The Vaginal-PVPA: A Vaginal Mucosa-Mimicking In Vitro Permeation Tool for Evaluation of Mucoadhesive Formulations

 ,
,  and
and
Abstract
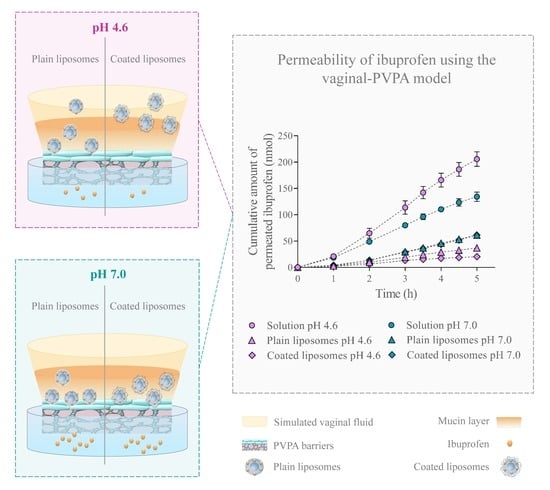
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Preparation of Plain Liposomes
2.3. Entrapment Efficiency and Recovery
2.4. Preparation of Chitosan-Coated Liposomes
2.5. Size and Zeta Potential Measurements
2.6. PVPA Barriers Preparation
2.7. Preparation of Mucus and Simulated Vaginal Fluid
2.8. In Vitro Permeability Study Using the PVPA Barriers
2.9. Mucoadhesive Properties of Coated Liposomes
2.10. Statistical Evaluation
3. Results and Discussion
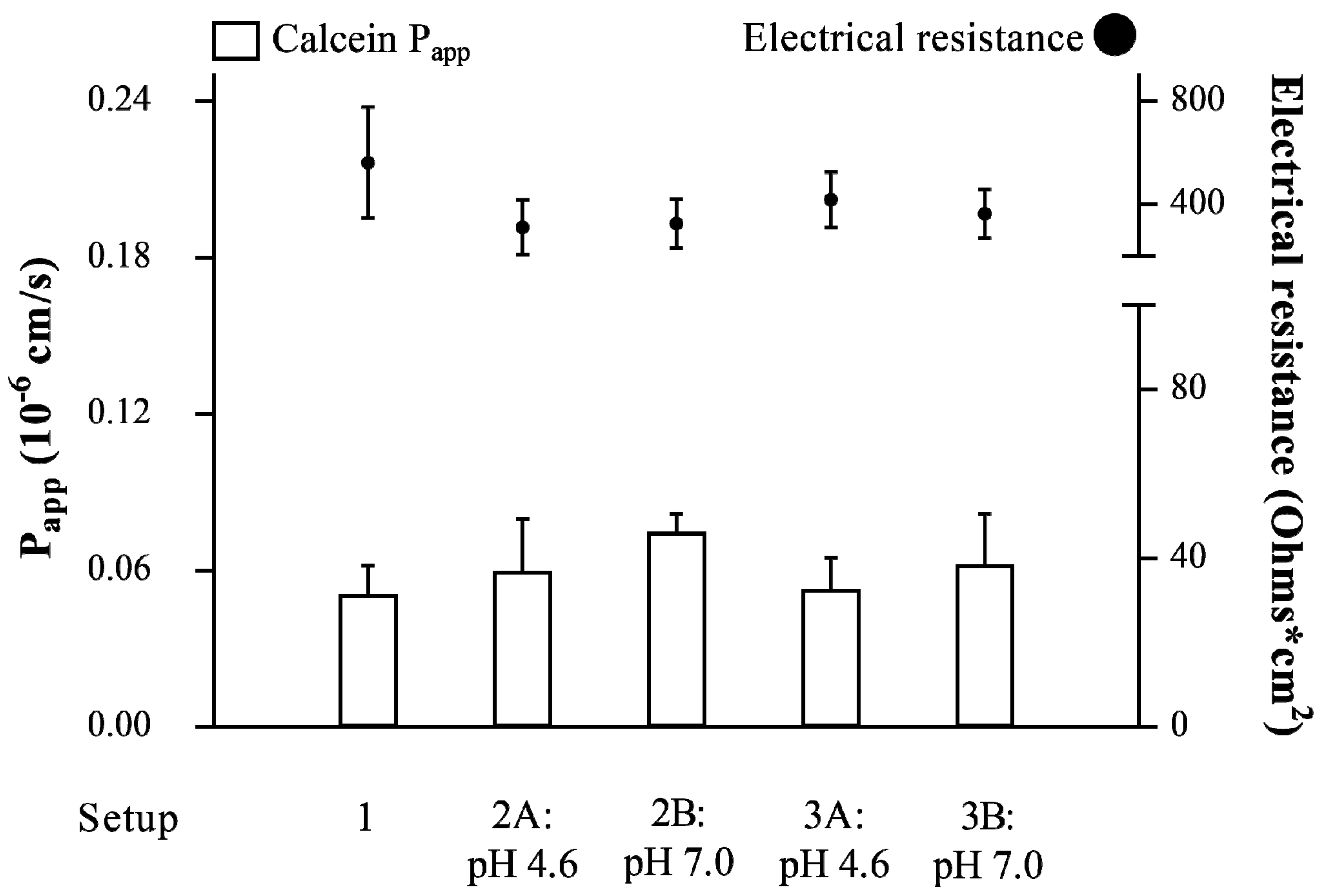
3.1. Validation of the Vaginal-PVPA Model in Terms of Barrier Integrity
3.2. Characterization of the Liposomal Formulations
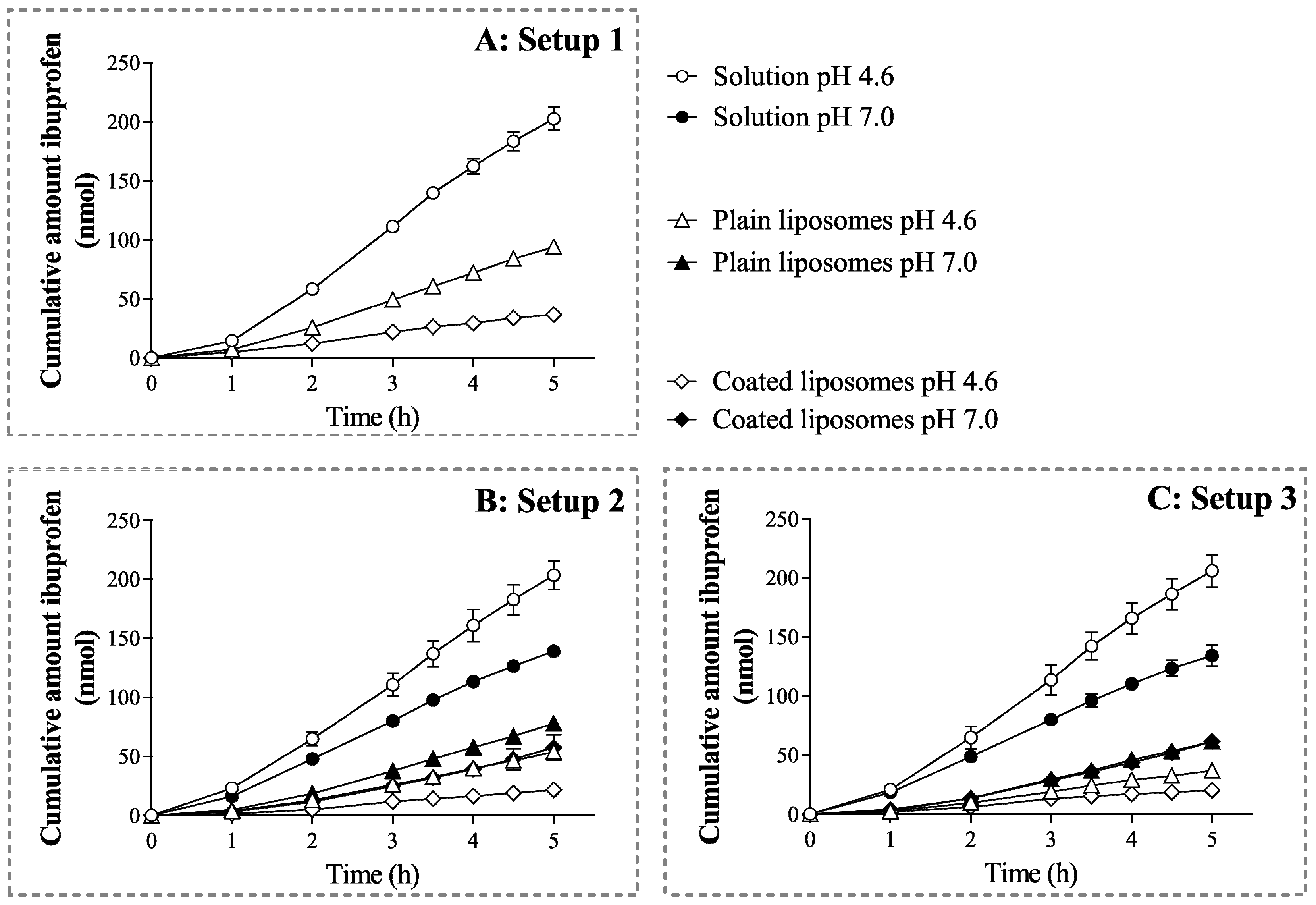
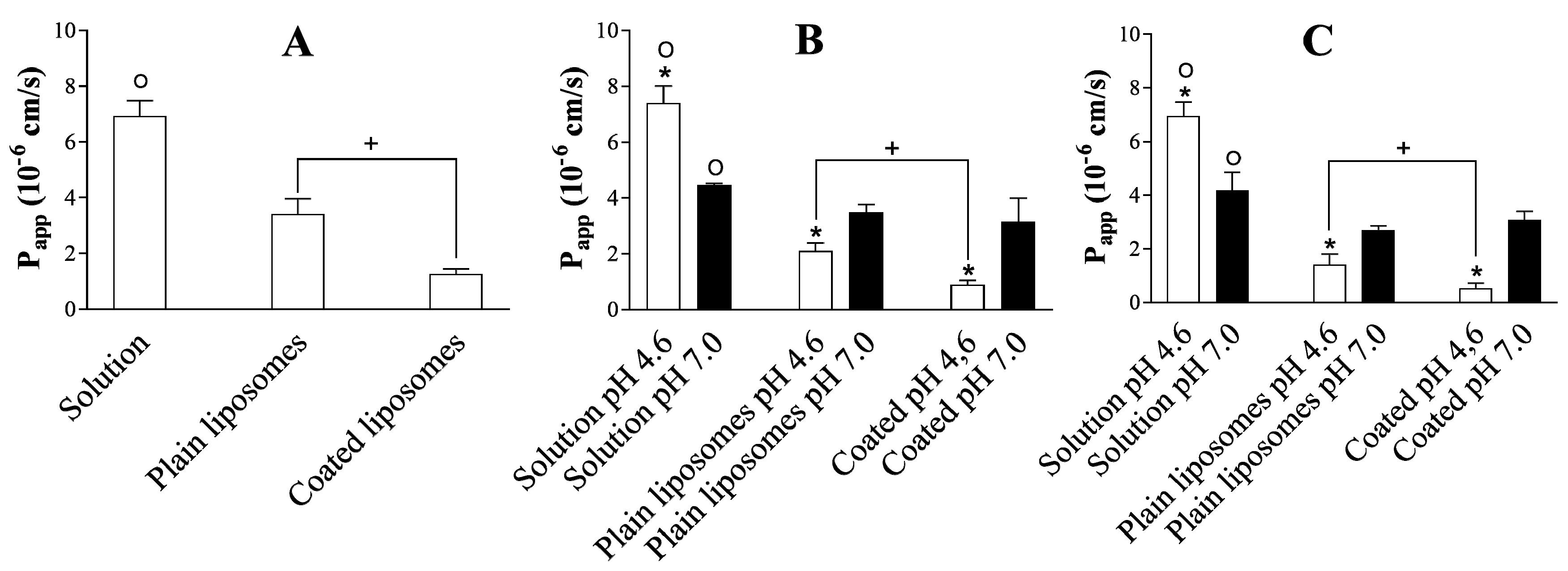
3.3. Dependence of Ibuprofen Permeability on the Formulation Type, Experimental Setup, and pH
Recovery of Ibuprofen in the Donor and Acceptor Compartment
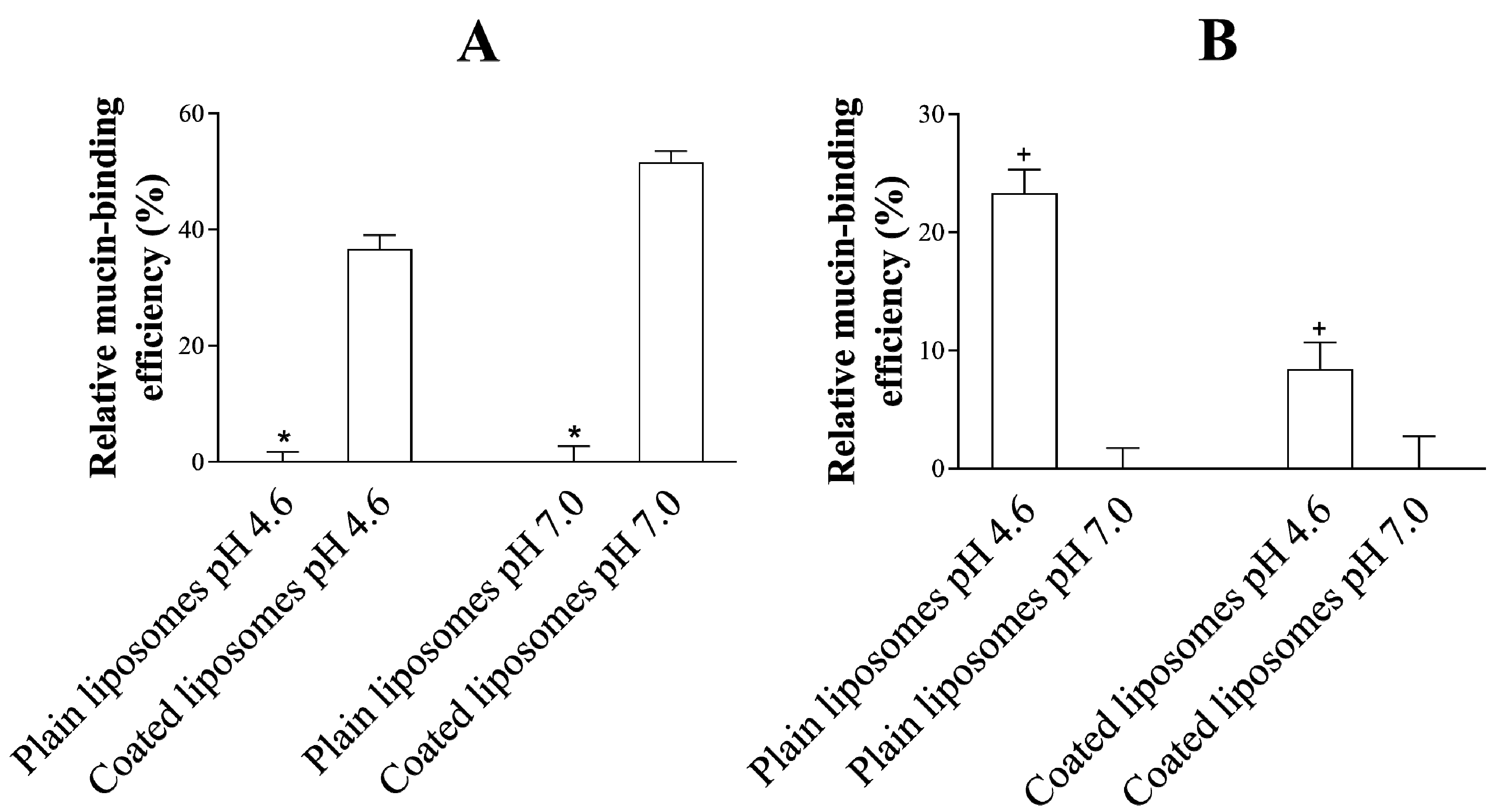
3.4. Mucoadhesive Properties of the Liposomes
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Rossi, S.; Vigani, B.; Sandri, G.; Bonferoni, M.C.; Caramella, C.M.; Ferrari, F.; Carla, C. Recent advances in the mucus-interacting approach for vaginal drug delivery: From mucoadhesive to mucus-penetrating nanoparticles. Expert Opin. Drug Deliv. 2019, 16, 777–781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tho, I.; Škalko-Basnet, N. Cell-based in vitro models for vaginal permeability studies. In Concepts and Models for Drug Permeability Studies—Cell Tissue Based in Vitro Culture Models; Sarmento, B., Ed.; Woodhead Publishing: Cambridge, UK, 2016; pp. 115–128. [Google Scholar]
- Vanic, Z.; Škalko-Basnet, N. Nanoformulations for vaginal therapy. In Nanotechnology Applied to Pharmaceutical Technology; Rai, M., dos Santos, C.A., Eds.; Springer International Publishing AG: Basel, Switzerland, 2017; pp. 183–221. [Google Scholar]
- Palmeira-De-Oliveira, R.; Palmeira-De-Oliveira, A.; De Oliveira, J.M. New strategies for local treatment of vaginal infections. Adv. Drug Deliv. Rev. 2015, 92, 105–122. [Google Scholar] [CrossRef] [PubMed]
- Das Neves, J.; Rocha, C.; Gonçalves, M.; Carrier, R.L.; Amiji, M.; Bahia, M.F.; Sarmentocde, B. Interactions of Microbicide Nanoparticles with a Simulated Vaginal Fluid. Mol. Pharm. 2012, 9, 3347–3356. [Google Scholar] [CrossRef] [PubMed]
- Jøraholmen, M.W.; Bhargava, A.; Julin, K.; Johannessen, M.; Skalko-Basnet, N. The Antimicrobial Properties of Chitosan Can Be Tailored by Formulation. Mar. Drugs 2020, 18, 96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, N.B.; Krieger, K.; Khan, F.M.; Huffman, W.; Chang, M.; Naik, A.; Yongle, R.; Hameed, I.; Krieger, K.; Girardi, L.N.; et al. The current state of animal models in research: A review. Int. J. Surg. 2019, 72, 9–13. [Google Scholar] [CrossRef]
- Grießinger, J.; Dünnhaupt, S.; Cattoz, B.; Griffiths, P.; Oh, S.; I Gómez, S.B.; Wilcox, M.; Pearson, J.; Gumbleton, M.; Abdulkarim, M.; et al. Methods to determine the interactions of micro- and nanoparticles with mucus. Eur. J. Pharm. Biopharm. 2015, 96, 464–476. [Google Scholar] [CrossRef] [Green Version]
- Owen, D.H.; Katz, D.F. A vaginal fluid simulant. Contraception 1999, 59, 91–95. [Google Scholar] [CrossRef]
- Vanic, Z.; Rukavina, Z.; Manner, S.; Fallarero, A.; Uzelac, L.; Kralj, M.; Klarić, D.A.; Bogdanov, A.; Raffai, T.; Virok, D.P.; et al. Azithromycin-liposomes as a novel approach for localized therapy of cervicovaginal bacterial infections. Int. J. Nanomed. 2019, 14, 5957–5976. [Google Scholar] [CrossRef] [Green Version]
- Falavigna, M.; Klitgaard, M.; Brase, C.; Ternullo, S.; Škalko-Basnet, N.; Flaten, G.E. Mucus-PVPA (mucus Phospholipid Vesicle-based Permeation Assay): An artificial permeability tool for drug screening and formulation development. Int. J. Pharm. 2017, 537, 213–222. [Google Scholar] [CrossRef] [Green Version]
- Falavigna, M.; Klitgaard, M.; Steene, E.; Flaten, G.E. Mimicking regional and fasted/fed state conditions in the intestine with the mucus-PVPA in vitro model: The impact of pH and simulated intestinal fluids on drug permeability. Eur. J. Pharm. Sci. 2019, 132, 44–54. [Google Scholar] [CrossRef]
- Naderkhani, E.; Erber, A.; Škalko-Basnet, N.; Flaten, G.E. Improved Permeability of Acyclovir: Optimization of Mucoadhesive Liposomes Using the Phospholipid Vesicle-Based Permeation Assay. J. Pharm. Sci. 2014, 103, 661–668. [Google Scholar] [CrossRef] [Green Version]
- Cook, M.T.; Brown, M.B. Polymeric gels for intravaginal drug delivery. J. Control. Release 2018, 270, 145–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donders, G.; Bellen, G.; Grincevičienė, Š.; Ruban, K.; Vieira-Baptista, P. Aerobic vaginitis: No longer a stranger. Res. Microbiol. 2017, 168, 845–858. [Google Scholar] [CrossRef] [PubMed]
- Berginc, K.; Suljaković, S.; Skalko-Basnet, N.; Kristl, A. Mucoadhesive liposomes as new formulation for vaginal delivery of curcumin. Eur. J. Pharm. Biopharm. 2014, 87, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Jøraholmen, M.W.; Vanic, Z.; Tho, I.; Skalko-Basnet, N. Chitosan-coated liposomes for topical vaginal therapy: Assuring localized drug effect. Int. J. Pharm. 2014, 472, 94–101. [Google Scholar] [CrossRef] [Green Version]
- Flaten, G.E.; Dhanikula, A.B.; Luthman, K.; Brandl, M. Drug permeability across a phospholipid vesicle based barrier: A novel approach for studying passive diffusion. Eur. J. Pharm. Sci. 2006, 27, 80–90. [Google Scholar] [CrossRef] [Green Version]
- Jøraholmen, M.W.; Skalko-Basnet, N.; Acharya, G.; Basnet, P. Resveratrol-loaded liposomes for topical treatment of the vaginal inflammation and infections. Eur. J. Pharm. Sci. 2015, 79, 112–121. [Google Scholar] [CrossRef] [Green Version]
- Berben, P.; Bauer-Brandl, A.; Brandl, M.; Faller, B.; Flaten, G.E.; Jacobsen, A.-C.; Brouwers, J.; Augustijns, P. Drug permeability profiling using cell-free permeation tools: Overview and applications. Eur. J. Pharm. Sci. 2018, 119, 219–233. [Google Scholar] [CrossRef]
- Izgü, F.; Bayram, G.; Tosun, K.; Izgü, D. Stratum corneum lipid liposome-encapsulated panomycocin: Preparation, characterization, and the determination of antimycotic efficacy against Candida spp. isolated from patients with vulvovaginitis in an in vitro human vaginal epithelium tissue model. Int. J. Nanomed. 2017, 12, 5601–5611. [Google Scholar] [CrossRef] [Green Version]
- Jøraholmen, M.W.; Basnet, P.; Tostrup, M.J.; Moueffaq, S.; Skalko-Basnet, N. Localized Therapy of Vaginal Infections and Inflammation: Liposomes-In-Hydrogel Delivery System for Polyphenols. Pharmaceutics 2019, 11, 53. [Google Scholar] [CrossRef] [Green Version]
- Andersen, T.; Mishchenko, E.; Flaten, G.E.; Sollid, J.U.E.; Mattsson, S.; Tho, I.; Skalko-Basnet, N. Chitosan-Based Nanomedicine to Fight Genital Candida Infections: Chitosomes. Mar. Drugs 2017, 15, 64. [Google Scholar] [CrossRef] [Green Version]
- Giordani, B.; Basnet, P.; Mishchenko, E.; Luppi, B.; Skalko-Basnet, N. Utilizing Liposomal Quercetin and Gallic Acid in Localized Treatment of Vaginal Candida Infections. Pharmaceutics 2019, 12, 9. [Google Scholar] [CrossRef] [Green Version]
- Major, I.; McConville, C. Vaginal drug delivery for the localised treatment of cervical cancer. Drug Deliv. Transl. Res. 2017, 7, 817–828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brako, F.; Mahalingam, S.; Rami-Abraham, B.; Craig, D.Q.; Edirisinghe, M. Application of nanotechnology for the development of microbicides. Nanotechnology 2016, 28, 52001. [Google Scholar] [CrossRef] [PubMed]
- Benet, L.Z.; Broccatelli, F.; Oprea, T.I. BDDCS Applied to Over 900 Drugs. AAPS J. 2011, 13, 519–547. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H. Thin-Film Hydration Followed by Extrusion Method for Liposome Preparation. In Advanced Structural Safety Studies; Humana Press: New York, NY, USA, 2016; Volume 1522, pp. 17–22. [Google Scholar]
- Mohammed, A.R.; Weston, N.; Coombes, A.; Fitzgerald, M.; Perrie, Y. Liposome formulation of poorly water soluble drugs: Optimisation of drug loading and ESEM analysis of stability. Int. J. Pharm. 2004, 285, 23–34. [Google Scholar] [CrossRef]
- Das Neves, J.; Amiji, M.; Sarmentocde, B. Mucoadhesive nanosystems for vaginal microbicide development: Friend or foe? Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2011, 3, 389–399. [Google Scholar] [CrossRef] [PubMed]
- Andersen, T.; Bleher, S.; Flaten, G.E.; Tho, I.; Mattsson, S.; Skalko-Basnet, N. Chitosan in Mucoadhesive Drug Delivery: Focus on Local Vaginal Therapy. Mar. Drugs 2015, 13, 222–236. [Google Scholar] [CrossRef] [Green Version]
- Takeuchi, H.; Matsui, Y.; Sugihara, H.; Yamamoto, H.; Kawashima, Y. Effectiveness of submicron-sized, chitosan-coated liposomes in oral administration of peptide drugs. Int. J. Pharm. 2005, 303, 160–170. [Google Scholar] [CrossRef]
- Naderkhani, E.; Isaksson, J.; Ryzhakov, A.; Flaten, G.E. Development of a Biomimetic Phospholipid Vesicle-based Permeation Assay for the Estimation of Intestinal Drug Permeability. J. Pharm. Sci. 2014, 103, 1882–1890. [Google Scholar] [CrossRef] [Green Version]
- Vieira, A.P.; Badshah, S.; Airoldi, C. Ibuprofen-loaded chitosan and chemically modified chitosans—Release features from tablet and film forms. Int. J. Biol. Macromol. 2013, 52, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Avdeef, A. Charge State. In Absorption and Drug Development: Solubility, Permeability, and Charge State; Avdeef, A., Ed.; John Wiley & Sons Inc.: Hoboken, NJ, USA, 2003; pp. 22–41. [Google Scholar]
- Eliyahu, S.; Almeida, A.; Macedo, M.H.; Das Neves, J.; Sarmentocde, B.; Bianco-Peled, H. The effect of freeze-drying on mucoadhesion and transport of acrylated chitosan nanoparticles. Int. J. Pharm. 2020, 573, 118739. [Google Scholar] [CrossRef] [PubMed]
- Schattling, P.; Taipaleenmäki, E.; Zhang, Y.; Städler, B. A Polymer Chemistry Point of View on Mucoadhesion and Mucopenetration. Macromol. Biosci. 2017, 17, 1700060. [Google Scholar] [CrossRef] [PubMed]
- Vimr, E.R.; Kalivoda, K.A.; Deszo, E.L.; Steenbergen, S.M. Diversity of Microbial Sialic Acid Metabolism. Microbiol. Mol. Biol. Rev. 2004, 68, 132–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, X.; Bansil, R.; Bhaskar, K.R.; Turner, B.S.; Lamont, J.T.; Niu, N.; Afdhal, N. pH-dependent conformational change of gastric mucin leads to sol-gel transition. Biophys. J. 1999, 76, 1250–1258. [Google Scholar] [CrossRef] [Green Version]
- Kumirska, J.; Weinhold, M.X.; Thöming, J.; Stepnowski, P. Biomedical Activity of Chitin/Chitosan Based Materials—Influence of Physicochemical Properties Apart from Molecular Weight and Degree of N-Acetylation. Polymers 2011, 3, 1875–1901. [Google Scholar] [CrossRef]
- Falavigna, M.; Stein, P.C.; Flaten, G.E.; Di Cagno, M.P. Impact of Mucin on Drug Diffusion: Development of a Straightforward In Vitro Method for the Determination of Drug Diffusivity in the Presence of Mucin. Pharmaceutics 2020, 12, 168. [Google Scholar] [CrossRef] [Green Version]
- Naderkhani, E.; Vasskog, T.; Flaten, G.E. Biomimetic PVPA in vitro model for estimation of the intestinal drug permeability using fasted and fed state simulated intestinal fluids. Eur. J. Pharm. Sci. 2015, 73, 64–71. [Google Scholar] [CrossRef] [Green Version]
- Baloglu, E.; Bernkop-Schnürch, A.; Karavana, S.Y.; Senyigit, Z.A. Strategies to prolong the intravaginal residence time of drug delivery systems. J. Pharm. Pharm. Sci. 2009, 12, 312–336. [Google Scholar] [CrossRef]
- Meng, J.; Sturgis, T.F.; Youan, B.-B.C. Engineering tenofovir loaded chitosan nanoparticles to maximize microbicide mucoadhesion. Eur. J. Pharm. Sci. 2011, 44, 57–67. [Google Scholar] [CrossRef] [Green Version]
- Rossi, S.; Vigani, B.; Bonferoni, M.C.; Sandri, G.; Caramella, C.; Ferrari, F. Rheological analysis and mucoadhesion: A 30 year-old and still active combination. J. Pharm. Biomed. Anal. 2018, 156, 232–238. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Concentration (g/L) |
|---|---|
| Sodium chloride | 3.510 |
| Potassium hydroxide | 1.400 |
| Calcium hydroxide | 0.222 |
| Bovine serum albumin | 0.018 |
| Lactic acid | 2.000 |
| Acetic acid | 1.000 |
| Glycerol | 0.160 |
| Urea | 0.400 |
| Glucose | 5.000 |
| pH | 4.6/7.0 |
| Setup | SVF (10 µL) | Mucus (50 µL) |
|---|---|---|
| 1 | ✕ | ✕ |
| 2 A: pH 4.6 B: pH 7.0 | ✕ ✕ | ✓ ✓ |
| 3 A: pH 4.6 B: pH 7.0 | ✓ ✓ | ✓ ✓ |
| Name | Diameter (nm) | PdI | Zeta Potential (mV) |
|---|---|---|---|
| Empty plain liposomes | 192.6 ± 4.57 | 0.094 ± 0.014 | −1.99 ± 3.53 |
| Plain liposomes | 193.4 ± 3.53 | 0.123 ± 0.020 | −14.0 ± 1.87 |
| Empty coated liposomes | 203.3 ± 3.51 | 0.097 ± 0.015 | 3.06 ±. 3.87 |
| Coated liposomes | 337.9 ± 21.9 | 0.322 ± 0.05 | 42.17 ± 5.34 (Area: 50%) 64.70 ± 2.40 (Area: 50%) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Falavigna, M.; Pattacini, M.; Wibel, R.; Sonvico, F.; Škalko-Basnet, N.; Flaten, G.E. The Vaginal-PVPA: A Vaginal Mucosa-Mimicking In Vitro Permeation Tool for Evaluation of Mucoadhesive Formulations. Pharmaceutics 2020, 12, 568. https://doi.org/10.3390/pharmaceutics12060568
Falavigna M, Pattacini M, Wibel R, Sonvico F, Škalko-Basnet N, Flaten GE. The Vaginal-PVPA: A Vaginal Mucosa-Mimicking In Vitro Permeation Tool for Evaluation of Mucoadhesive Formulations. Pharmaceutics. 2020; 12(6):568. https://doi.org/10.3390/pharmaceutics12060568
Chicago/Turabian StyleFalavigna, Margherita, Martina Pattacini, Richard Wibel, Fabio Sonvico, Natasa Škalko-Basnet, and Gøril Eide Flaten. 2020. "The Vaginal-PVPA: A Vaginal Mucosa-Mimicking In Vitro Permeation Tool for Evaluation of Mucoadhesive Formulations" Pharmaceutics 12, no. 6: 568. https://doi.org/10.3390/pharmaceutics12060568