Oral Administration of Artemisone for the Treatment of Schistosomiasis: Formulation Challenges and In Vivo Efficacy

 , ,
, , 
Abstract
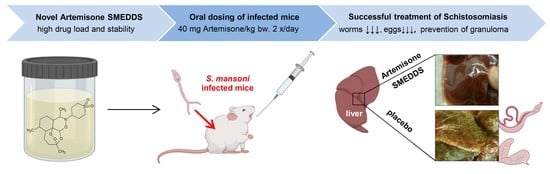
:
1. Introduction
- Chemical and thermodynamic instability due to the use of aqueous dispersions
- Low drug load (the major volume is water)
- Use of deleterious organic solvents
- Difficult to scale-up technologies (ultrasonication)
- Special requirements/high sensitivity to oxidation (for example in Pheroid™ technology)
- High drug load and in vivo efficacy
- Easy manufacturing process without special equipment
- High storage stability (thermodynamic and chemical)
- High potential for translation into clinics
- Pre-screening of drug solubility in different excipients.
- Selection of components by ternary phase diagrams.
- Optimization of the composition based on physicochemical properties.
- Stability studies.
- In vitro activity.
- In vivo activity.
2. Materials and Methods
2.1. Materials
2.2. Methods
2.2.1. Formulation Development
Solubility of Artemisone in Organic Solvents, Excipients and SMEDDS
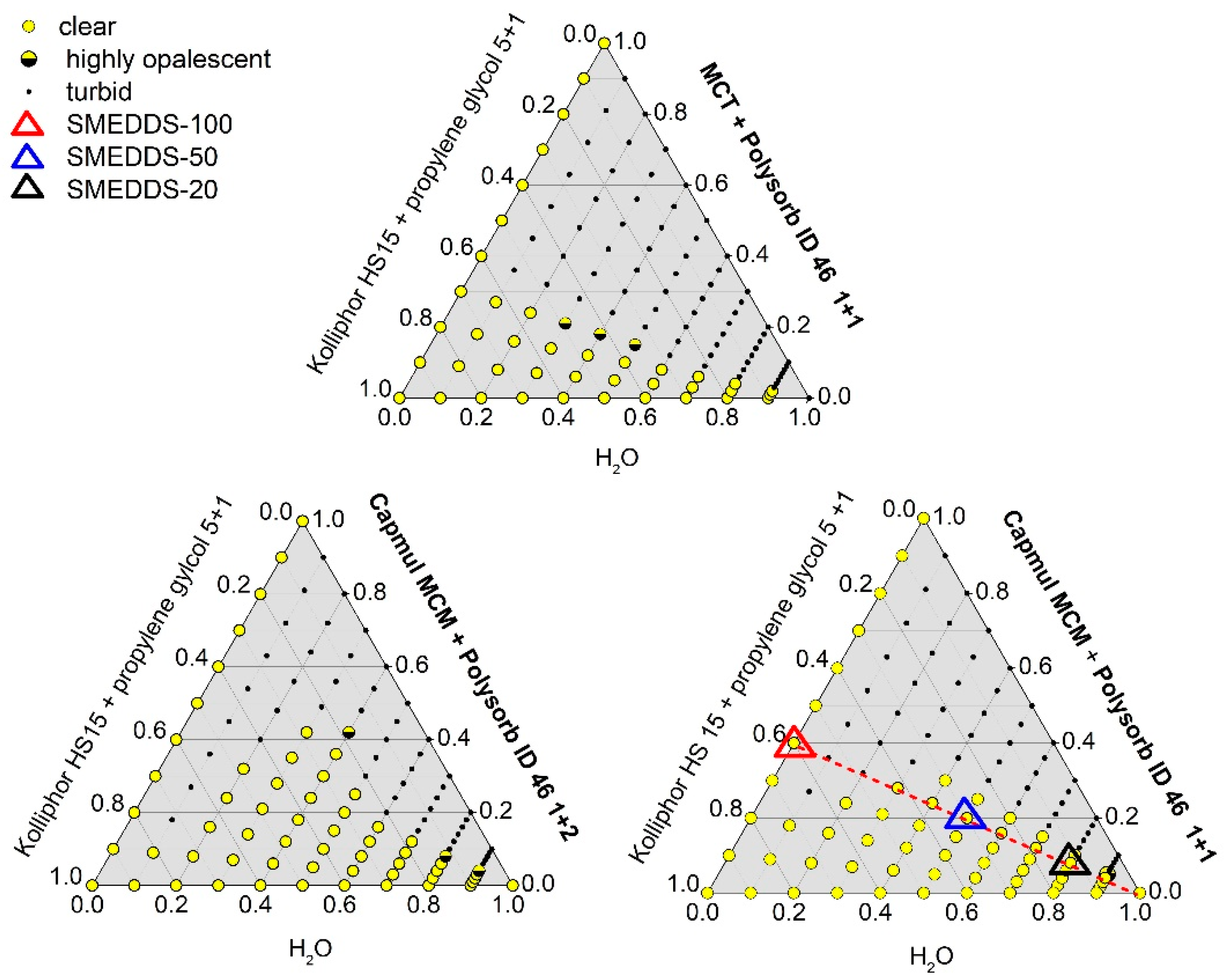
Ternary Phase Diagrams
Formulation Preparation
Assessment of Self-Microemulsifying Potential of Lipid Formulations
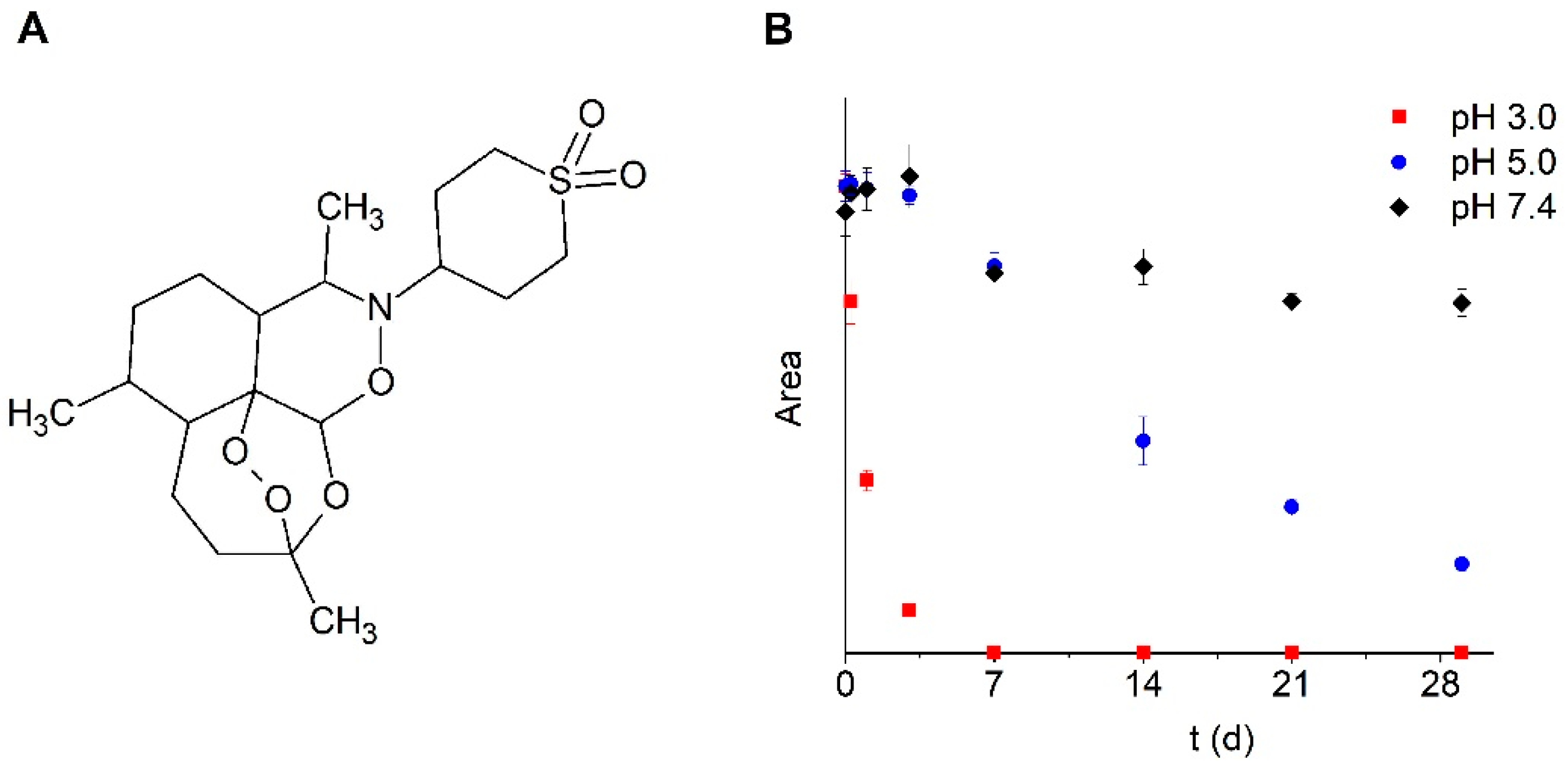
Stability of Artemisone in SMEDDS and PBS
Stability at Different Temperatures and Ionic Strength
Rheology
Dynamic Light Scattering (DLS)
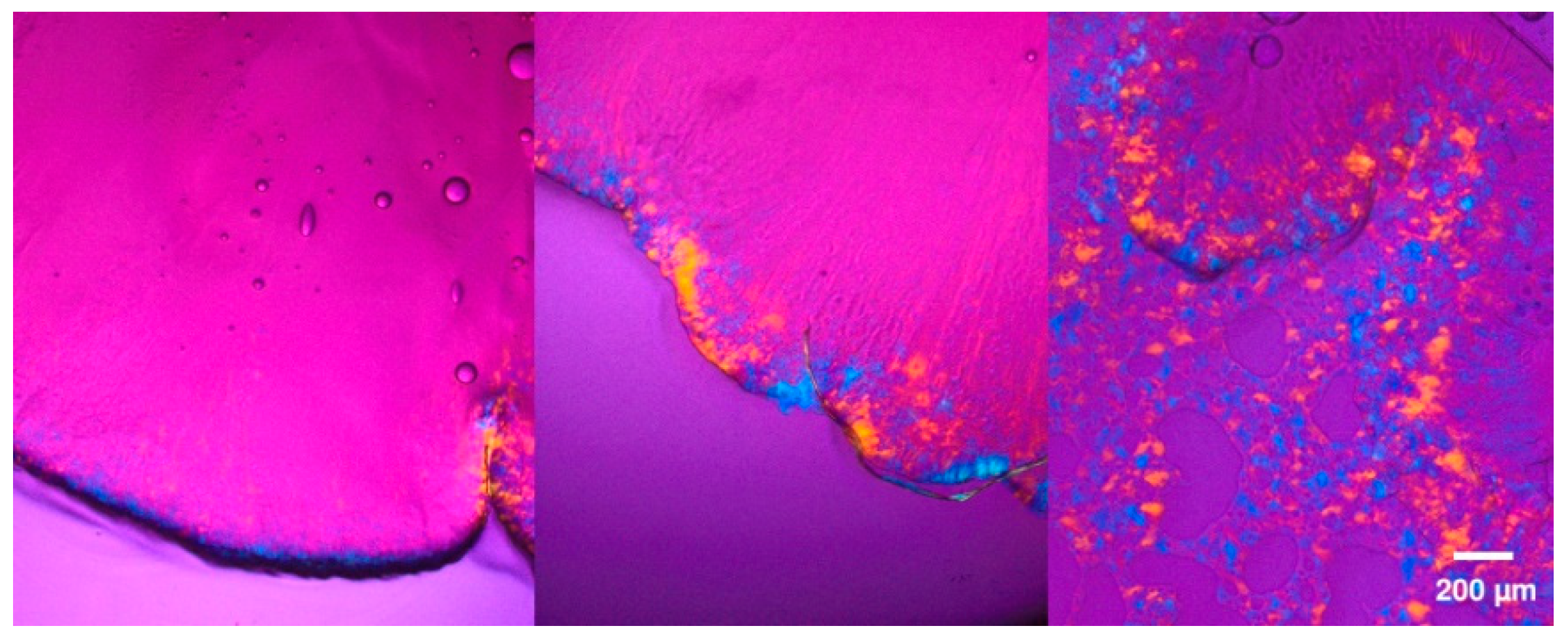
Cryogenic Electron Microscopy
2.2.2. Measurement of Biological Activity in Culture
Caenorhabditis elegans
Plasmodium falciparum
2.2.3. Measurement of Biological Activity in Vivo
S. mansoni
Mice
Treatment of Schistosoma Infected Mice
Assessments of Treatments
3. Results
3.1. Formulation Development
3.1.1. Physical Stability
3.1.2. Storage Stability
3.2. Biological Activity
3.2.1. In Vitro Examinations
P. Falciparum
C. elegans
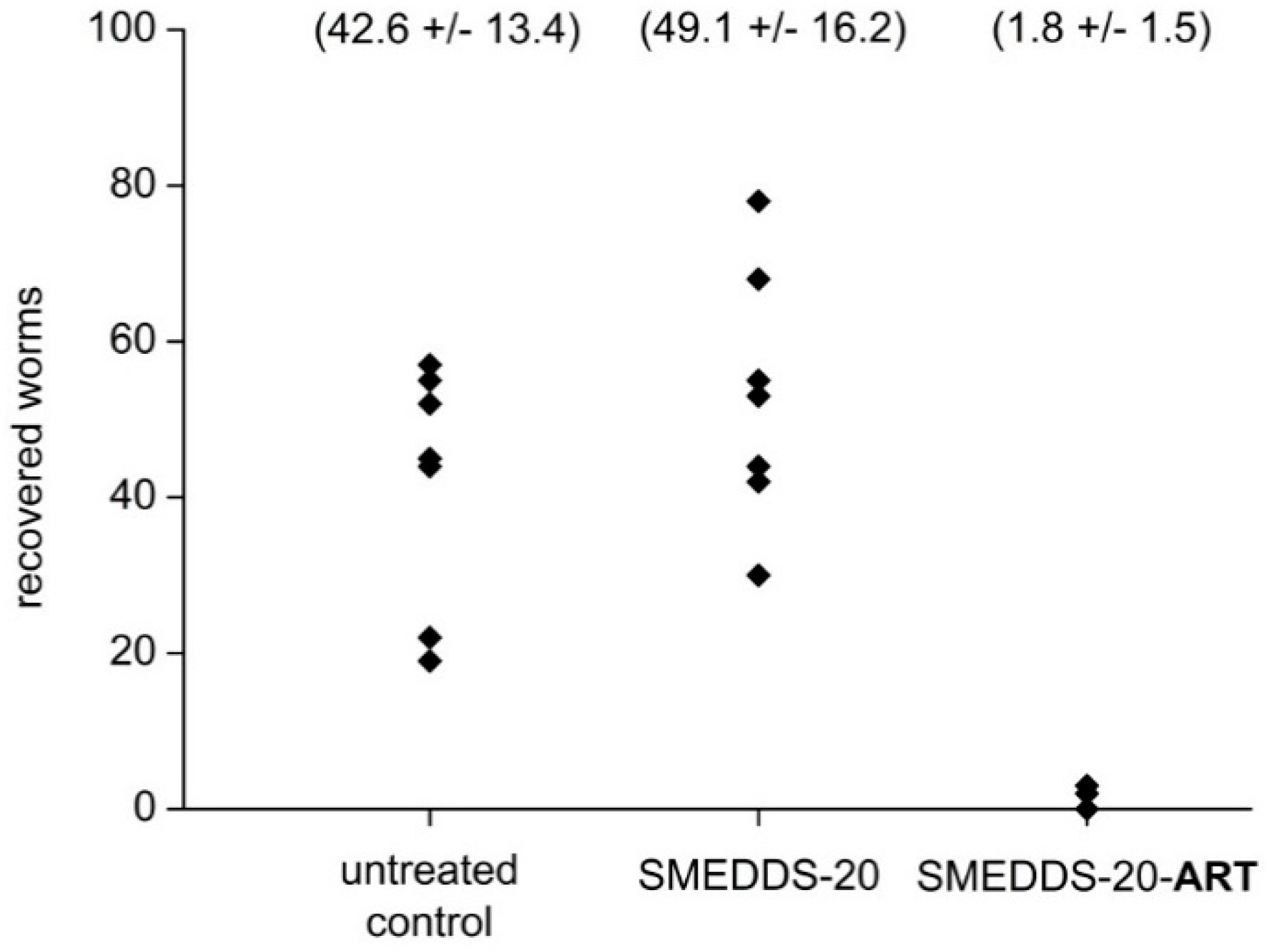
3.2.2. In Vivo Examinations
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ACN | acetonitrile |
| ART | artemisone |
| MCM | medium chain monoglycerides |
| MCT | medium chain triglycerides |
| ME | microemulsion |
| PBS | phosphate-buffered saline |
| SMEDDS | self-microemulsifying drug delivery systems |
References
- McManus, D.P.; Bergquist, R.; Cai, P.; Ranasinghe, S.; Tebeje, B.M.; You, H. Schistosomiasis-from immunopathology to vaccines. Semin. Immunopathol. 2020, 19, 1–17. [Google Scholar]
- Da Silva, V.B.R.; Campos, B.R.K.L.; de Oliveira, J.F.; Decout, J.L.; do Carmo Alves de Lima, M. Medicinal chemistry of antischistosomal drugs: Praziquantel and oxamniquine. Bioorganic Med. Chem. 2017, 25, 3259–3277. [Google Scholar] [CrossRef] [PubMed]
- Thomas, C.M.; Timson, D.J. The Mechanism of Action of Praziquantel: Can New Drugs Exploit Similar Mechanisms? Curr. Med. Chem. 2018, 27, 676–696. [Google Scholar] [CrossRef] [PubMed]
- Gemma, S.; Federico, S.; Brogi, S.; Brindisi, M.; Butini, S.; Campiani, G. Dealing with schistosomiasis: Current drug discovery strategies. In Annual Reports in Medicinal Chemistry; Elsevier: Amsterdam, The Netherland, 2019; Volume 53, pp. 107–138. ISBN 9780128198667. [Google Scholar]
- Gold, D.; Alian, M.; Domb, A.; Karawani, Y.; Jbarien, M.; Chollet, J.; Haynes, R.K.; Wong, H.N.; Buchholz, V.; Greiner, A.; et al. Elimination of Schistosoma mansoni in infected mice by slow release of artemisone. Int. J. Parasitol. Drugs Drug Resist. 2017, 7, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Vale, N.; Gouveia, M.J.; Rinaldi, G.; Brindley, P.J.; Gärtner, F.; Da Costa, J.M.C. Praziquantel for schistosomiasis: Single-drug metabolism revisited, mode of action, and resistance. Antimicrob. Agents Chemother. 2017, 61, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Bergquist, R.; Elmorshedy, H. Artemether and Praziquantel: Origin, Mode of Action, Impact, and Suggested Application for Effective Control of Human Schistosomiasis. Trop. Med. Infect. Dis. 2018, 125, 125. [Google Scholar] [CrossRef] [Green Version]
- Saeed, M.E.M.; Krishna, S.; Greten, H.J.; Kremsner, P.G.; Efferth, T. Antischistosomal activity of artemisinin derivatives in vivo and in patients. Pharmacol. Res. 2016, 110, 216–226. [Google Scholar] [CrossRef]
- Inyang-Etoh, P.C.; Ejezie, G.C.; Useh, M.F.; Inyang-Etoh, E.C. Efficacy of artesunate in the treatment of urinary schistosomiasis, in an endemic community in Nigeria. Ann. Trop. Med. Parasitol. 2004, 98, 491–499. [Google Scholar] [CrossRef]
- Hegazy, L.A.M.; Al Motiam, M.H.; Abd El-Aal, N.F.; Ibrahim, S.M.; Mohamed, H.K. Evaluation of artesunate and praziquantel combination therapy in murine schistosomiasis mansoni. Iran. J. Parasitol. 2018, 13, 193–203. [Google Scholar]
- Pérez del Villar, L.; Burguillo, F.J.; López-Abán, J.; Muro, A. Systematic Review and Meta-Analysis of Artemisinin based therapies for the treatment and prevention of schistosomiasis. PLoS ONE 2012, 7, e45867. [Google Scholar] [CrossRef]
- de Corrêa, S.A.P.; de Oliveira, R.N.; Mendes, T.M.F.; dos Santos, K.R.; Boaventura, S.; Garcia, V.L.; de Jeraldo, V.L.S.; Allegretti, S.M. In vitro and in vivo evaluation of six artemisinin derivatives against Schistosoma mansoni. Parasitol. Res. 2019, 118, 505–516. [Google Scholar]
- Haynes, R.K.; Fugmann, B.; Stetter, J.; Rieckmann, K.; Heilmann, H.D.; Chan, H.W.; Cheung, M.K.; Lam, W.L.; Wong, H.N.; Croft, S.L.; et al. Artemisone-A highly active antimalarial drug of the artemisinin class. Angew. Chem. Int. Ed. 2006, 45, 2082–2088. [Google Scholar] [CrossRef] [PubMed]
- Dwivedi, A.; Mazumder, A.; du Plessis, L.; du Preez, J.L.; Haynes, R.K.; du Plessis, J. In vitro anti-cancer effects of artemisone nano-vesicular formulations on melanoma cells. Nanomed. Nanotechnol. Biol. Med. 2015, 11, 2041–2050. [Google Scholar] [CrossRef]
- Mazuz, M.L.; Shkap, V.; Wollkomirsky, R.; Leibovich, B.; Savitsky, I.; Fleiderovitz, L.; Noam, S.; Elena, B.; Molad, T.; Golenser, J. Neospora caninum: Chronic and congenital infection in consecutive pregnancies of mice. Vet. Parasitol. 2016, 219, 66–70. [Google Scholar] [CrossRef] [PubMed]
- Oiknine-Djian, E.; Bar-On, S.; Laskov, I.; Lantsberg, D.; Haynes, R.K.; Panet, A.; Wolf, D.G. Artemisone demonstrates synergistic antiviral activity in combination with approved and experimental drugs active against human cytomegalovirus. Antivir. Res. 2019, 172, 104639. [Google Scholar] [CrossRef]
- Steyn, J.D.; Wiesner, L.; du Plessis, L.H.; Grobler, A.F.; Smith, P.J.; Chan, W.-C.C.; Haynes, R.K.; Kotzé, A.F. Absorption of the novel artemisinin derivatives artemisone and artemiside: Potential application of PheroidTM technology. Int. J. Pharm. 2011, 414, 260–266. [Google Scholar] [CrossRef]
- Hartwig, C.L.; Rosenthal, A.S.; D’Angelo, J.; Griffin, C.E.; Posner, G.H.; Cooper, R.A. Accumulation of artemisinin trioxane derivatives within neutral lipids of Plasmodium falciparum malaria parasites is endoperoxide-dependent. Biochem. Pharmacol. 2009, 77, 322–336. [Google Scholar] [CrossRef] [Green Version]
- Burger, C.; Aucamp, M.; du Preez, J.; Haynes, R.K.; Ngwane, A.; du Plessis, J.; Gerber, M. Formulation of Natural Oil Nano-Emulsions for the Topical Delivery of Clofazimine, Artemisone and Decoquinate. Pharm. Res. 2018, 35, 186. [Google Scholar] [CrossRef]
- Van Zyl, L.; Viljoen, J.M.; Haynes, R.K.; Aucamp, M.; Ngwane, A.H.; du Plessis, J. Topical Delivery of Artemisone, Clofazimine and Decoquinate Encapsulated in Vesicles and Their In vitro Efficacy Against Mycobacterium tuberculosis. AAPS PharmSciTech 2019, 20, 33. [Google Scholar] [CrossRef]
- Zech, J.; Timoracky, M.; Syrowatka, F.; Mäder, K. Artemisone in Electrosprayed Solid Lipid Microparticles. In Proceedings of the PBP World Meeting, Granada, Spain, 19–22 March 2018; pp. 1–2. [Google Scholar]
- Häusler, O.; Palmieri, J.; Wyart, H. New isosorbide derivates for drug solubilization. Roquette Frères 2016, 8, 2016. [Google Scholar]
- Cui, C.; Zhen, Y.; Qu, J.; Chen, B.; Tan, T. Synthesis of biosafe isosorbide dicaprylate ester plasticizer by lipase in a solvent-free system and its sub-chronic toxicity in mice. RSC Adv. 2016, 6, 11959–11966. [Google Scholar] [CrossRef]
- Isosorbide Diesters-Brief Profile-ECHA. Available online: https://echa.europa.eu/brief-profile/-/briefprofile/100.121.882 (accessed on 1 April 2020).
- International Conference on Harmonisation ICH. Q2(R1): Validation of analytical procedures: Text and methodology. Fed. Regist. 1997, 62, 27463–27467. [Google Scholar]
- Abdalla, A.; Klein, S.; Mäder, K. A new self-emulsifying drug delivery system (SEDDS) for poorly soluble drugs: Characterization, dissolution, in vitro digestion and incorporation into solid pellets. Eur. J. Pharm. Sci. 2008, 35, 457–464. [Google Scholar] [CrossRef] [PubMed]
- BASF. Kolliphor® HS 15. Available online: https://pharmaceutical.basf.com/global/en/drug-formulation/products/kolliphor-hs-15.html (accessed on 31 May 2020).
- Çilek, A.; Çelebi, N.; Tirnaksiz, F.; Tay, A. A lecithin-based microemulsion of rh-insulin with aprotinin for oral administration: Investigation of hypoglycemic effects in non-diabetic and STZ-induced diabetic rats. Int. J. Pharm. 2005, 298, 176–185. [Google Scholar] [CrossRef]
- Lawrence, M.J.; Rees, G.D. Microemulsion-based media as novel drug delivery systems. Adv. Drug Deliv. Rev. 2012, 64, 175–193. [Google Scholar] [CrossRef]
- Bloch, E.H. Inflammation in schistosomiasis. Bibl. Anat. 1979, 17, 105–114. [Google Scholar]
- Costain, A.H.; MacDonald, A.S.; Smits, H.H. Schistosome Egg Migration: Mechanisms, Pathogenesis and Host Immune Responses. Front. Immunol. 2018, 9, 3042. [Google Scholar] [CrossRef] [Green Version]
- Efferth, T.; Kaina, B. Toxicity of the antimalarial artemisinin and its dervatives. Crit. Rev. Toxicol. 2010, 40, 405–421. [Google Scholar] [CrossRef]
- Gordi, T.; Lepist, E.I. Artemisinin derivatives: Toxic for laboratory animals, safe for humans? Toxicol. Lett. 2004, 147, 99–107. [Google Scholar] [CrossRef]
- Guiguemde, W.A.; Hunt, N.H.; Guo, J.; Marciano, A.; Haynes, R.K.; Clark, J.; Guy, R.K.; Golenser, J. Treatment of murine cerebral malaria by artemisone in combination with conventional antimalarial drugs: Antiplasmodial effects and immune responses. Antimicrob. Agents Chemother. 2014, 58, 4745–4754. [Google Scholar] [CrossRef] [Green Version]
- Gomes, C.; Boareto, A.C.; Dalsenter, P.R. Clinical and non-clinical safety of artemisinin derivatives in pregnancy. Reprod. Toxicol. 2016, 65, 194–203. [Google Scholar] [CrossRef] [PubMed]
- Maronpot, R.R. Liver–Inflammation. Available online: https://ntp.niehs.nih.gov/nnl/hepatobiliary/liver/inflamm/index.htm (accessed on 1 April 2020).
- Reimers, N.; Homann, A.; Höschler, B.; Langhans, K.; Wilson, R.A.; Pierrot, C.; Khalife, J.; Grevelding, C.G.; Chalmers, I.W.; Yazdanbakhsh, M.; et al. Drug-Induced Exposure of Schistosoma mansoni Antigens SmCD59a and SmKK7. PLoS Negl. Trop. Dis. 2015, 9, 1–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buchter, V.; Hess, J.; Gasser, G.; Keiser, J. Assessment of tegumental damage to Schistosoma mansoni and S. haematobium after in vitro exposure to ferrocenyl, ruthenocenyl and benzyl derivatives of oxamniquine using scanning electron microscopy. Parasites Vectors 2018, 11, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Sirois, P. Leukotrienes: One step in our understanding of asthma. Respir. Investig. 2019, 57, 97–110. [Google Scholar] [CrossRef]
- McManus, D.P.; Dunne, D.W.; Sacko, M.; Utzinger, J.; Vennervald, B.J.; Zhou, X.-N. Schistosomiasis. Nat. Rev. Dis. Prim. 2018, 4, 13. [Google Scholar] [CrossRef]
- Nair, A.; Jacob, S. A simple practice guide for dose conversion between animals and human. J. Basic Clin. Pharm. 2016, 7, 27. [Google Scholar] [CrossRef] [Green Version]
- Bergquist, R.; Utzinger, J.; Keiser, J. Controlling schistosomiasis with praziquantel: How much longer without a viable alternative? Infect. Dis. Poverty 2017, 6, 1–10. [Google Scholar] [CrossRef] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| t [min] | Mobile Phase V/V/V | Flow Rate | Sample Injection Volume | T Column | UV Detection | ||
|---|---|---|---|---|---|---|---|
| ACN | H2O | Formic Acid | |||||
| 0–10 gradient | 52.5→100 | 47.5→0 | 0.1 | 0.3 mL/min | 10 µL | 35 °C | 200 nm |
| 10–20 isocratic | 100 | 0 | 0.1 | ||||
| 20–30 isocratic | 52.5 | 47.5 | 0.1 | ||||
| Excipient | SMEDDS-100 | SMEDDS-50 | SMEDDS-20 |
|---|---|---|---|
| Kolliphor® HS15 | 50 | 25 | 10 |
| Propylene glycol | 10 | 5 | 2 |
| Polysorb® ID 46 | 20 | 10 | 4 |
| Capmul® MCM | 20 | 10 | 4 |
| PBS | 0 | 50 | 80 |
| Treatment | # Mouse | Eggs/cm2 | Granulomas/cm2 |
|---|---|---|---|
| SMEDDS-20 | 1 | 47 | 41 |
| 2 | 72 | 20 | |
| 3 | 172 | 76 | |
| 4 | 63 | 68 | |
| 5 | 70 | 50 | |
| SMEDDS-20-ART | 1–6 | 0 | 0 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zech, J.; Gold, D.; Salaymeh, N.; Sasson, N.C.; Rabinowitch, I.; Golenser, J.; Mäder, K. Oral Administration of Artemisone for the Treatment of Schistosomiasis: Formulation Challenges and In Vivo Efficacy. Pharmaceutics 2020, 12, 509. https://doi.org/10.3390/pharmaceutics12060509
Zech J, Gold D, Salaymeh N, Sasson NC, Rabinowitch I, Golenser J, Mäder K. Oral Administration of Artemisone for the Treatment of Schistosomiasis: Formulation Challenges and In Vivo Efficacy. Pharmaceutics. 2020; 12(6):509. https://doi.org/10.3390/pharmaceutics12060509
Chicago/Turabian StyleZech, Johanna, Daniel Gold, Nadeen Salaymeh, Netanel Cohen Sasson, Ithai Rabinowitch, Jacob Golenser, and Karsten Mäder. 2020. "Oral Administration of Artemisone for the Treatment of Schistosomiasis: Formulation Challenges and In Vivo Efficacy" Pharmaceutics 12, no. 6: 509. https://doi.org/10.3390/pharmaceutics12060509