRef]
- Zhao, P.; Xia, J.; Cao, M.; Xu, H. Wavelength-Controlled Light-Responsive Polymer Vesicle Based on Se–S Dynamic Chemistry. ACS Macro Lett. 2020, 10, 163–168. [Google Scholar] [CrossRef]
- Sun, T.; Jin, Y.; Qi, R.; Peng, S.; Fan, B. Oxidation responsive mono-cleavable amphiphilic di-block polymer micelles labeled with a single diselenide. Polym. Chem. 2013, 4, 4017–4023. [Google Scholar] [CrossRef]
- Li, T.; Pan, S.; Gao, S.; Xiang, W.; Sun, C.; Cao, W.; Xu, H. Diselenide-Pemetrexed Assemblies for Combined Cancer Immuno-, Radio-, and Chemotherapies. Angew. Chem. Int. Ed. 2020, 132, 2722–2726. [Google Scholar] [CrossRef]
- Xiong, D.; Yao, N.; Gu, H.; Wang, J.; Zhang, L. Stimuli-responsive shell cross-linked micelles from amphiphilic four-arm star copolymers as potential nanocarriers for “pH/redox-triggered” anticancer drug release. Polymer 2017, 114, 161–172. [Google Scholar] [CrossRef]
- Kim, Y.; Pourgholami, M.H.; Morris, D.L.; Lu, H.; Stenzel, M.H. Effect of shell-crosslinking of micelles on endocytosis and exocytosis: Acceleration of exocytosis by crosslinking. Biomater. Sci. 2013, 1, 265–275. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Mn a (gmol−1) | Mn b (gmol−1) | Mw b (gmol−1) | PDI b |
|---|---|---|---|---|
| mPEG-PBLA | 8485 | 8396 | 9797 | 1.16 |
| mPEG-PBLA-PCL | 15,904 | 14,692 | 20,623 | 1.37 |
| mPEG-PLA-PCL | 13,355 | 11,783 | 14,953 | 1.27 |
| Samples | Blank PMs | DOX-Loaded PMs | ||||
|---|---|---|---|---|---|---|
| Dh (nm) | ζ-potential | PI | Dh (nm) | ζ-potential | PI | |
| NCMs | 171.55 ± 1.88 | −9.68 ± 3.03 | 0.19 ± 0.04 | 188.13 ± 4.90 | 1.55 ± 4.09 | 0.27 ± 0.03 |
| CCMs | 156.57 ± 4.42 | −13.1 ± 1.17 | 0.23 ± 0.06 | 168.14 ± 1.72 | −3.71 ± 3.51 | 0.21 ± 0.01 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Birhan, Y.S.; Darge, H.F.; Hanurry, E.Y.; Andrgie, A.T.; Mekonnen, T.W.; Chou, H.-Y.; Lai, J.-Y.; Tsai, H.-C. Fabrication of Core Crosslinked Polymeric Micelles as Nanocarriers for Doxorubicin Delivery: Self-Assembly, In Situ Diselenide Metathesis and Redox-Responsive Drug Release. Pharmaceutics 2020, 12, 580. https://doi.org/10.3390/pharmaceutics12060580
Birhan YS, Darge HF, Hanurry EY, Andrgie AT, Mekonnen TW, Chou H-Y, Lai J-Y, Tsai H-C. Fabrication of Core Crosslinked Polymeric Micelles as Nanocarriers for Doxorubicin Delivery: Self-Assembly, In Situ Diselenide Metathesis and Redox-Responsive Drug Release. Pharmaceutics. 2020; 12(6):580. https://doi.org/10.3390/pharmaceutics12060580
Chicago/Turabian StyleBirhan, Yihenew Simegniew, Haile Fentahun Darge, Endiries Yibru Hanurry, Abegaz Tizazu Andrgie, Tefera Worku Mekonnen, Hsiao-Ying Chou, Juin-Yih Lai, and Hsieh-Chih Tsai. 2020. "Fabrication of Core Crosslinked Polymeric Micelles as Nanocarriers for Doxorubicin Delivery: Self-Assembly, In Situ Diselenide Metathesis and Redox-Responsive Drug Release" Pharmaceutics 12, no. 6: 580. https://doi.org/10.3390/pharmaceutics12060580