1. Introduction
Pancreatic Ductal Adenocarcinoma (PDAC) is one of the most aggressive and dangerous cancerous diseases with a high mortality rate [
1]. In the USA, more than 55,000 new cases were estimated in 2018 which is 3–4% of the all newly diagnosed cancer cases. Approximately 80% of these incidences will lead to death within a year [
2]. The average 5-year survival rate is less than 5% [
3]. The main reason for the high mortality of pancreatic cancer patients could be very poor prognosis. The early diagnosis of PDAC is still difficult, and most patients have already progressed to not operable and incurable statuses at the recognition of the disease [
4]. In addition, the chemotherapy applied to treat pancreatic cancers is usually ineffective due to the fast development of resistance. Chemotherapy causes many side effects because of the low selectivity of the currently used drugs [
5]. Furthermore, the hypovascularity of PDAC and a dense desmoplastic stroma that create barriers restrict drug delivery to the tumor site [
6]. Therefore, the design of efficient anticancer agents against PADC is one of the most challenging tasks for scientists working on cancer research [
7]. Targeted tumor therapy could be a promising strategy to overcome these drawbacks in pancreatic cancer treatment—similar to other types of cancers [
8]. Targeted tumor therapy is based on targeting tumor-specific or overexpressed receptors or other cell surface compartments on tumor cells that can be recognized selectively by antibodies or small molecules like folic acid or peptides [
9,
10]. Drug molecules attached to these homing moieties can enter specifically into tumor cells, resulting in selective toxicity without causing toxic side effects in healthy tissues. The application of small molecule drug conjugates (SMDCs) over antibody-drug conjugate (ADCs) may have an advantage in the treatment of PDAC because SMDCs have higher tissue permeability [
11].
Several homing peptides that recognize pancreatic cancer cells and could be used for drug targeting directly or as a part of nanoparticles have been described in the literature [
12,
13,
14,
15]. One of them is the CKAAKNK oligopeptide that was selected by phage display technique and which can specifically bind to tumor vessels in RIP-Tag2 transgenic mice, a prototypical mouse model of multistage pancreatic islet cell carcinoma [
16]. Valetti et al. attached the CKAAKN homing peptide to a squalene (SQ) molecule via thiol-maleimide Michael addition coupling [
17]. The conjugate was co-nanoprecipitated with the squalenoyl prodrug of gemcitabine (SQdFdC) resulting in nanoparticles. The construct was tested on MIA PaCa-2 human pancreatic adenocarcinoma cells, which overexpress frizzled-5 (FZD-5) receptors compared to NIH/3T3 fibroblasts. It was indicated that these cells selectively took up the nanoparticles decorated with the homing peptide by a receptor-mediated way [
18]. Frizzled receptors as Wnt binding 7TM GPCRs are key players in the Wnt/β-catenin signal pathway that is commonly hyperactivated in pancreatic cancers, leading to enhanced cell proliferation [
19]. In the presence of appropriate ligands, the FZD-5 receptor can be internalized, usually in a heterodimeric form [
20]. Therefore, this protein is a promising target for drug targeting to tumor cells. In addition, these data indicated the efficacy of the CKAAKN peptide as a homing device—related to the Wnt-2 sequence—for targeted tumor therapy.
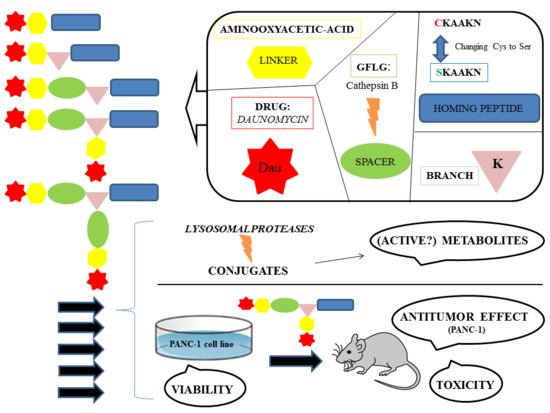
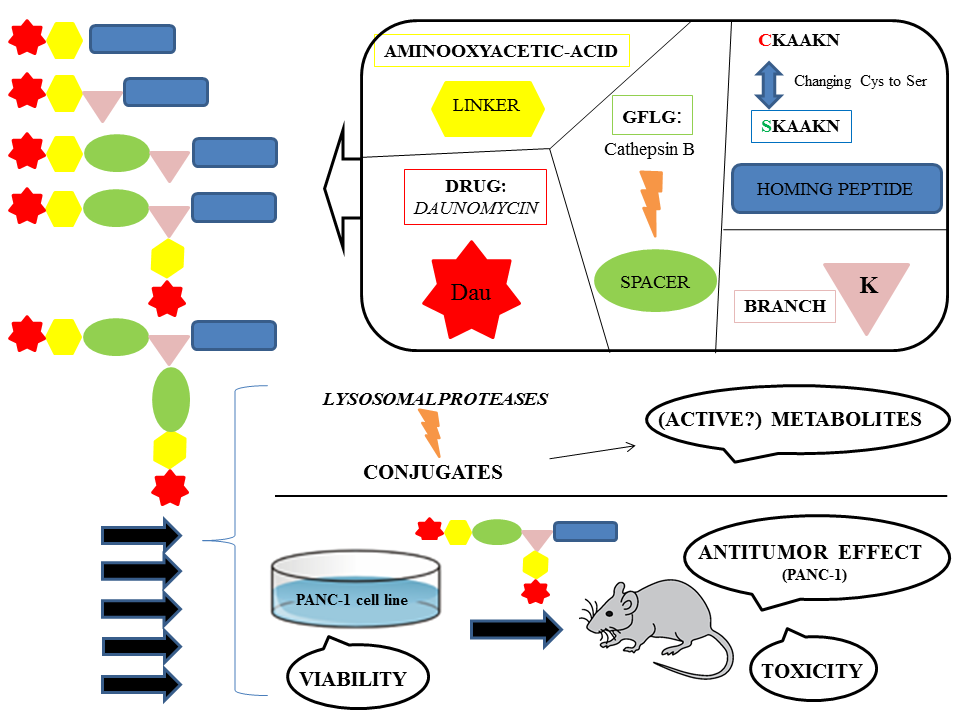
In our research, several SMDCs were developed, derived from the CKAAKN oligopeptide. The structure–activity relationship was investigated as well. In this work, cysteine was replaced by serine to remove the unnecessary thiol group at the conjugation site. This exchange is widely used to eliminate reactive thiol group when it is not essential for the biological activity. In addition, the substitution of Cys by Ser improves the hydrophilicity and solubility of the peptide and its conjugates. Daunomycin (Dau), as an anticancer agent was attached to the homing peptides via oxime linkage, which shows proper stability in the circulation and allows the release of an active metabolite in lysosomes [
21,
22]. This active metabolite contains an amino acid (Aaa) to which daunomycin is connected through an aminooxyacetyl moiety (
Dau=Aoa-Aaa-OH). This metabolite was proved to bind to DNA; however, the binding efficacy highly depends on the type of the amino acid [
22].
The PANC-1 cell line, originally derived from head pancreatic carcinoma, was applied in our studies, and has an invasive phenotype and the ability to give metastasis to the peripancreatic lymph node; thus, this cell line can be considered as an in vitro model of lymph-node-positive PDAC. The invasiveness and metastatic potential of pancreatic cancer cells has been shown to be influenced by the Wnt/β-catenin pathway. This Wnt/β-catenin pathway has also been reported as a central element of immune-escape mechanisms of pancreatic tumors by providing an environment with immune-tolerogenic cytokine and chemokine [
23]. Moreover, the expression level of β-catenin, a key protein of the Wnt pathway, has been found to be well-correlated with the gemcitabine-resistance of different pancreatic cell lines, including PANC-1 cells [
24]. These three characteristics of pancreatic tumor cells like PANC-1 seem to be interrelated and orchestrated by the Wnt/β-catenin pathway.
The influence of the number of drugs as well as the presence of an enzyme cleavable spacer on antitumor activity was studied in PANC-1 pancreatic cancer cells. The antitumor effect is influenced by several cellular factors; therefore, the binding, the cellular uptake and the metabolism of the conjugates were also investigated. The best compounds identified in the in vitro studies were applied in vivo experiment using subcutan (s.c.) developed PANC-1 tumor-bearing SCID mice. The results were compared with free drug administration.
2. Materials and Methods
2.1. Chemicals
All amino acid derivatives and Wang resin were purchased from Iris Biotech GmbH (Marktredwitz, Germany), whereas reagents for coupling and cleavage (N,N′-diisopropylcarbodiimide (DIC), 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU), 1-hydroxybenzotriazole hydrate (HOBt), 4-(dimethylamino)pyridine (DMAP), triisopropylsilane (TIS), trifluoroacetic acid (TFA)) and ninhydrin were delivered by Sigma-Aldrich Kft. (Budapest, Hungary). Aminooxyacetic acid (Aoa) and methoxyamine were TCI (Tokyo, Japan) products. The solvents (dichloromethane (DCM), N,N-dimethylformamide (DMF), acetonitrile (CH3CN)) for synthesis and purification were obtained from Reanal (Budapest, Hungary) or VWR International Kft. (Debrecen, Hungary). Daunomycin (Dau) was donated from IVAX (Budapest, Hungary).
2.2. Synthesis of Peptides
Peptides were synthesized manually by solid-phase peptide synthesis on Wang resin (0.25 g, 0.52 mmol/g) using the standard protocol of Fmoc/
tBu strategy. The first amino acid derivative (5 equivalent to the resin capacity) was attached to the resin with a DIC coupling agent in the presence of 0.5 equivalent DMAP in DMF. The Fmoc group was removed with 2% DBU and 2% piperidine in DMF (four times; 2, 2, 5, 10 min, respectively). For the coupling of the following amino acid derivatives, DIC-HOBt mixture (3 equivalent each) were applied in DMF for 60 min. The
ε-amino group of lysine used for the development of a branch was protected with 4-methyltrityl (Mtt) group that could be removed selectively next to the
tert-butyl type protecting groups with 2% TFA and 2% TIS in DCM (7 times; 1, 1, 3, 3, 5, 10, 30 min, respectively). The aminooxyacetic acid used for the development of an oxime linkage was incorporated in its isopropylidene protected form [
25] either to the
N-terminus or to both
N-termini (backbone and branch) of the peptides. DIC and HOBt coupling agents were used for this purpose, similar to the coupling of amino acid derivatives. The peptides were removed from the resin by cleavage with 5 mL TFA, containing 0.125 mL distilled water and 0.125 mL TIS (as scavengers). The crude product was precipitated by dry diethyl ether, dissolved in 10% acetic acid, freeze-dried and purified by RP-HPLC (Gradient I.).
2.3. Synthesis of Daunomycin Conjugates
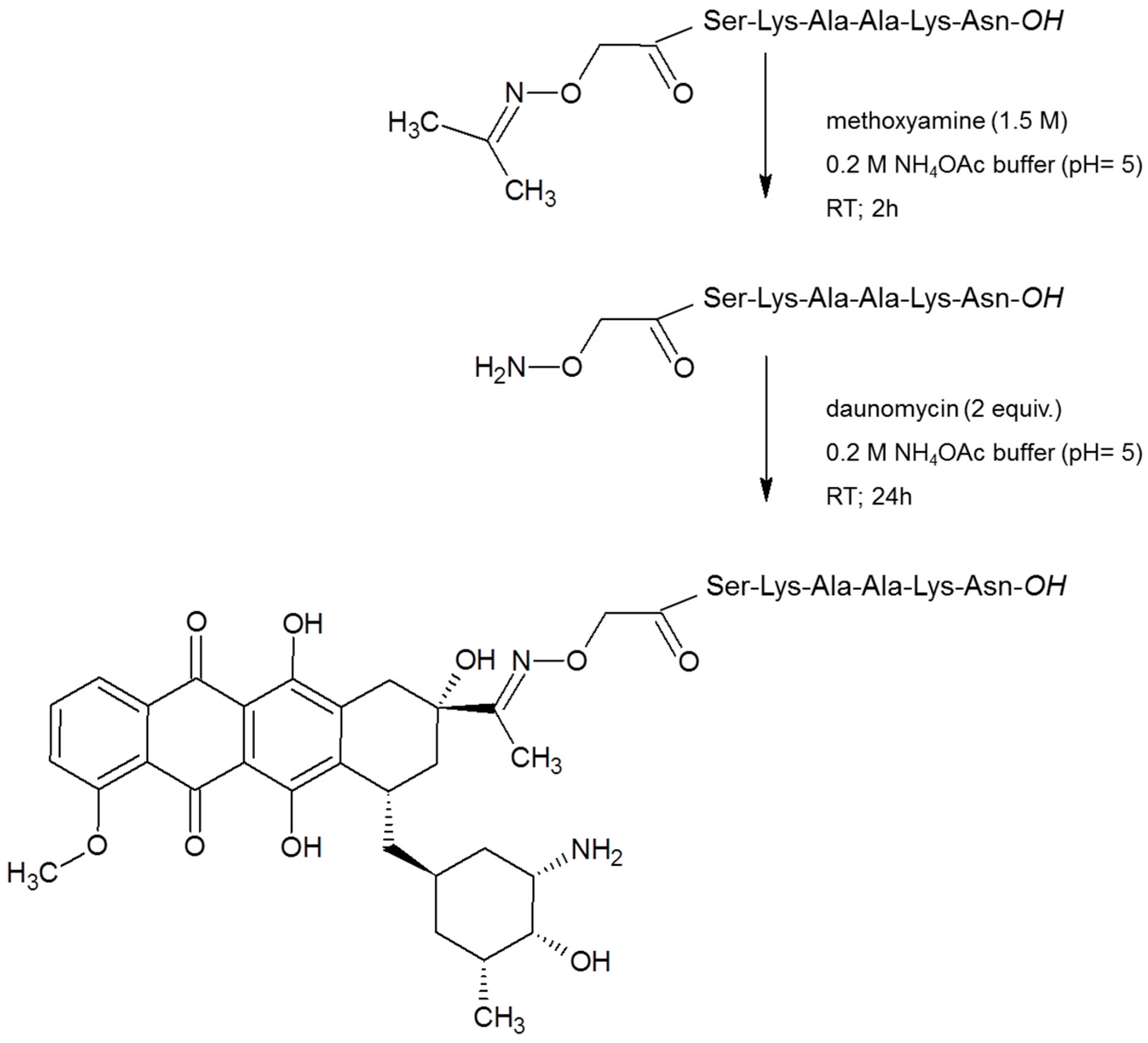
In the first step, the isopropylidene protecting group was removed from the aminooxyacetyl moiety of the purified peptide derivatives by methoxyamine (in 1.5 M concentration) in 0.2 M NH4OAc buffer solution (pH = 5) at RT for 2 h. The reaction took place quantitatively. The unprotected products were isolated by RP-HPLC (Gradient I.). Prior to the conjugation, the solvent was evaporated and then the rest was re-dissolved in 0.2 M NH4OAc buffer solution (pH = 5) and 2 equivalent Dau to the peptide was added to the mixture. The reaction was carried out overnight. The reaction mixture was injected directly to the HPLC in all cases and the conjugates were separated from the excess of Dau by RP-HPLC (Gradient II.)
2.4. Reverse Phase High-Performance Liquid Chromatography (RP-HPLC)
The purification of the crude products was carried out by RP-HPLC using KNAUER 2501 HPLC system (Bad Homburg, Germany) and Phenomenex Luna (Torrance, CA, USA) C18 column (250 × 21.2 mm I.D.) with 10 µm silica (100 Å pore size). Experiments were carried out at a flow rate of 14 mL/min at room temperature. Linear gradient elution was applied. Gradient I: 0 min 5% B, 10 min 5% B, 10.1 min 20% B, 50 min 80% B. Gradient II: 0 min 20% B, 5 min 20% B, 50 min 80% B. Eluent A was 0.1% TFA in distilled water and eluent B was 0.1% TFA in CH3CN-water (80:20, v/v). Peaks were detected at λ = 220 nm.
Analytical RP-HPLC was performed on a Waters Symmetry (WAT 045905) C18 column (150 × 4.6 mm I.D.) with 5 µm silica (100 Å pore size) as a stationary phase. A linear gradient elution was developed: 0 min 0% B; 2 min 0% B; 22 min 90% B with eluent A (0.1% TFA in water) and eluent B (0.1% TFA in acetonitrile-water (80: 20, v/v)). A flow rate of 1 mL/min was used at ambient temperature. Samples were applied dissolved in eluent A and 20 μL was injected. Peaks were detected at λ = 220 nm.
2.5. Mass Spectrometry (MS)
The identification of the peptide analogues and conjugates was achieved by electrospray ionization mass spectrometry (ESI-MS) on a Bruker Daltonics Esquire 3000 Plus (Bremen, Germany) ion trap mass spectrometer, operating in continuous sample injection at 4 µL/min flow rate. Samples were dissolved in ACN-water (50:50 v/v%) mixture containing 0.1 v/v% AcOH. Mass spectra were recorded in positive ion mode in the m/z 50–2000 range.
For the stability and metabolism studies of the conjugates, liquid chromatography–mass spectrometry (LC-MS) analyses were performed on a Q ExactiveTM Focus, high resolution and high mass accuracy, hybrid quadrupole-orbitrap mass spectrometer (Thermo Fisher Scientific, Bremen, Germany) using on-line UHPLC coupling. UHPLC separation was performed on a Dionex 3000 UHPLC system using a Supelco Ascentis C18 column (2.1 × 150 mm, 3 µm). Linear gradient elution (0 min 2% B, 1 min 2% B, 17 min 90% B) with eluent A (0.1% HCOOH in water, v/v) and eluent B (0.1% HCOOH in acetonitrile/water, 80:20, v/v) was used at a flow rate of 0.2 mL/min at 40 °C. High-resolution mass spectra were acquired in the 200–1600 m/z range. LC-MS data were analyzed by XcaliburTM software (Thermo Fisher Scientific) and with Origin Pro 8 (OriginLab Corp., Northampton, MA, USA).
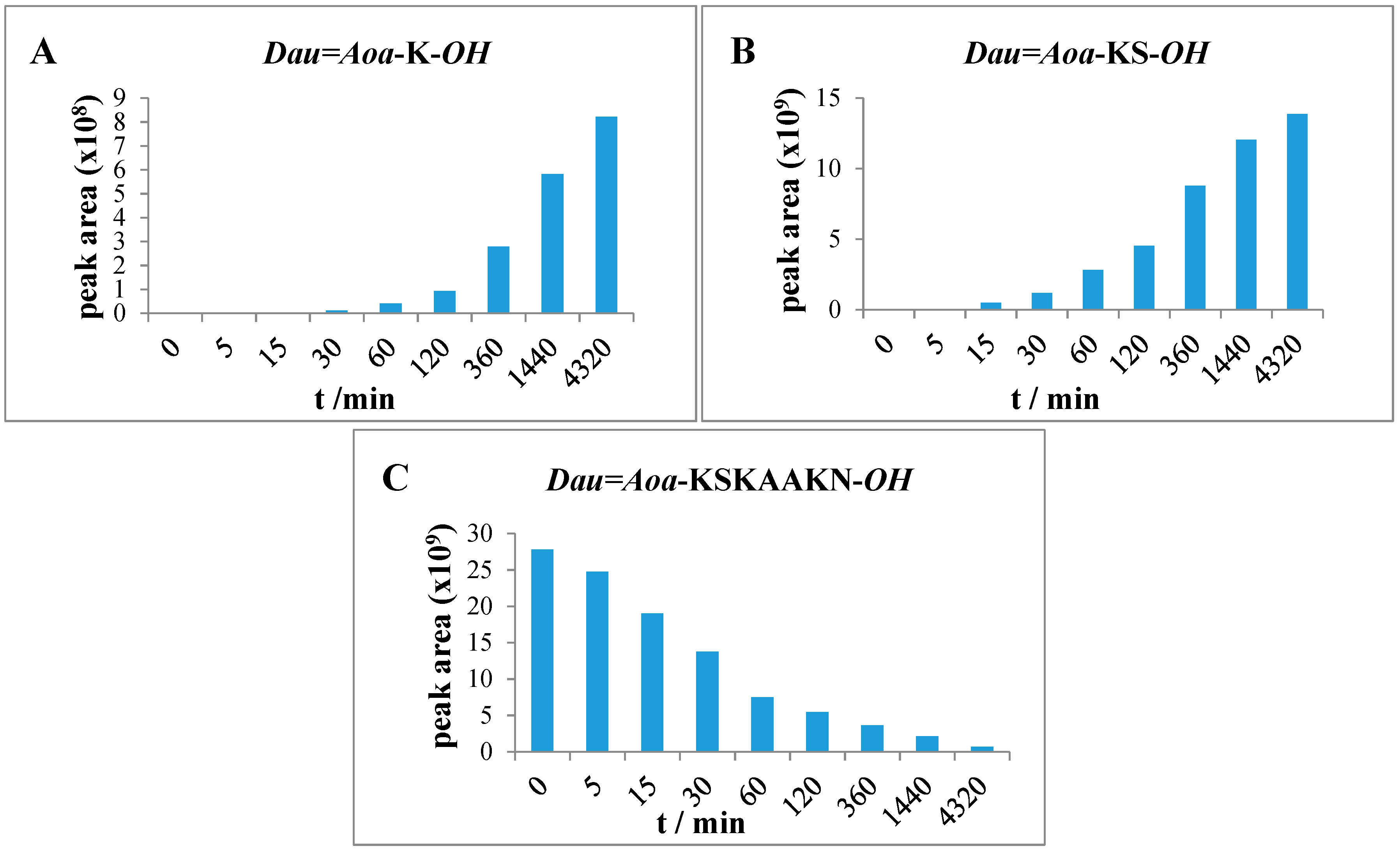
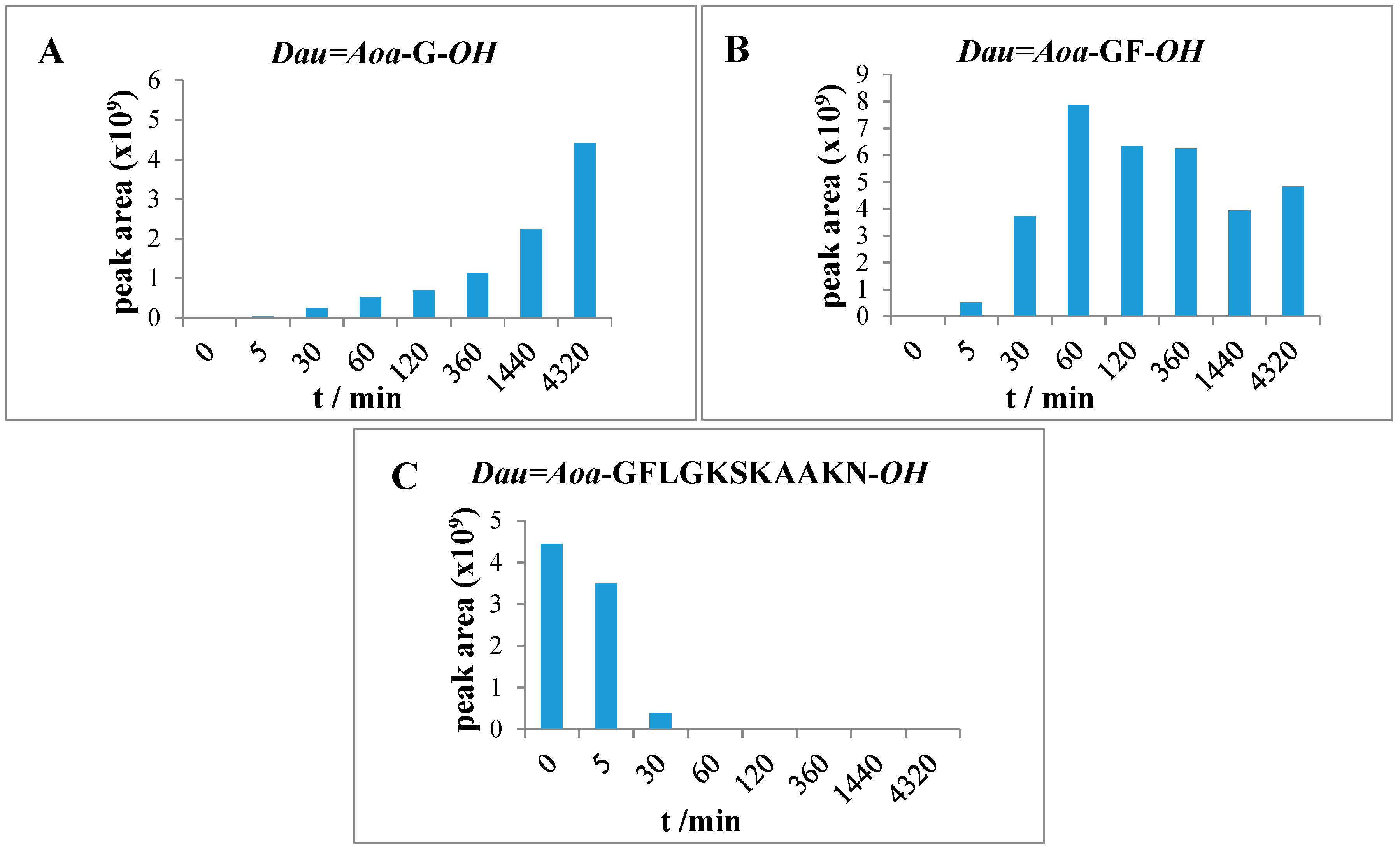
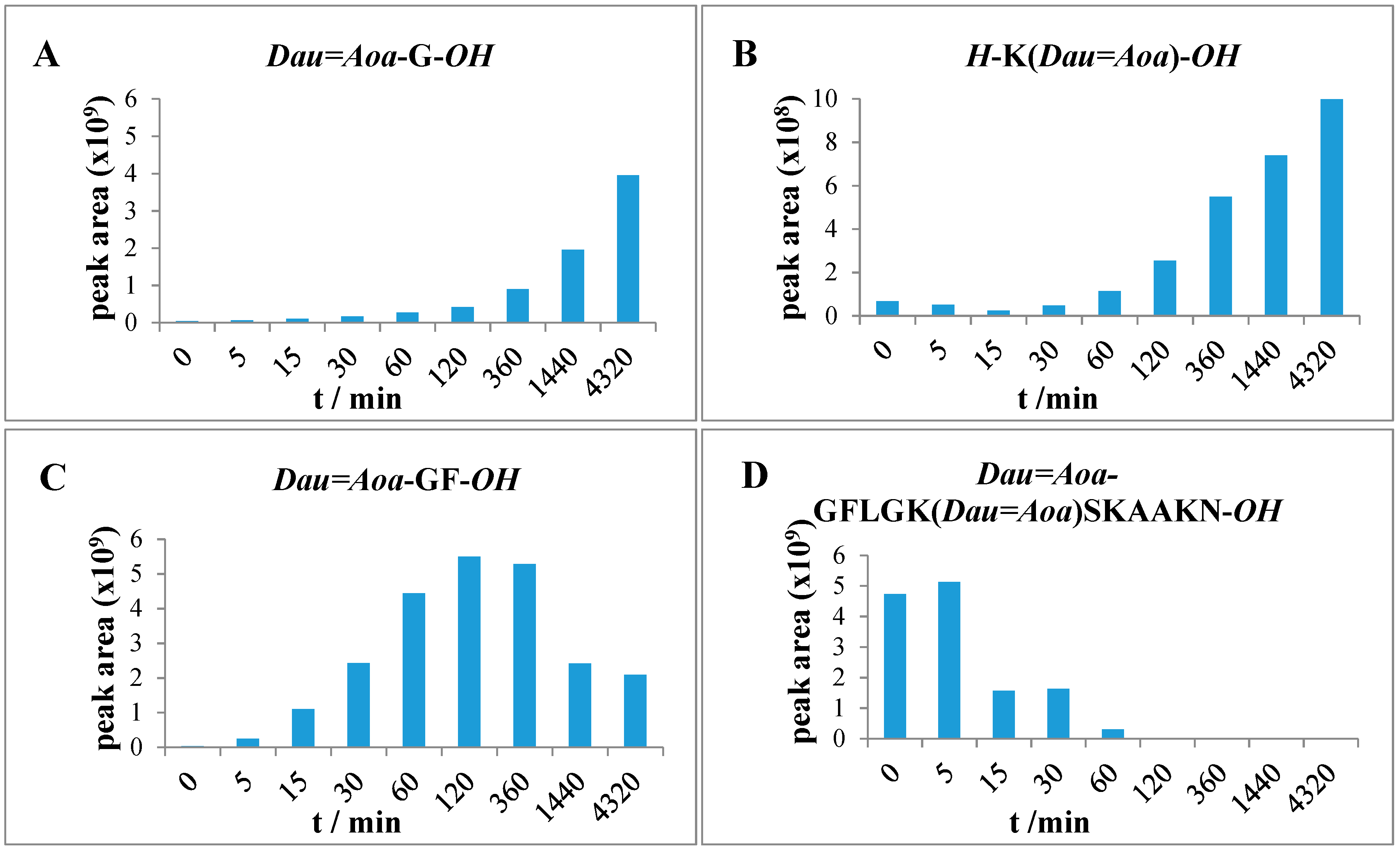
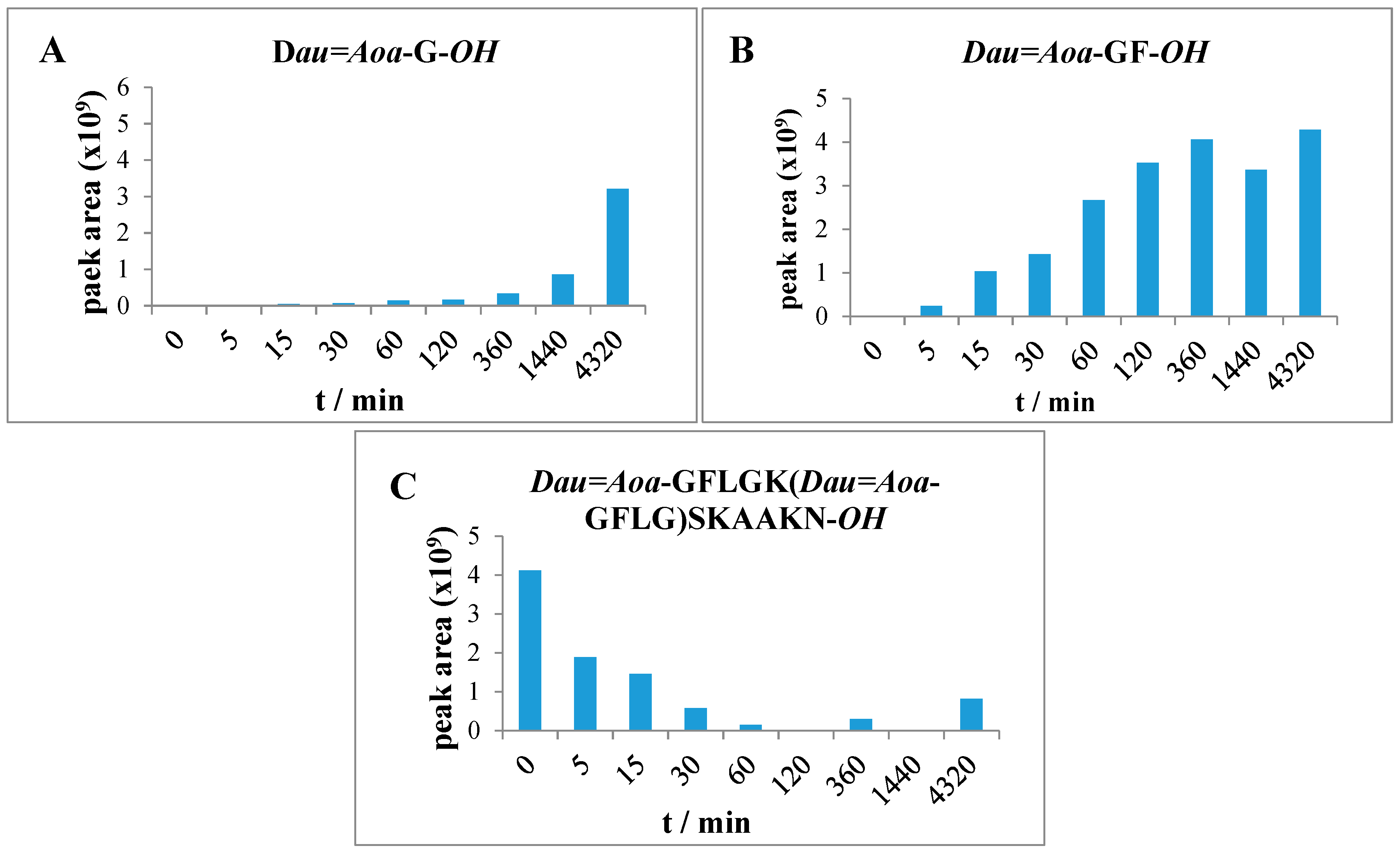
2.6. Measurement of Lysosomal Degradation of Conjugates by LC-MS
Conjugates were dissolved in distilled water in 2.5 μg/μL concentration followed by dilution with 0.2 M NaOAc solution (pH = 5.03) to 0.025 μg/μL. The lysosome-homogenate was prepared from rat liver and contained proteins in 16.6 μg/μL concentration. An aliquot (20 μL) of this stock solution was further diluted with 190 μL 0.2 M NaOAc solution, therefore the final protein concentration was 0.83 μg/μL. To prepare the reaction mixture, 15 μL (0.83 μg/μL) lysosome homogenate was added to 500 μL (0.025 μg/μL) conjugate solution. Furthermore, a control reaction mixture was always prepared which contained 500 μL conjugate solution and 15 μL NaOAc solution only. The solutions were stirred on 600 rpm at 37 °C and samples (50 μL) were taken out at 0 min, 5 min, 15 min, 30 min, 1 h, 2 h, 6 h, 24 h, and 72 h. The enzymatic activity was quenched by adding 5 μL formic acid to the samples. After this procedure, samples were frozen immediately at −25 °C. Control samples were taken at 0 min, 15 min, 1 h, 6 h, 24 h and 72 h. Composition of the samples was determined by HPLC-MS as described above.
2.7. Cell Cultures
For the in vitro characterization of conjugates four different tumor cell lines were used: PANC-1 (human pancreatic carcinoma of ductal origin), Colo-205 (human colorectal adenocarcinoma), A2058 (human metastatic melanoma) obtained from the European Collection of Authenticated Cell Cultures (ECACC, Salisbury, UK) and EBC-1 (human lung squamous cell carcinoma) purchased from the Japanese Research Resources Bank (Tokyo, Japan). Normal Human Dermal Fibroblasts (NHDF; Promocell, Heidelberg, Germany) as non-tumorous control cells were also investigated in order to determine the tumor selectivity of the proposed conjugates.
Dulbecco’s Modified Eagle Medium (DMEM, Lonza, Basel, Switzerland) was used for the culturing of the PANC-1, Colo-205 and EBC-1 cell lines, while the A2058 cell line was maintained in RPMI 1640 (Lonza). These basal media were supplemented with 10% fetal bovine serum (FBS, Gibco®/Invitrogen Corporation, New York, NY, USA), L-glutamine (2 mmol/L) (Lonza) and 100 µg/mL penicillin/streptomycin (Gibco®/Invitrogen Corporation). The medium of the Colo-205 cell line also contained 4500 mg/L D-glucose (Sigma-Aldrich, St. Louis, MO, USA), while, in case of EBC-1 cells, 1% non-essential amino acids (NEAA, Gibco®/Invitrogen Corporation) and 1 mM sodium pyruvate (Sigma-Aldrich) were also added to the culturing medium. For the cultivation of NHDF cells, Promocell Fibroblast Growth Medium (Promocell, Heidelberg, Germany) was used after adding SupplementMix (supplements necessary for the optimal growth of human fibroblasts, Promocell, Heidelberg, Germany) and the aforementioned antibiotics. All cell lines were grown in a T25 culture flask (Sigma-Aldrich or Eppendorf AG, Hamburg, Germany) in an incubator providing an atmosphere of 37 °C and 5% CO2.
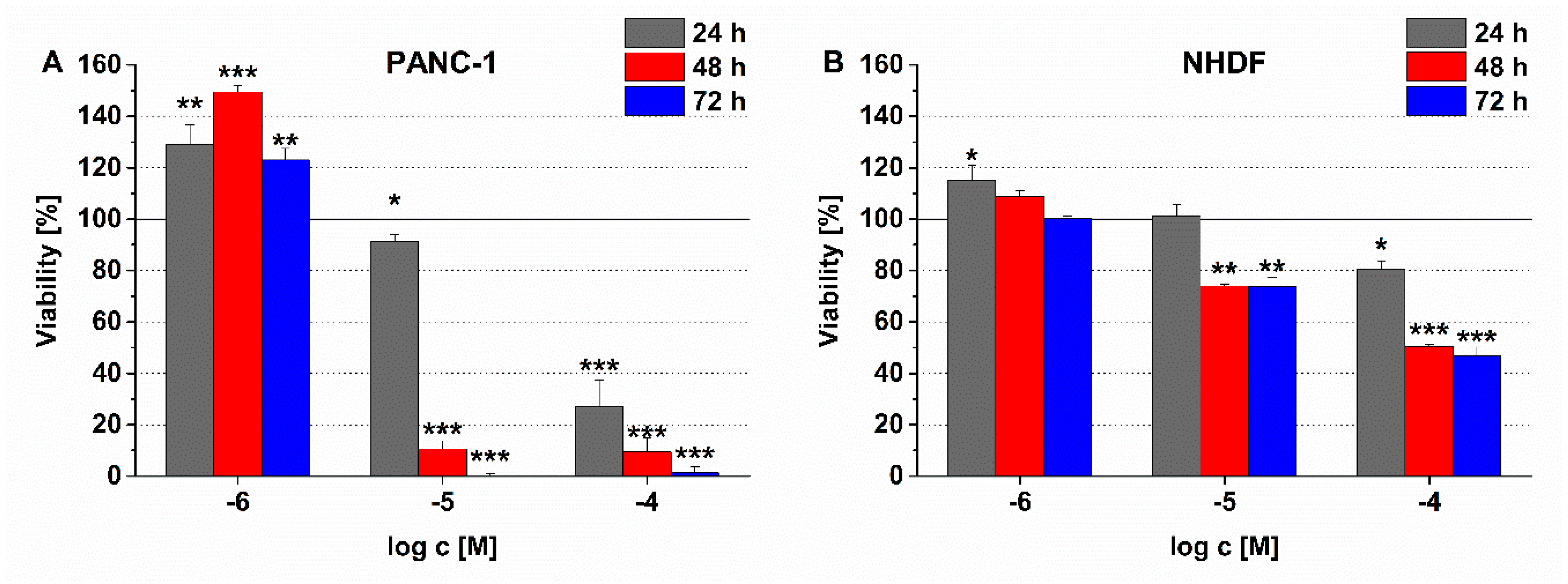
2.8. Measurement of the In Vitro Cytotoxicity of Conjugates
The PANC-1 model cell line exhibits adherent properties under laboratory conditions; therefore, the potential effects of novel antitumor conjugates on cell viability were measured by impedimetry allowing the real-time detection of cell adhesion. This measurement is based on the registration of electrical resistance (impedance, Z) in alternating current (AC) field. The living cells transplanted to the gold measuring electrodes are physically insulated by phospholipid bilayer that covers them. This instrumentally measurable property changes (decreases) in response to cellular cytotoxic agents. Our measurements were performed on xCELLigence single plate (ACEA Biosciences, San Diego, CA, USA) dedicated for impedimetric analysis of cellular samples at 37 °C and 5% CO2.
During the initial phase of the experiments—the baseline recording—a special 96-well cell culture plate, E-plate (ACEA Biosciences), equipped with measuring electrodes, was pretreated with freshly prepared cell culture medium (for 60 min; sampling frequency: 1 min). Subsequently, PANC-1 cells were plated at a cell density of 104 cells/well. During the 24 h incubation, the cells evenly covered the electrodes at the bottom of the wells of the E-plate. The resulting confluent cell cultures were then treated with the test substances at the following final concentrations: 10−6, 10−5, 10−4 M. Total treatment duration was 72 h and the sampling rate was 1 min (0–24 h); and then 15 min (48–72 h). In our measurements, three replicates were used, the control was the drug-free medium. The device displays the impedance change in the form of a cell index (CI), which is a relative (to the start of the experiment) and dimensionless index. The CI results were analyzed with xCELLigence RTCA 2.0 software and Origin Pro 8.0 software. Normalized CI values, expressed as a percentage of control, were used to characterize the cell viability and hence the effect of conjugates.
EBC-1 and Colo-205 model cells have weak/negligible adherent properties, and A2058 cells could not produce constant cell index values. Therefore, a colorimetric assay (alamarBlue-assay) was chosen instead of the xCELLigence system to investigate the viability of these model cells treated with the conjugates. Due to the growth characteristics of NHDF cells, alamarBlue-assay was performed also on this cell line.
The protocol for the alamarBlue-assay was similar to the method which was published earlier [
26], with some minor modifications. Briefly, the cell seeding occurred on 96-well cell culture plates (Sarstedt AG, Nümbrecht, Germany) at 10
4 cells/well concentration. After a 24 h long culturing period, the treatment was carried out with the conjugates at 10
−4, 10
−5 and 10
−6 M final concentrations for 24, 48 and 72 h. In the next steps, the alamarBlue reagent (0.15 mg/mL, Sigma-Aldrich) dissolved in phosphate-buffered saline (PBS; pH = 7.2), was added to the wells. After 6 h incubation with the reagent, the fluorescence intensity of the samples was obtained by an LS-50B Luminescence Spectrometer (Perkin Elmer Ltd., Buckinghamshire, UK) or a Fluoroskan
TM FL Microplate Fluorometer and Luminometer (Thermo Scientific, Waltham, MA, USA) by using the following settings:
λex = 560 nm and
λem = 590 nm.
Three parallels were performed per treatment group. In the case of controls, an equivalent volume of cell culture media was added to the cell. Fluorescence intensities of the samples treated with various concentrations of conjugates were expressed as a percentage of the fluorescence of control.
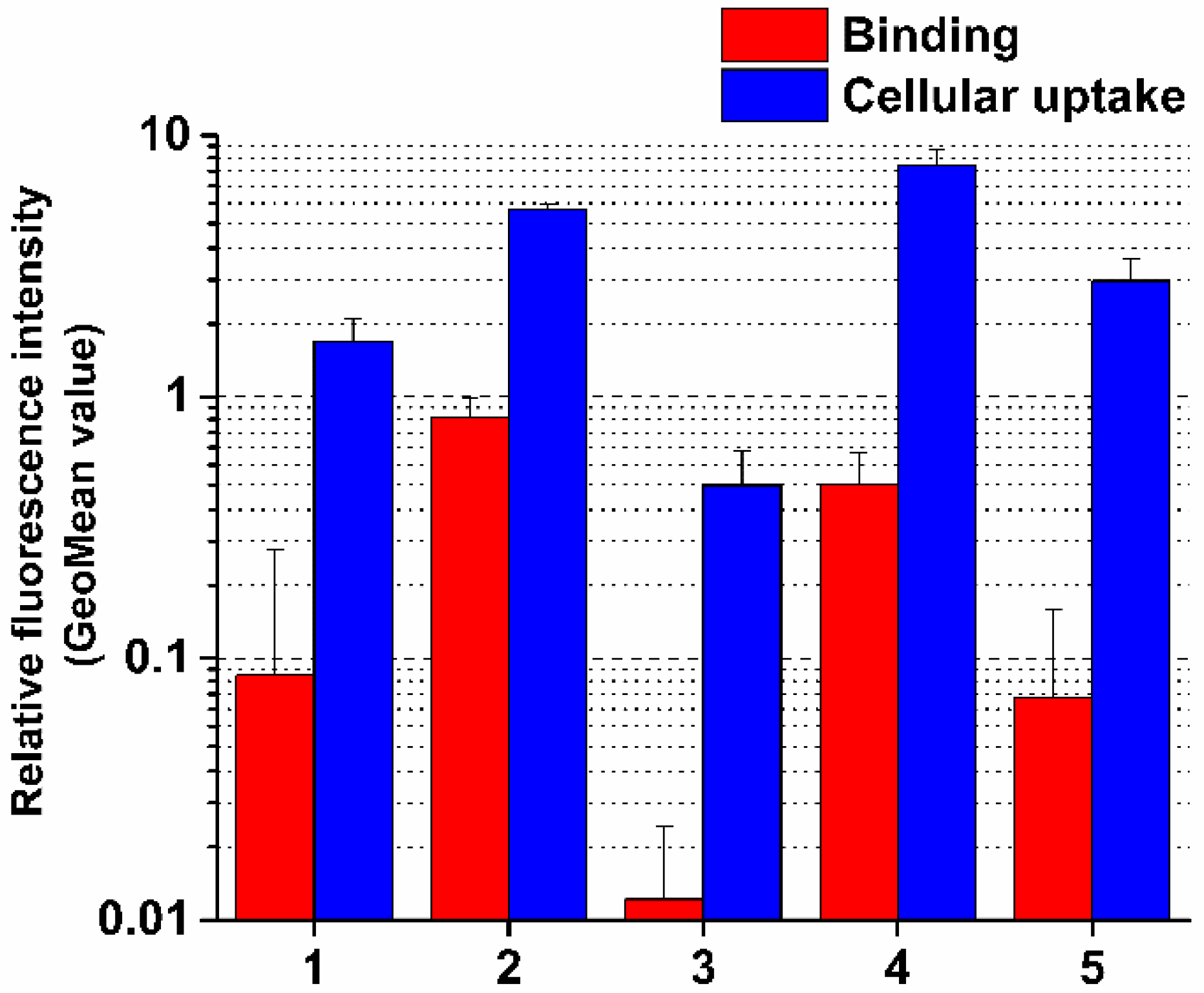
2.9. Flow Cytometric Measurement of Cell Surface Binding and Internalization
Cell surface binding and internalization of the conjugates were performed by flow cytometry (FACS-Calibur, Becton Dickinson, San Jose, CA, USA) based on the detection of the fluorescence activity of Dau (λex = 488 nm, λem = 585 nm) linked to the peptides. Studies of binding and uptake were performed on PANC-1 cells.
Cells were seeded (2.5 × 105 cells/mL, 900 μL/well) on 12-well plates, 24 h prior to the treatment with conjugates and free Dau. To distinguish the cell surface binding and internalization of conjugates, the cells were treated with the conjugate solutions at a final concentration of 10−5 M at two temperatures (37 °C and 4 °C) in parallel. After the incubation period of 30 min, cells were washed with PBS and were removed from the plate using TrypLE (Thermo Fisher Scientific, Waltham, MA, USA) cell-dissociation reagent, thus avoiding cell surface protein degradation. To stop the enzymatic dissociation, 500 μL of fresh medium was added to the wells after 3–5 min and the cells were transferred to FACS-tubes. After the centrifugation of the cell suspension, the cell pellets were resuspended in PBS (400 μL/tube) and the samples were measured by a flow cytometer.
To determine the fluorescence intensity and to evaluate the results CellQuest Pro (Becton Dickinson) and Flowing2.5.1. (Turku Center of Biotechnology, Turku, Finland) software were used. The measurement was carried out twice with two parallels per treatment group. Samples containing cells treated with fresh cell culture medium at 37 °C and 4 °C were used as negative controls. The instrument determines the relative fluorescence intensity of Dau built in the conjugates as geometric mean channel (GeoMean) value.
GeoMean values of the 4 °C and 37 °C samples were corrected with GeoMean values representing the autofluorescence of negative control samples. The fluorescence intensity of cells treated at 4 °C is proportional to the amount of conjugates bound to the cell surface, whereas the fluorescence intensity of cells treated at 37 °C is composed of the signal of conjugates internalized by the cells and those bound to the cell surface, too. The fluorescence intensity specific for the amount of conjugates internalized by the cells was calculated by subtracting GeoMean values of the cells incubated at 4 °C from GeoMean values of the samples incubated at 37 °C.
2.10. Experimental Animals
The Balb/c mice and immunodeficient SCID mice used in these studies were kept as described previously [
27] and cared for according to the “Guiding Principles for the Care and Use of Animals” based upon the Helsinki Declaration, and they were approved by the local ethical committee. The permission license for breeding and performing experiments with laboratory animals: PEI/001/1738-3/2015 and PEI/001/2574-6/2015.
2.11. Acute and Chronic Toxicity Studies
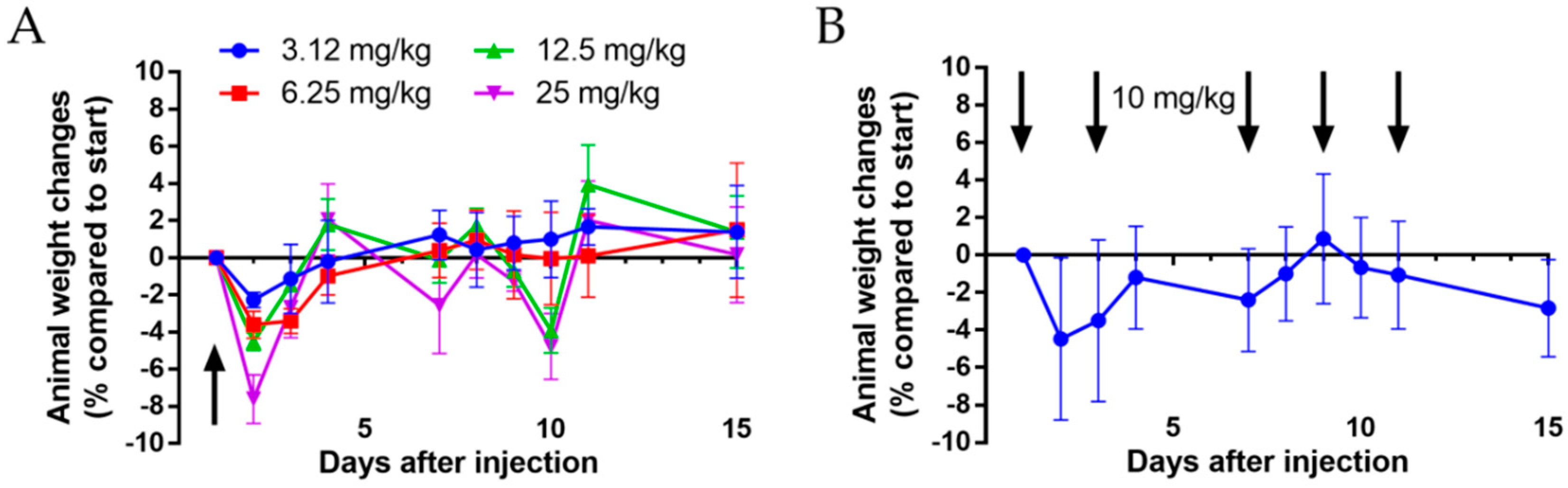
Prior to the determination of in vivo antitumor activity, the acute and chronic toxicity studies of conjugate 4 (Dau=Aoa-GFLG-K(Dau=Aoa)-SKAAKN-OH) were investigated. Healthy Balb/c male mice (3 animals in each group with 29–33 g body weight) were used for these experiments. The conjugate was dissolved in sterile water for injection (Pharmamagist Kft., Budapest, Hungary) and injected in a volume of 0.1 mL/10 g body weight using the appropriate concentrations. In acute toxicity study, intraperitoneal (i.p.) administration of the conjugate 4 was carried out in 4 different doses: 3.125, 6.25, 12.5 and 25 mg/kg Dau-content. In chronic toxicity study, mice were treated with a dose of 10 mg/kg Dau-content of the conjugate on days 1, 3, 7, 9 and 11 (5 treatments). The toxicity was evaluated on the basis of life span, behavior and looking of the mice, as well as body weight. These parameters were followed for 14 days.
2.12. Mouse Model of Subcutaneous Human Pancreatic Cancer, Doses of Treatments and Measurements
For establishing pancreatic tumor in experimental animals, PANC-1 cells were used, which were cultured in RPMI 1640 medium (Lonza), supplemented with 10% heat-inactivated FBS (Biosera, Nuaille, France), and 1% penicillin/streptomycin (Lonza). They were cultured in sterile T175 flasks (Sarstedt AG) with a ventilation cap at 37 °C in a humidified atmosphere with 5% CO2.
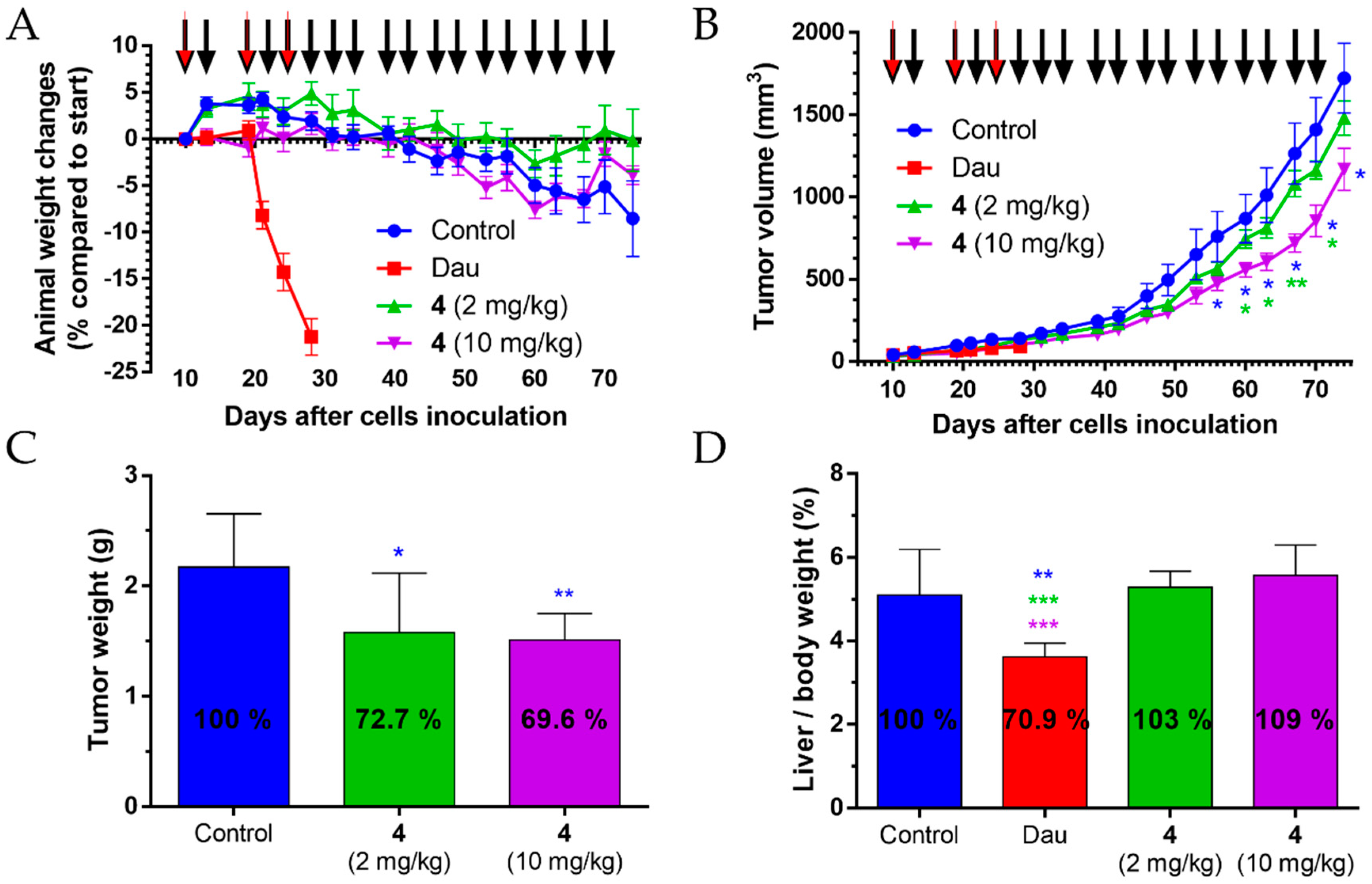
Pancreatic cancer (PANC-1) cells were injected into SCID male mice (22–34 g) subcutaneously (s.c.), 3 × 106 cells per animal in 200 µL M199 (Sigma-Aldrich) per animal. The (i.p.) administration of treatment started 10 days after cells inoculation when the average tumor volume was 36 mm3. Four groups with 7 animals per group were established and treated. The doses and schedule were as follows: control group was treated with sterile water used for the solubilization of Dau and the conjugates, while animals in the group administered with free Dau were treated on days 10, 19 and 24 after cell inoculation with a dose of 1 mg/kg. Mice administered with the conjugate 4 were separated in two groups and treated either with a dose of 10 mg/kg Dau-content (21.6 mg/kg conjugate) or with a dose of 2 mg/kg Dau-content (4.3 mg/kg conjugate) on days 10, 13, 19, 21, 24, 28, 31, 34, 39, 42, 46, 49, 53, 56, 60, 63, 67 and 70 after cells inoculation. Animal weight and tumor volumes were measured initially when the treatment started and at periodic intervals according to the treatment schedule. A digital caliper was used to measure the longest (a) and the shortest diameter (b) of a given tumor. The tumor volume was calculated using the formula V = ab2 × π/6, where a and b represent the measured parameters (length and width). The experiment was terminated on day 74 after cells inoculation (day 65 of treatment). Animals treated with free Dau had to be terminated after 3 treatments on day 28 after cells inoculation (day 19 of treatment) due to significant weight-loss. Animals were sacrificed by cervical dislocation; primary tumors and livers were harvested and weighted.
2.13. Statistical Analysis
For data on the cell viability assay, a one-way ANOVA algorithm of OriginPro 8.0 (OriginLab Corporation, Northampton, MA, USA) was used to assess the significance and calculate p-values. To compare the difference for all means, a Tukey’s post-hoc test was performed. In the case of the in vivo studies, statistical analyses were performed by GraphPad Prism 6 (GraphPad Software, San Diego, CA, USA) using the non-parametric Mann–Whitney test, where p-values lower or equal than 0.05 were considered statistically significant. The symbols *, **, and *** mean significant at p ≤ 0.05, p ≤ 0.01, and p ≤ 0.001, respectively.

 ,
, 











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}