Development of a Self-Emulsifying Drug Delivery System for Optimized Topical Delivery of Clofazimine


Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Pre-Formulation Studies
2.2.1. Solubility
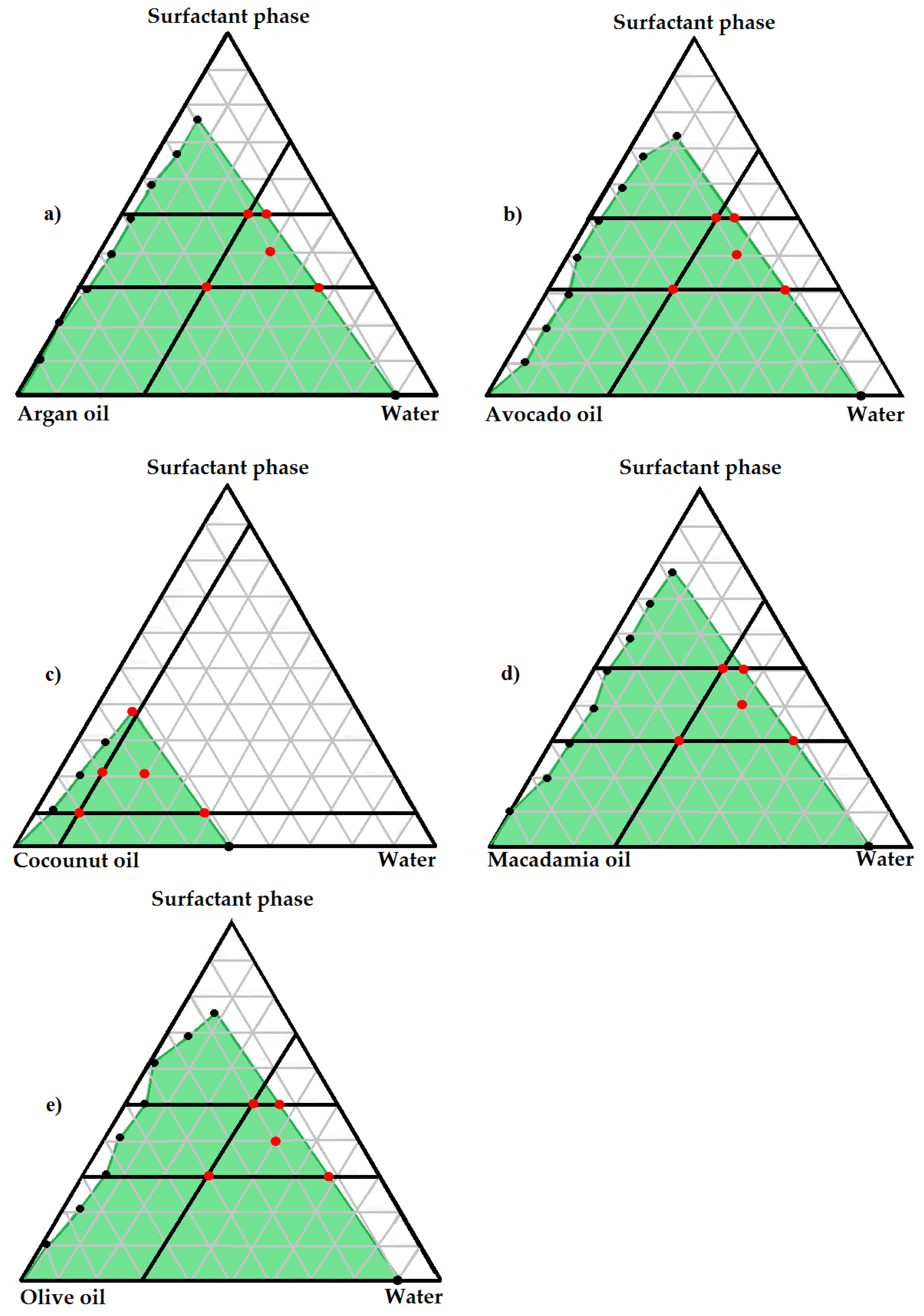
2.2.2. Pseudo-Ternary Phase Diagrams
2.3. Preparation of Topical SEDDSs
2.4. Characterization of Topical SEDDSs
2.4.1. Droplet Size, Zeta-Potential and Size Distribution
2.4.2. Robustness to Dilution
2.4.3. Efficacy and Self-Emulsification Time
2.4.4. Viscosity and pH
2.4.5. Cloud Point
2.4.6. Thermodynamic Stability Studies
2.5. Topical Delivery
2.5.1. Encapsulation Efficiency
2.5.2. Drug Release Experiments
2.5.3. Skin Preparation
2.5.4. Skin Diffusion Studies
2.5.5. Tape Stripping
2.6. Isothermal Calorimetry
3. Results and Discussion
3.1. Pre-Formulation and Characterization
3.1.1. Solubility Studies
3.1.2. Pseudo-Ternary Phase Diagrams and Topical SEDDSs Preparation
3.1.3. Droplet Size, Zeta-Potential and Size Distribution
3.1.4. Robustness to Dilution
3.1.5. Efficacy and Self-Emulsification Time
3.1.6. Viscosity and pH
3.1.7. Cloud Point
3.1.8. Thermodynamic Stability
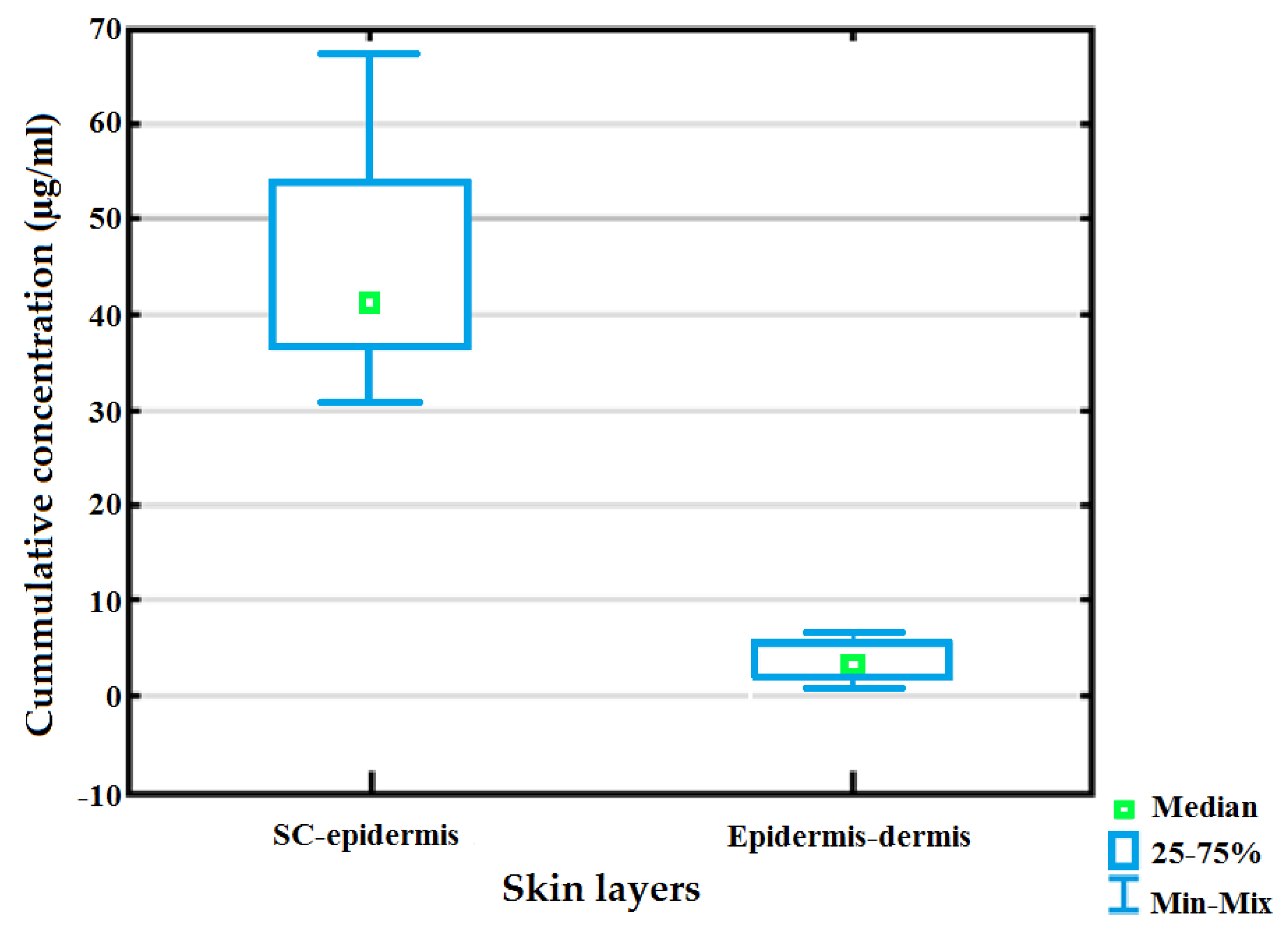
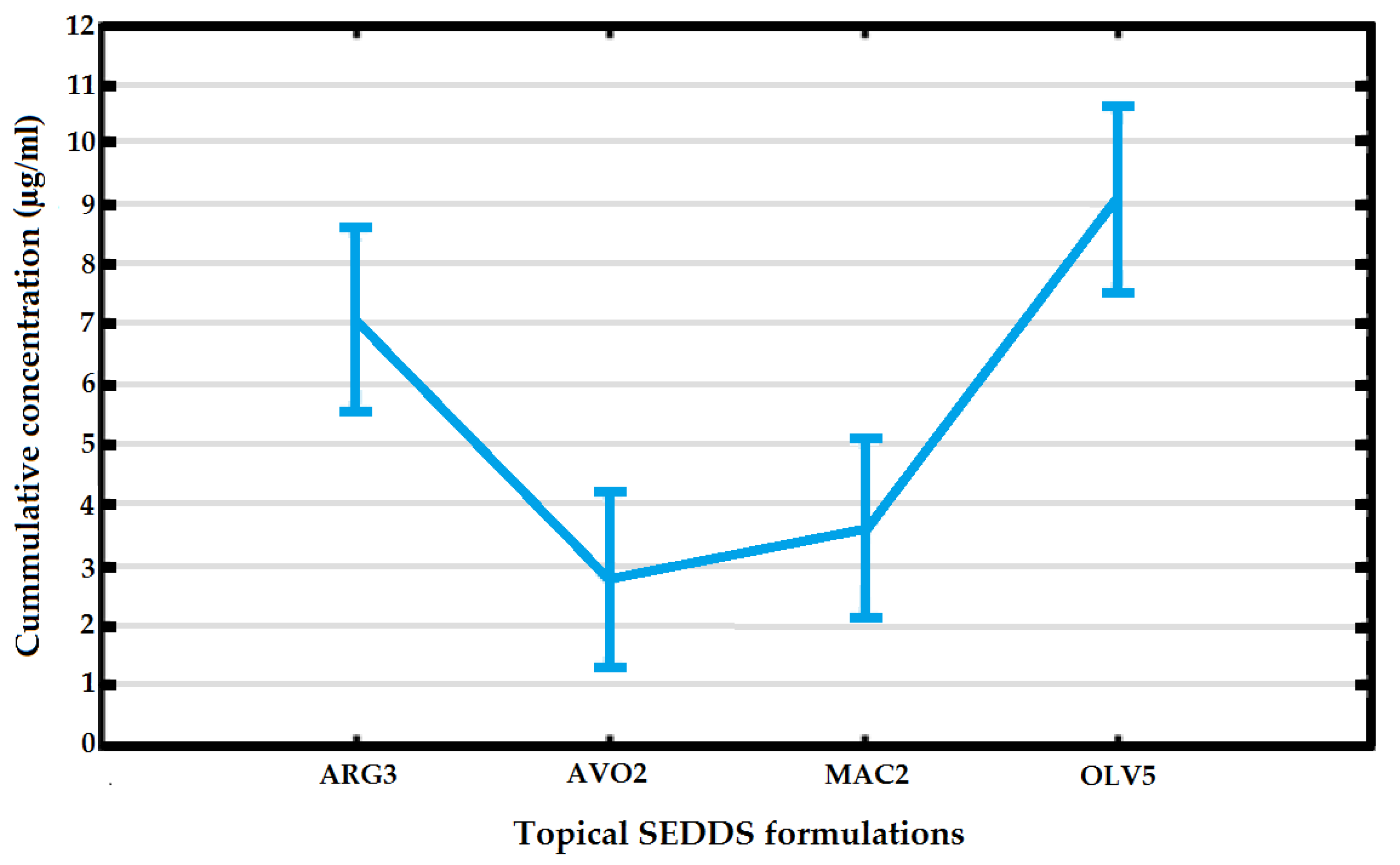
3.2. Topical Clofazimine Delivery
3.3. Isothermal Microcalometry
3.4. Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Churchyard, G.J. A Short Regimen for Rifampin-Resistant Tuberculosis. N. Engl. J. Med. 2019, 380, 1279–1280. [Google Scholar] [CrossRef] [PubMed]
- Nunn, A.J.; Phillips, P.P.; Meredith, S.K.; Chiang, C.-Y.; Conradie, F.; Dalai, D.; Van Deun, A.; Dat, P.-T.; Langley, I.; Master, I.; et al. A Trial of a Shorter Regimen for Rifampin-Resistant Tuberculosis. N. Engl. J. Med. 2019, 380, 1201–1213. [Google Scholar] [CrossRef] [PubMed]
- Van Zyl, L.; Viljoen, J.M.; Haynes, R.K.; Aucamp, M.; Ngwane, A.H.; Du Plessis, J. Topical Delivery of Artemisone, Clofazimine and Decoquinate Encapsulated in Vesicles and Their In vitro Efficacy against Mycobacterium tuberculosis. AAPS PharmSciTech. 2019, 20, 33. [Google Scholar] [CrossRef] [PubMed]
- Van Zyl, L.; Du Plessis, J.; Viljoen, J. Cutaneous tuberculosis overview and current treatment regimens. Tuberculosis 2015, 95, 629–638. [Google Scholar] [CrossRef]
- Bellefroid, C.; Lechanteur, A.; Evrard, B.; Piel, G. Lipid gene nanocarriers for the treatment of skin diseases: Current state-of-the-art. Eur. J. Pharm. Biopharm. 2019, 137, 95–111. [Google Scholar] [CrossRef]
- Chen, Q.; Chen, W.; Hao, F. Cutaneous tuberculosis: A great imitator. Clin. Dermatol. 2019, 37, 192–199. [Google Scholar] [CrossRef]
- Rao, M.; Valentini, D.; Zumla, A.; Maeurer, M. Evaluation of the efficacy of valproic acid and suberoylanilide hydroxamic acid (vorinostat) in enhancing the effects of first-line tuberculosis drugs against intracellular Mycobacterium tuberculosis. Int. J. Infect. Dis. 2018, 69, 78–84. [Google Scholar] [CrossRef] [Green Version]
- Li, G.-L.; Xu, Z.; Jiang, Y.; Liu, H.-C.; Zhao, L.-L.; Li, M.-C.; Xu, D.; Zhao, X.-Q.; Liu, Z.-G.; Wang, R.; et al. Synergistic activities of clofazimine with moxifloxacin or capreomycin against Mycobacterium tuberculosis in China. Int. J. Antimicrob. Agents 2019, 54, 642–646. [Google Scholar] [CrossRef]
- Jagannath, C.; Reddy, M.V.; Kailasam, S.; O’Sullivan, J.F.; Gangadharam, P.R. Chemotherapeutic activity of clofazimine and its analogues against Mycobacterium tuberculosis. In vitro, intracellular, and in vivo studies. Am. J. Respir. Crit. Care Med. 1995, 151, 1083–1086. [Google Scholar] [CrossRef]
- Dey, T.; Brigden, G.; Cox, H.; Shubber, Z.; Cooke, G.; Ford, N. Outcomes of clofazimine for the treatment of drug-resistant tuberculosis: a systematic review and meta-analysis. J. Antimicrob. Chemother. 2012, 68, 284–293. [Google Scholar] [CrossRef] [Green Version]
- Mirnejad, R.; Asadi, A.; Khoshnood, S.; Mirzaei, H.; Heidary, M.; Fattorini, L.; Ghodousi, A.; Darban-Sarokhalil, D. Clofazimine: A useful antibiotic for drug-resistant tuberculosis. Biomed. Pharmacother. 2018, 105, 1353–1359. [Google Scholar] [CrossRef] [PubMed]
- PubChem. Clofazimine (Compound). Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Clofazimine#section=Solubility (accessed on 17 May 2020).
- Singh, B.; Beg, S.; Khurana, R.K.; Sandhu, P.S.; Kaur, R.; Katare, O.P. Recent advances in self-emulsifying drug delivery systems (SEDDS). Crit. Rev. Ther. Drug Carr. Syst. 2014, 31, 121–185. [Google Scholar] [CrossRef] [PubMed]
- Pouton, C.W. Lipid formulations for oral administration of drugs: Non-emulsifying, self-emulsifying and ‘self-microemulsifying’ drug delivery systems. Eur. J. Pharm. Sci. 2000, 11, S93–S98. [Google Scholar] [CrossRef]
- Pouton, C.W.; Porter, C.J. Formulation of lipid-based delivery systems for oral administration: Materials, methods and strategies. Adv. Drug Deliv. Rev. 2008, 60, 625–637. [Google Scholar] [CrossRef] [PubMed]
- Pouton, C.W. Formulation of poorly water-soluble drugs for oral administration: Physicochemical and physiological issues and the lipid formulation classification system. Eur. J. Pharm. Sci. 2006, 29, 278–287. [Google Scholar] [CrossRef] [PubMed]
- Leichner, C.; Baus, R.A.; Jelkmann, M.; Plautz, M.; Barthelmes, J.; Dünnhaupt, S.; Bernkop-Schnürch, A. In vitro evaluation of a self-emulsifying drug delivery system (SEDDS) for nasal administration of dimenhydrinate. Drug Deliv. Transl. Res. 2019, 9, 945–955. [Google Scholar] [CrossRef] [Green Version]
- Kauss, T.; Gaubert, A.; Tabaran, L.; Tonelli, G.; Phoeung, T.; Langlois, M.-H.; White, N.; Cartwright, A.; Gomes, M.; Gaudin, K. Development of rectal self-emulsifying suspension of a moisture-labile water-soluble drug. Int. J. Pharm. 2018, 536, 283–291. [Google Scholar] [CrossRef]
- Rohrer, J.; Zupančič, O.; Hetényi, G.; Kurpiers, M.; Bernkop-Schnürch, A. Design and evaluation of SEDDS exhibiting high emulsifying properties. J. Drug Deliv. Sci. Technol. 2018, 44, 366–372. [Google Scholar] [CrossRef]
- Köllner, S.; Nardin, I.; Markt, R.; Griesser, J.; Prüfert, F.; Bernkop-Schnürch, A. Self-emulsifying drug delivery systems: Design of a novel vaginal delivery system for curcumin. Eur. J. Pharm. Biopharm. 2017, 115, 268–275. [Google Scholar] [CrossRef]
- Pattewar, S.V.; Kasture, S.B.; Pande, V.V.; Sharma, S. A New Self Microemulsifying Mouth Dissolving Film. Indian J. Pharm. Educ. Res. 2016, 50, 191–199. [Google Scholar] [CrossRef] [Green Version]
- Elkasabgy, N.A. Ocular supersaturated self-nanoemulsifying drug delivery systems (S-SNEDDS) to enhance econazole nitrate bioavailability. Int. J. Pharm. 2014, 460, 33–44. [Google Scholar] [CrossRef] [PubMed]
- Xiao, L.; Yi, T.; Liu, Y. A new self-microemulsifying mouth dissolving film to improve the oral bioavailability of poorly water soluble drugs. Drug Dev. Ind. Pharm. 2012, 39, 1284–1290. [Google Scholar] [CrossRef] [PubMed]
- Abdulkarim, M.; Sharma, P.K.; Gumbleton, M. Self-emulsifying drug delivery system: Mucus permeation and innovative quantification technologies. Adv. Drug Deliv. Rev. 2019, 142, 62–74. [Google Scholar] [CrossRef] [PubMed]
- El Khayat, N.W.; Donia, A.A.; Mady, O.Y.; El Maghraby, G.M. Optimization of eugenol microemulsion for transdermal delivery of indomethacin. J. Drug Deliv. Sci. Technol. 2018, 48, 311–318. [Google Scholar] [CrossRef]
- Vaughn, A.R.; Clark, A.; Sivamani, R.K.; Shi, V. Natural Oils for Skin-Barrier Repair: Ancient Compounds Now Backed by Modern Science. Am. J. Clin. Dermatol. 2017, 19, 103–117. [Google Scholar] [CrossRef] [PubMed]
- Van Zyl, L.; Du Preez, J.; Gerber, M.; Du Plessis, J.; Viljoen, J. Essential Fatty Acids as Transdermal Penetration Enhancers. J. Pharm. Sci. 2016, 105, 188–193. [Google Scholar] [CrossRef] [PubMed]
- Viljoen, J.; Cowley, A.; Du Preez, J.; Gerber, M.; Du Plessis, J. Penetration enhancing effects of selected natural oils utilized in topical dosage forms. Drug Dev. Ind. Pharm. 2015, 41, 2045–2054. [Google Scholar] [CrossRef]
- Lane, M.E. Skin penetration enhancers. Int. J. Pharm. 2013, 447, 12–21. [Google Scholar] [CrossRef]
- Rani, S.; Rana, R.; Saraogi, G.K.; Kumar, V.; Gupta, U. Self-Emulsifying Oral Lipid Drug Delivery Systems: Advances and Challenges. AAPS PharmSciTech 2019, 20, 129. [Google Scholar] [CrossRef]
- Haque, T.; Talukder, M.U. Chemical Enhancer: A Simplistic Way to Modulate Barrier Function of the Stratum Corneum. Adv. Pharm. Bull. 2018, 8, 169–179. [Google Scholar] [CrossRef] [Green Version]
- Jones, J.; White, K.; Delgado-Charro, M.B. A mechanistic approach to modelling the formation of a drug reservoir in the skin. Math. Biosci. 2016, 281, 36–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ita, K. Transdermal drug delivery: Progress and challenges. J. Drug Deliv. Sci. Technol. 2014, 24, 245–250. [Google Scholar] [CrossRef]
- Cicero, N.; Albergamo, A.; Salvo, A.; Bua, G.D.; Bartolomeo, G.; Mangano, V.; Rotondo, A.; Di Stefano, V.; Di Bella, G.; Dugo, G. Chemical characterization of a variety of cold-pressed gourmet oils available on the Brazilian market. Food Res. Int. 2018, 109, 517–525. [Google Scholar] [CrossRef] [PubMed]
- El Kharrassi, Y.; Maata, N.; Mazri, M.A.; El Kamouni, S.; Talbi, M.; El Kebbaj, R.; Moustaid, K.; Essamadi, A.K.; Andreoletti, P.; El Mzouri, E.H.; et al. Chemical and phytochemical characterizations of argan oil (Argania spinosa L. skeels), olive oil (Olea europaea L. cv. Moroccan picholine), cactus pear (Opuntia megacantha salm-dyck) seed oil and cactus cladode essential oil. J. Food Meas. Charact. 2017, 12, 747–754. [Google Scholar] [CrossRef]
- Rueda, A.; Seiquer, I.; Olalla-Herrera, M.; Gimenez, R.; Lara, L.; Cabrera, C. Characterization of Fatty Acid Profile of Argan Oil and Other Edible Vegetable Oils by Gas Chromatography and Discriminant Analysis. J. Chem. 2014, 2014, 1–8. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, L.; Zhang, M.; Pang, Y.; Li, Z.; Zhao, A.; Feng, J. Self-emulsifying drug delivery system and the applications in herbal drugs. Drug Deliv. 2013, 22, 475–486. [Google Scholar] [CrossRef] [Green Version]
- Lemery, E.; Briançon, S.; Chevalier, Y.; Bordes, C.; Oddos, T.; Gohier, A.; Bolzinger, M.-A. Skin toxicity of surfactants: Structure/toxicity relationships. Colloids Surfaces A Physicochem. Eng. Asp. 2015, 469, 166–179. [Google Scholar] [CrossRef]
- Ghanbarzadeh, S.; Khorrami, A.; Arami, S. Nonionic surfactant-based vesicular system for transdermal drug delivery. Drug Deliv. 2014, 22, 1071–1077. [Google Scholar] [CrossRef] [Green Version]
- Prajapat, M.D.; Patel, N.J.; Bariya, A.; Patel, S.S.; Butani, S.B. Formulation and evaluation of self-emulsifying drug delivery system for nimodipine, a BCS class II drug. J. Drug Deliv. Sci. Technol. 2017, 39, 59–68. [Google Scholar] [CrossRef]
- Balata, G.F.; A Essa, E.; A Shamardl, H.; Zaidan, S.H.; Abourehab, M.A. Self-emulsifying drug delivery systems as a tool to improve solubility and bioavailability of resveratrol. Drug Des. Dev. Ther. 2016, 10, 117–128. [Google Scholar] [CrossRef] [Green Version]
- Ke, Z.; Hou, X.; Jia, X.-B. Design and optimization of self-nanoemulsifying drug delivery systems for improved bioavailability of cyclovirobuxine D. Drug Des. Dev. Ther. 2016, 10, 2049–2060. [Google Scholar] [CrossRef] [Green Version]
- Czajkowska-Kośnik, A.; Szekalska, M.; Amelian, A.; Szymańska, E.; Winnicka, K. Development and Evaluation of Liquid and Solid Self-Emulsifying Drug Delivery Systems for Atorvastatin. Molecules 2015, 20, 21010–21022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Syed, H.K.; Peh, K.K. Identification of phases of various oil, surfactant/ co-surfactants and water system by ternary phase diagram. Acta Pol. Pharm. Drug Res. 2014, 71, 301–309. [Google Scholar]
- Kang, B.K.; Lee, J.S.; Chon, S.K.; Jeong, S.Y.; Yuk, S.H.; Khang, G.; Lee, H.B.; Cho, S.H. Development of self-microemulsifying drug delivery systems (SMEDDS) for oral bioavailability enhancement of simvastatin in beagle dogs. Int. J. Pharm. 2004, 274, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Solans, C.; Morales, D.; Homs, M. Spontaneous emulsification. Curr. Opin. Colloid Interface Sci. 2016, 22, 88–93. [Google Scholar] [CrossRef]
- Sanka, K.; Suda, D.; Bakshi, V. Optimization of solid-self nanoemulsifying drug delivery system for solubility and release profile of clonazepam using simplex lattice design. J. Drug Deliv. Sci. Technol. 2016, 33, 114–124. [Google Scholar] [CrossRef]
- Parmar, K.; Patel, J.; Sheth, N. Self nano-emulsifying drug delivery system for Embelin: Design, characterization and in-vitro studies. Asian J. Pharm. Sci. 2015, 10, 396–404. [Google Scholar] [CrossRef] [Green Version]
- Sheshala, R.; Anuar, N.K.; Abu Samah, N.H.; Wong, T.W. In Vitro Drug Dissolution/Permeation Testing of Nanocarriers for Skin Application: a Comprehensive Review. AAPS PharmSciTech 2019, 20, 164. [Google Scholar] [CrossRef]
- Heylings, J.R.; Davies, D.J.; Burton, R. Dermal absorption of testosterone in human and pig skin in vitro. Toxicol. Vitr. 2018, 48, 71–77. [Google Scholar] [CrossRef]
- Bharadwaj, R.; Haloi, J.; Medhi, S. Topical delivery of methanolic root extract of Annona reticulata against skin cancer. S. Afr. J. Bot. 2019, 124, 484–493. [Google Scholar] [CrossRef]
- Clausen, M.-L.; Slotved, H.-C.; Krogfelt, K.; Agner, T. Tape Stripping Technique for Stratum Corneum Protein Analysis. Sci. Rep. 2016, 6, 19918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zupančič, O.; Rohrer, J.; Lam, H.T.; Grießinger, J.A.; Bernkop-Schnürch, A. Development and in vitro characterization of self-emulsifying drug delivery system (SEDDS) for oral opioid peptide delivery. Drug Dev. Ind. Pharm. 2017, 43, 1694–1702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suvarna, V. Development and characterization of solid self-emulsifying drug delivery system containing nateglinide. Asian J. Pharm. Sci. 2017, 11, 27–36. [Google Scholar] [CrossRef]
- Trombino, S.; Russo, R.; Mellace, S.; Varano, G.P.; Laganà, A.S.; Marcucci, F.; Cassano, R. Solid lipid nanoparticles made of trehalose monooleate for cyclosporin-A topic release. J. Drug Deliv. Sci. Technol. 2019, 49, 563–569. [Google Scholar] [CrossRef]
- Esposito, E.; Carducci, F.; Mariani, P.; Huang, N.; Simelière, F.; Cortesi, R.; Romeo, G.; Puglia, C. Monoolein liquid crystalline phases for topical delivery of crocetin. Colloids Surfaces B Biointerfaces 2018, 171, 67–74. [Google Scholar] [CrossRef]
- Ibrahim, T.; Abdallah, M.H.; El-Megrab, N.A.; Elnahas, H. Upgrading of dissolution and anti-hypertensive effect of Carvedilol via two combined approaches: self-emulsification and liquisolid techniques. Drug Dev. Ind. Pharm. 2017, 44, 873–885. [Google Scholar] [CrossRef]
- Mojeiko, G.; De Brito, M.; Salata, G.; Lopes, L.B. Combination of microneedles and microemulsions to increase celecoxib topical delivery for potential application in chemoprevention of breast cancer. Int. J. Pharm. 2019, 560, 365–376. [Google Scholar] [CrossRef]
- Kiselmann, C.; Dobler, D.; Schmidts, T.; Eicher, A.; Möbs, C.; Pfützner, W.; Runkel, F. Development of a skin-friendly microemulsion for dermal allergen-specific immunotherapy. Int. J. Pharm. 2018, 550, 463–469. [Google Scholar] [CrossRef]
- Hegde, R.R.; Verma, A.; Ghosh, A. Microemulsion: New Insights into the Ocular Drug Delivery. ISRN Pharm. 2013, 2013. [Google Scholar] [CrossRef]
- Abdel-Messih, H.A.; Ishak, R.A.H.; Geneidi, A.S.; Mansour, S. Tailoring novel soft nano-vesicles ‘Flexosomes’ for enhanced transdermal drug delivery: Optimization, characterization and comprehensive ex vivo–in vivo evaluation. Int. J. Pharm. 2019, 560, 101–115. [Google Scholar] [CrossRef]
- El Zaafarany, G.M.; Awad, G.A.; Holayel, S.M.; Mortada, N. Role of edge activators and surface charge in developing ultradeformable vesicles with enhanced skin delivery. Int. J. Pharm. 2010, 397, 164–172. [Google Scholar] [CrossRef] [PubMed]
- Ujilestari, T.; Dono, N.D.; Ariyadi, B.; Martien, R.; Zuprizal, Z. Formulation and characterization of self-nano emulsifying drug delivery systems of lemongrass (cymbopogon citratus) essential oil. Malays. J. Fundam. Appl. Sci. 2018, 14, 360–363. [Google Scholar] [CrossRef] [Green Version]
- Mahapatra, A.K.; Murthy, P.N.; Swadeep, B.; Swain, R.P. Self-emulsifying drug delivery systems (SEDDS): An update from formulation development to therapeutic strategies. Int. J. PharmTech Res. 2014, 6, 546–568. [Google Scholar]
- Zaichik, S.; Steinbring, C.; Menzel, C.; Knabl, L.; Orth-Höller, D.; Ellemunter, H.; Niedermayr, K.; Bernkop-Schnurch, A. Development of self-emulsifying drug delivery systems (SEDDS) for ciprofloxacin with improved mucus permeating properties. Int. J. Pharm. 2018, 547, 282–290. [Google Scholar] [CrossRef]
- Kaur, R.; Ajitha, M. Transdermal delivery of fluvastatin loaded nanoemulsion gel: Preparation, characterization and in vivo anti-osteoporosis activity. Eur. J. Pharm. Sci. 2019, 136, 104956. [Google Scholar] [CrossRef]
- Danaei, M.; Dehghankhold, M.; Ataei, S.; Davarani, F.H.; Javanmard, R.; Dokhani, A.; Khorasani, S.; Mozafari, M.R. Impact of Particle Size and Polydispersity Index on the Clinical Applications of Lipidic Nanocarrier Systems. Pharm. 2018, 10, 57. [Google Scholar] [CrossRef] [Green Version]
- Sharma, S.; Shukla, P.; Misra, A.; Mishra, P.R. Interfacial and Colloidal Properties of Emulsified Systems: Pharmaceutical and Biological Perspective, 1st ed.; Oshima, H., Makino, K., Eds.; Elsevier: Amsterdam, The Netherlands, 2014; pp. 149–172. [Google Scholar]
- Gupta, V.; Trivedi, P. Lipid Nano Carriers for Drug Targeted Therapy, 1st ed.; Grumezescu, A.M., Ed.; William Andrew Applied Science Publishers: Kidlington, UK, 2018; pp. 563–627. [Google Scholar]
- Zafeiri, I.; Norton, J.E.; Smith, P.; Norton, I.T.; Spyropoulos, F. The role of surface active species in the fabrication and functionality of edible solid lipid particles. J. Colloid Interface Sci. 2017, 500, 228–240. [Google Scholar] [CrossRef]
- Umerska, A.; Cassisa, V.; Matougui, N.; Joly-Guillou, M.L.; Eveillard, M.; Saulnier, P. Antibacterial action of lipid nanocapsules containing fatty acids or monoglycerides as co-surfactants. Eur. J. Pharm. Biopharm. 2016, 108, 100–110. [Google Scholar] [CrossRef]
- Carter, P.; Narasimhan, B.; Wang, Q. Biocompatible nanoparticles and vesicular systems in transdermal drug delivery for various skin diseases. Int. J. Pharm. 2019, 555, 49–62. [Google Scholar] [CrossRef]
- Gumustas, M.; Sengel-Turk, C.T.; Gumustas, A.; Ozkan, S.A.; Uslu, B. Multifunctional Systems for Combined Delivery, Biosensing and Diagnostics, 1st ed.; Grumezescu, A.M., Ed.; Elsevier: Amsterdam, The Netherlands, 2017; pp. 67–108. [Google Scholar]
- Salvo, P.; Pingitore, A.; Barbini, A.; Di Francesco, F. A wearable sweat rate sensor to monitor the athletes’ performance during training. Sci. Sports 2018, 33, e51–e58. [Google Scholar] [CrossRef]
- Williams, H.D.; Sassene, P.; Kleberg, K.; Bakala-N’Goma, J.-C.; Calderone, M.; Jannin, V.; Igonin, A.; Partheil, A.; Marchaud, D.; Jule, E.; et al. Toward the Establishment of Standardized In Vitro Tests for Lipid-Based Formulations, Part 1: Method Parameterization and Comparison of In Vitro Digestion Profiles Across a Range of Representative Formulations. J. Pharm. Sci. 2012, 101, 3360–3380. [Google Scholar] [CrossRef] [PubMed]
- Chaudhari, K.S.; Akamanchi, K.G. Novel bicephalous heterolipid based self-microemulsifying drug delivery system for solubility and bioavailability enhancement of efavirenz. Int. J. Pharm. 2019, 560, 205–218. [Google Scholar] [CrossRef] [PubMed]
- Vasconcelos, T.; Araújo, F.; Lopes, C.; Loureiro, A.; Das Neves, J.; Marques, S.; Sarmento, B. Multicomponent self nano emulsifying delivery systems of resveratrol with enhanced pharmacokinetics profile. Eur. J. Pharm. Sci. 2019, 137, 105011. [Google Scholar] [CrossRef] [PubMed]
- Nasr, A.M.; Gardouh, A.R.M.; Ghorab, M. Novel Solid Self-Nanoemulsifying Drug Delivery System (S-SNEDDS) for Oral Delivery of Olmesartan Medoxomil: Design, Formulation, Pharmacokinetic and Bioavailability Evaluation. Pharmaceutics 2016, 8, 20. [Google Scholar] [CrossRef]
- Parhi, R.; Swain, S. Transdermal Evaporation Drug Delivery System: Concept to Commercial Products. Adv. Pharm. Bull. 2018, 8, 535–550. [Google Scholar] [CrossRef]
- Kathe, K.; Kathpalia, H. Film forming systems for topical and transdermal drug delivery. Asian J. Pharm. Sci. 2017, 12, 487–497. [Google Scholar] [CrossRef]
- Keurentjes, A.J.; Maibach, H.I. Percutaneous penetration of drugs applied in transdermal delivery systems: an in vivo based approach for evaluating computer generated penetration models. Regul. Toxicol. Pharmacol. 2019, 108, 104428. [Google Scholar] [CrossRef]
- Shahnaz, G.; Hartl, M.; Barthelmes, J.; Leithner, K.; Sarti, F.; Hintzen, F.; Rahmat, D.; Salvenmoser, W.; Bernkop-Schnürch, A. Uptake of phenothiazines by the harvested chylomicrons ex vivo model: Influence of self-nanoemulsifying formulation design. Eur. J. Pharm. Biopharm. 2011, 79, 171–180. [Google Scholar] [CrossRef]
- A Elmarzugi, N.; Eid, A.M.; A El-Enshasy, H.; Aziz, R. The preparation and evaluation of self-nanoemulsifying systems containing Swietenia oil and an examination of its anti-inflammatory effects. Int. J. Nanomed. 2014, 9, 4685–4695. [Google Scholar] [CrossRef] [Green Version]
- Perazzo, A.; Preziosi, V.; Guido, S. Phase inversion emulsification: Current understanding and applications. Adv. Colloid Interface Sci. 2015, 222, 581–599. [Google Scholar] [CrossRef]
- Rastogi, V.; Yadav, P. Transdermal drug delivery system: An overview. Asian J. Pharm. 2012, 6, 161. [Google Scholar] [CrossRef]
- Akula, S.; Gurram, A.K.; Devireddy, S.R. Self-Microemulsifying Drug Delivery Systems: An Attractive Strategy for Enhanced Therapeutic Profile. Int. Sch. Res. Not. 2014, 2014. [Google Scholar] [CrossRef] [PubMed]
- Roumpea, E.; Kovalchuk, N.M.; Chinaud, M.; Nowak, E.; Simmons, M.; Angeli, P. Experimental studies on droplet formation in a flow-focusing microchannel in the presence of surfactants. Chem. Eng. Sci. 2019, 195, 507–518. [Google Scholar] [CrossRef]
- Thomas, N.; Holm, R.; Garmer, M.; Karlsson, J.J.; Müllertz, A.; Rades, T. Supersaturated Self-Nanoemulsifying Drug Delivery Systems (Super-SNEDDS) Enhance the Bioavailability of the Poorly Water-Soluble Drug Simvastatin in Dogs. AAPS J. 2012, 15, 219–227. [Google Scholar] [CrossRef] [Green Version]
- Ganti, S.S.; Nguyen, H.X.; Murnane, K.S.; Blough, B.E.; Banga, A.K. Transdermal formulation of 4-benzylpiperidine for cocaine-use disorder. J. Drug Deliv. Sci. Technol. 2018, 47, 299–308. [Google Scholar] [CrossRef]
- Tshisevhe, V.; Mbelle, N.M.; Peters, R.P. Cutaneous tuberculosis in HIV-infected individuals: Lessons learnt from a case series. South. Afr. J. HIV Med. 2019, 20, 3. [Google Scholar] [CrossRef]
- Čižinauskas, V.; Elie, N.; Brunelle, A.; Briedis, V. Skin Penetration Enhancement by Natural Oils for Dihydroquercetin Delivery. Molecules 2017, 22, 1536. [Google Scholar] [CrossRef] [Green Version]
- Balázs, B.; Vizserálek, G.; Berkó, S.; Budai-Szűcs, M.; Kelemen, A.; Sinkó, B.; Takács-Novák, K.; Szabó-Révész, P.; Csányi, E. Investigation of the Efficacy of Transdermal Penetration Enhancers Through the Use of Human Skin and a Skin Mimic Artificial Membrane. J. Pharm. Sci. 2016, 105, 1134–1140. [Google Scholar] [CrossRef]
- Elmasry, S.R.; Hathout, R.M.; Abdel-Halim, M.; Mansour, S. In Vitro transdermal delivery of sesamol using oleic acid chemically-modified gelatin nanoparticles as a potential breast cancer medication. J. Drug Deliv. Sci. Technol. 2018, 48, 30–39. [Google Scholar] [CrossRef]
- Hadgraft, J.; Lane, M.E. Advanced topical formulations (ATF). Int. J. Pharm. 2016, 514, 52–57. [Google Scholar] [CrossRef] [Green Version]
- Watson, R.; Preedy, V. (Eds.) Omega Fatty Acids in brain and Neurological Health, 2nd ed.; Academic Press: London, UK, 2019. [Google Scholar]
- Jiménez-Peñalver, P.; Castillejos, M.; Koh, A.; Gross, R.; Sánchez, A.; Font, X.; Gea, T. Production and characterization of sophorolipids from stearic acid by solid-state fermentation, a cleaner alternative to chemical surfactants. J. Clean. Prod. 2018, 172, 2735–2747. [Google Scholar] [CrossRef] [Green Version]
- Gore, E.; Picard, C.; Savary, G. Spreading behavior of cosmetic emulsions: Impact of the oil phase. Biotribology 2018, 16, 17–24. [Google Scholar] [CrossRef]
- Vaz, S.; Silva, R.; Amaral, M.; Martins, E.; Lobo, J.M.S.; Silva, A. Evaluation of the biocompatibility and skin hydration potential of vitamin E-loaded lipid nanosystems formulations: In vitro and human in vivo studies. Colloids Surfaces B Biointerfaces 2019, 179, 242–249. [Google Scholar] [CrossRef] [PubMed]
- Warner, R.R.; Stone, K.J.; Boissy, Y.L. Hydration Disrupts Human Stratum Corneum Ultrastructure. J. Investig. Dermatol. 2003, 120, 275–284. [Google Scholar] [CrossRef] [Green Version]
- Mansour, R.S.; Sallam, A.A.; Hamdan, I.I.; Khalil, E.; Yousef, I. Elucidation of penetration enhancement mechanism of Emu oil using FTIR microspectroscopy at EMIRA laboratory of SESAME synchrotron. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2017, 185, 1–10. [Google Scholar] [CrossRef]
- Lundborg, M.; Wennberg, C.L.; Narangifard, A.; Lindahl, E.; Norlén, L. Predicting drug permeability through skin using molecular dynamics simulation. J. Control. Release 2018, 283, 269–279. [Google Scholar] [CrossRef]
- Kim, M.-J.; Doh, H.-J.; Choi, M.-K.; Chung, S.-J.; Shim, C.-K.; Kim, D.-D.; Kim, J.S.; Yong, C.S.; Choi, H.-G. Skin Permeation Enhancement of Diclofenac by Fatty Acids. Drug Deliv. 2008, 15, 373–379. [Google Scholar] [CrossRef]
- Cholo, M.C.; Mothiba, M.T.; Fourie, B.; Anderson, R. Mechanisms of action and therapeutic efficacies of the lipophilic antimycobacterial agents clofazimine and bedaquiline. J. Antimicrob. Chemother. 2016, 72, 338–353. [Google Scholar] [CrossRef] [Green Version]
- Cholo, M.C.; Steel, H.C.; Fourie, P.B.; Germishuizen, W.A.; Anderson, R. Clofazimine: Current status and future prospects. J. Antimicrob. Chemother. 2011, 67, 290–298. [Google Scholar] [CrossRef]
- Yano, T.; Kassovska-Bratinova, S.; Teh, J.S.; Winkler, J.; Sullivan, K.; Isaacs, A.; Schechter, N.M.; Rubin, H. Reduction of clofazimine by mycobacterial type 2 NADH: Quinone oxidoreductase a pathway for the generation of bactericidal levels of reactive oxygen species. J. Biol. Chem. 2011, 286, 10276–10287. [Google Scholar] [CrossRef] [Green Version]
- Cederbaum, A.I. Liver Pathophysiology, 1st ed.; Academic Press: London, UK, 2017; pp. 401–419. [Google Scholar]
- Patel, P.; Ahir, K.; Patel, V.; Manani, L.; Patel, C. Drug-Excipient compatibility studies: First step for dosage form development. J. Pharm. Innov. 2015, 4, 14–20. [Google Scholar]
- Tiberi, S.; Migliori, G.B.; Chakaya, J.M.; Kaesava, T.; Al Abri, S.S.; Wejse, C.; Goletti, D.; Kapata, N.; Sotgiu, G.; Bomanji, J.; et al. Commemorating World TB Day 2020:“IT’S TIME”—It’s time to End the Global TB Epidemic. Int. J. Infect. Dis. 2020, 92, S1–S4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Floyd, K.; Glaziou, P.; Houben, R.M.G.J.; Sumner, T.; White, R.; Raviglione, M. Global tuberculosis targets and milestones set for 2016–2035: Definition and rationale. Int. J. Tuberc. Lung Dis. 2018, 22, 723–730. [Google Scholar] [CrossRef] [PubMed]
- Khadke, S.; Roces, C.B.; Cameron, A.; Devitt, A.; Perrie, Y. Formulation and manufacturing of lymphatic targeting liposomes using microfluidics. J. Control. Release 2019, 307, 211–220. [Google Scholar] [CrossRef] [PubMed]
- Van Staden, D.; Du Plessis, J.; Viljoen, J. Development of Topical/Transdermal Self-Emulsifying Drug Delivery Systems, Not as Simple as Expected. Sci. Pharm. 2020, 88, 17. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Grading | Description |
|---|---|
| Grade A | Swift emulsion formation, demonstrating a clear/bluish appearance (60 s) |
| Grade B | Swift formation of emulsion, which appears bluish (60 s) |
| Grade C | Emulsion displays fine milky appearance (120 s) |
| Grade D | Dull, greyish white appearance with an additional oily appearance together with slow emulsification, (>120 s) |
| Grade E | Poor or minimal emulsification noted with large oil droplets noticed on the surface |
| Natural Oil | Solubility of Clofazimine (mg/mL) |
|---|---|
| Argan oil (ARG) | 2.23 ± 0.54 |
| Avocado oil (AVO) | 6.29 ± 0.44 |
| Coconut oil (CCT) | 1.78 ± 0.67 |
| Macadamia oil (MAC) | 1.25 ± 0.46 |
| Olive oil (OLV) | 1.09 ± 0.65 |
| SEDDS | Droplet Size (nm) | PDI | Zeta-Potential (mV) | Self-Emulsification Grading | Self-Emulsification Time (s) | Viscosity (mPa.s) | pH | Cloud Point (°C) |
|---|---|---|---|---|---|---|---|---|
| ARG1 | 66.24 | 0.39 | −30.60 | D | 499 | 230.53 | 6.95 | 27.00 |
| ARG3 | 107.32 | 0.62 | −29.90 | D | 482 | 9436.27 | 6.00 | 34.00 |
| ARG5 | 440.78 | 1.00 | −23.40 | D | 423 | 1060.14 | 6.60 | 46.90 |
| AVO2 | 64.11 | 0.34 | −32.80 | D | 423 | 4103.20 | 5.01 | 40.00 |
| AVO3 | 345.75 | 1.00 | −40.90 | D | 131 | 710.40 | 3.82 | 45.90 |
| AVO4 | 221.95 | 1.00 | −37.50 | D | 130 | 6971.10 | 5.02 | 44.00 |
| AVO5 | 59.013 | 0.55 | −38.20 | D | 430 | 1178.40 | 5.05 | 50.60 |
| MAC1 | 108.01 | 0.80 | −25.00 | D | 374 | 23,071.00 | 7.40 | 31.90 |
| MAC2 | 108.71 | 0.76 | −29.20 | D | 346 | 17,476.53 | 6.61 | 32.50 |
| MAC3 | 116.68 | 0.73 | −27.70 | D | 181 | 5748.37 | 7.01 | 31.40 |
| MAC5 | 462.32 | 1.00 | −26.00 | B | 59 | 993.60 | 7.02 | 36.00 |
| OLV1 | 360.25 | 1.00 | −21.30 | D | 393 | 2511.03 | 7.37 | 34.80 |
| OLV2 | 222.87 | 1.00 | −30.00 | D | 385 | 6006.17 | 7.13 | 34.80 |
| OLV3 | 502.08 | 1.00 | −31.00 | D | 274 | 11,740.00 | 7.13 | 33.80 |
| OLV5 | 154.80 | 0.80 | −23.60 | C | 90 | 7220.00 | 6.97 | 35.90 |
| Topical SEDDS Formulation | Average % Released | Average Cumulative Concentration (μg/mL) | Median of Cumulative Concentration (μg/mL) | Average % Released |
|---|---|---|---|---|
| ARG3 | 0.5 ± 0.002 | 0.729 ± 0.030 | 0.726 | 0.5 ± 0.002 |
| AVO2 | 2.5 ± 0.007 | 1.685 ± 0.161 | 1.707 | 2.5 ± 0.007 |
| MAC2 | 2 ± 0.002 | 0.586 ± 0.038 | 0.571 | 2 ± 0.002 |
| OLV5 | 7 ± 0.002 | 0.459 ± 0.350 | 0.445 | 7 ± 0.002 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
van Staden, D.; du Plessis, J.; Viljoen, J. Development of a Self-Emulsifying Drug Delivery System for Optimized Topical Delivery of Clofazimine. Pharmaceutics 2020, 12, 523. https://doi.org/10.3390/pharmaceutics12060523
van Staden D, du Plessis J, Viljoen J. Development of a Self-Emulsifying Drug Delivery System for Optimized Topical Delivery of Clofazimine. Pharmaceutics. 2020; 12(6):523. https://doi.org/10.3390/pharmaceutics12060523
Chicago/Turabian Stylevan Staden, Daniélle, Jeanetta du Plessis, and Joe Viljoen. 2020. "Development of a Self-Emulsifying Drug Delivery System for Optimized Topical Delivery of Clofazimine" Pharmaceutics 12, no. 6: 523. https://doi.org/10.3390/pharmaceutics12060523