Development and Evaluation of Matrices Composed of β-cyclodextrin and Biodegradable Polyesters in the Controlled Delivery of Pindolol

 , , , ,
, , , ,  and
and
Abstract
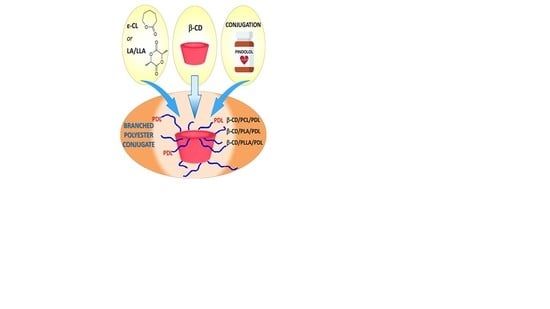
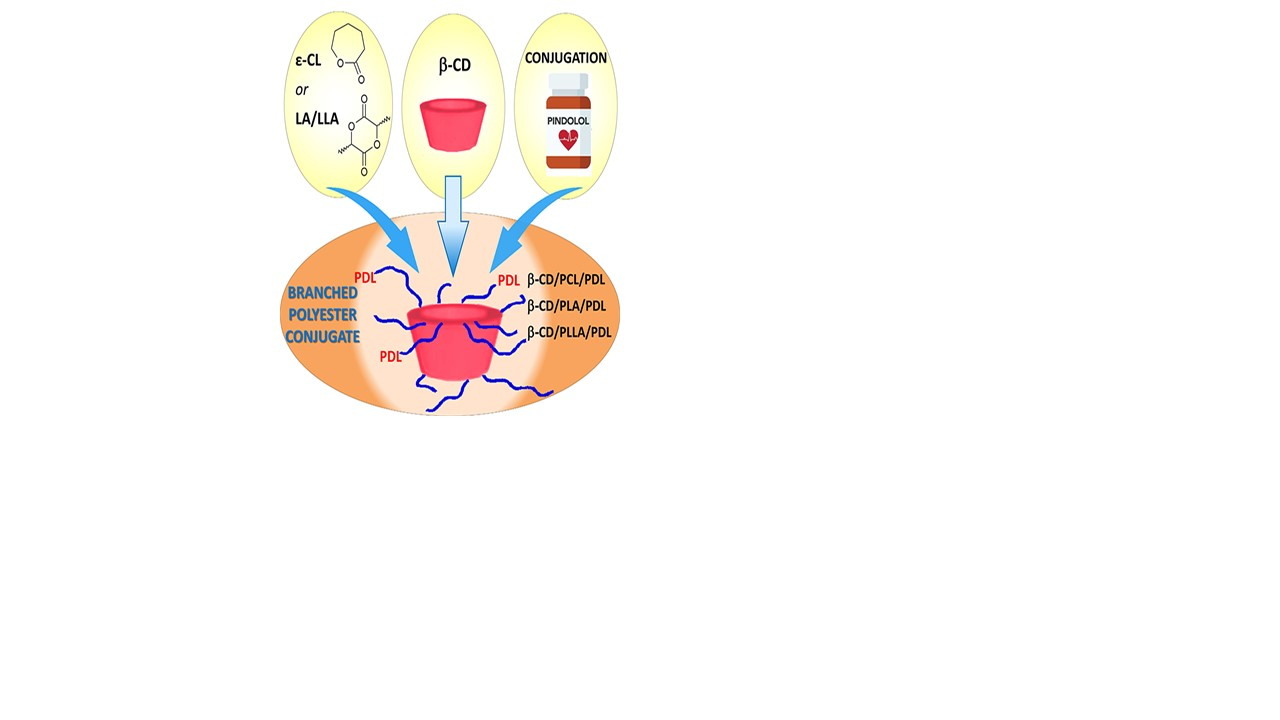
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. The ROP Procedure
2.3. Synthesis of the PDL Conjugates
2.4. Molecular Simulations
2.4.1. Structure Choice and Preparation
2.4.2. Conformational Search
2.5. Toxicity Assays
2.5.1. Microtox and Spirotox Tests
2.5.2. Umu-test
2.6. PDL Release Study from the Branched Conjugates
2.7. Measurements
3. Results and Discussion
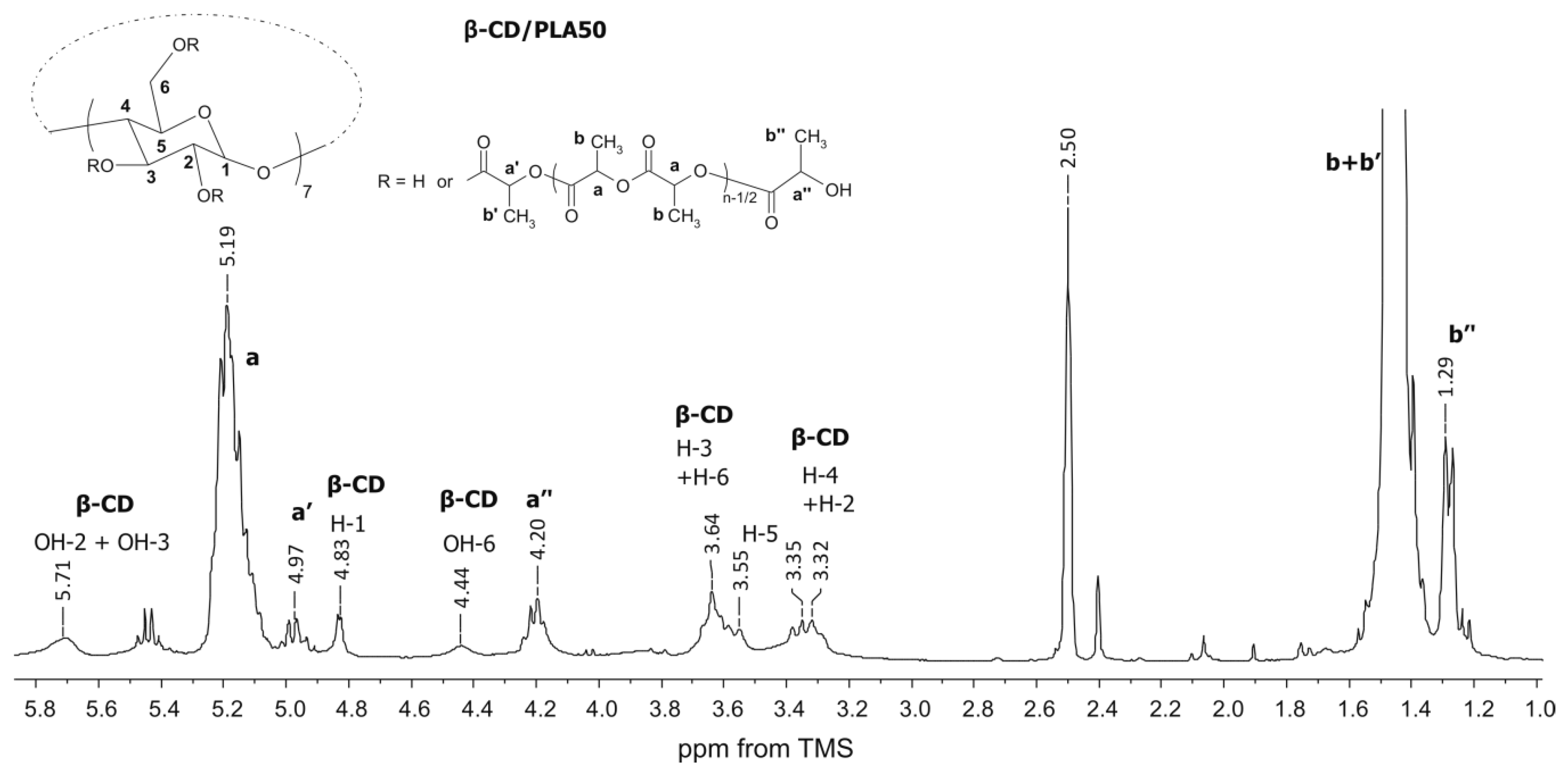
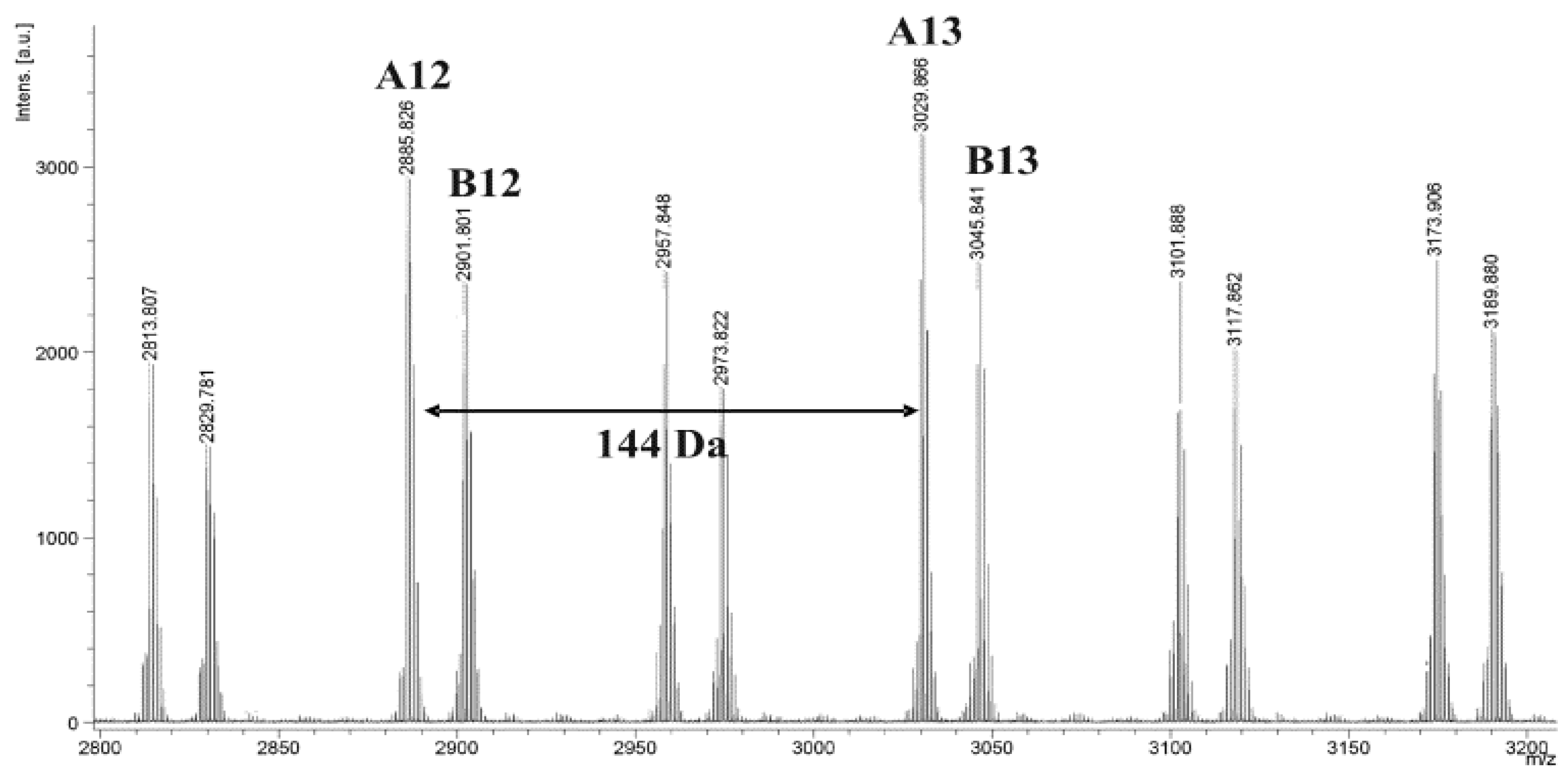
3.1. Structural Characterization of the Matrices
3.2. Molecular Modeling Studies
3.3. Toxicity Studies
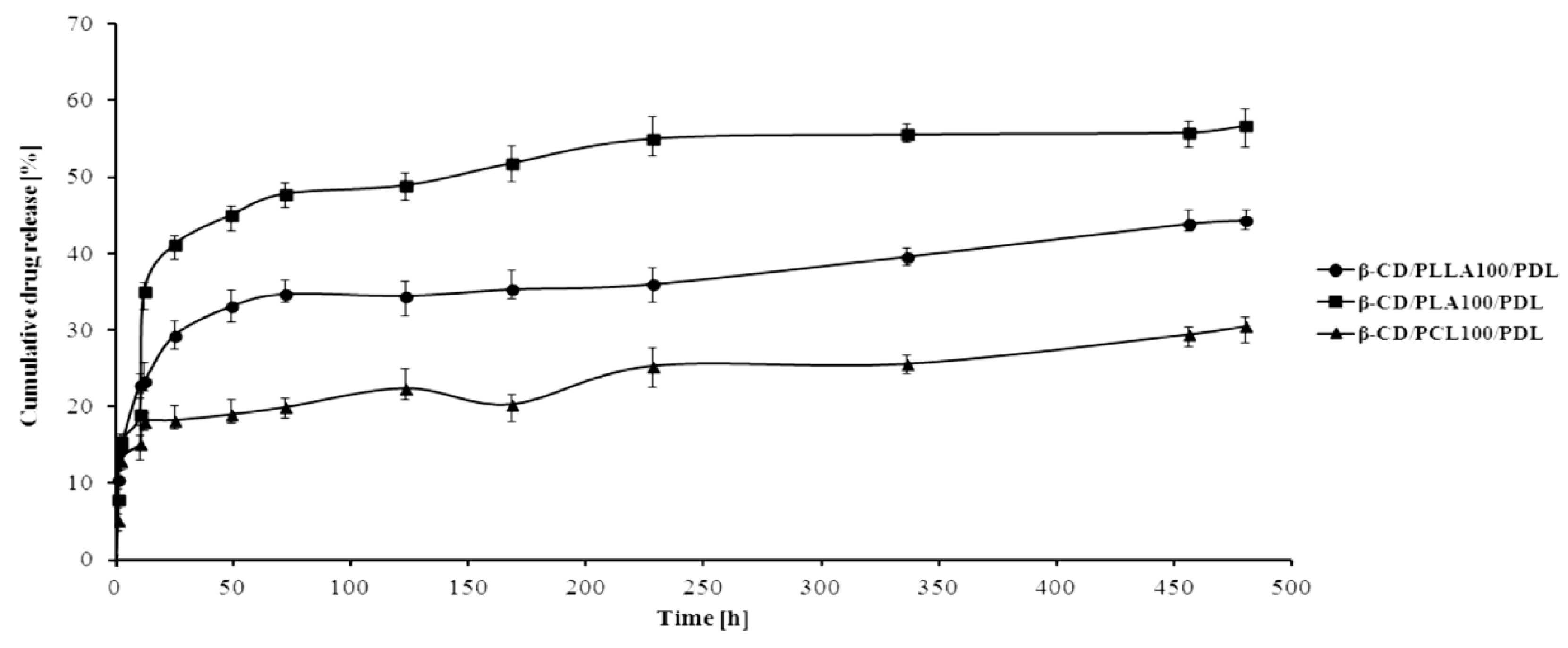
3.4. The PDL Conjugates’ Synthesis and Drug-Release Characteristics
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Mills, K.T.; Bundy, J.D.; Kelly, T.N.; Reed, J.E.; Patricia, M.; Kearney, P.M.; Reynolds, K.; Chen, J.; He, J. Global Disparities of Hypertension Prevalence and Control: A Systematic Analysis of Population-based Studies from 90 Countries. Circulation 2016, 134, 441–450. [Google Scholar] [CrossRef]
- World Health Organization. Cardiovascular Disease. A Global Brief on Hypertension; Silent Killer, Global Public Health Crisis. Available online: https://www.who.int/cardiovascular_diseases/publications/global_brief_hypertension/en/ (accessed on 5 March 2020).
- Guidelines Committee. 2003 European Society of Hypertension-European Society of Cardiology guidelines for the management of arterial hypertension. J. Hypertens. 2003, 21, 1011–1053. [Google Scholar] [CrossRef] [PubMed]
- Pasut, G.; Veronese, F.M. Polymer–drug conjugation, recent achievements and general strategies. Prog. Polym. Sci. 2007, 32, 933–961. [Google Scholar] [CrossRef]
- Oledzka, E.; Sobczak, M. Innovations in Biotechnology: Polymers in the Pharmaceutical Applications—Natural and Bioactive Initiators and Catalysts in the Synthesis of Biodegradable and Bioresorbable Polyesters and Polycarbonates; Agbo, E.C., Ed.; IntechOpen: London, UK, 2012; pp. 139–160. [Google Scholar]
- Kulshrestha, A.S.; Mahapatro, A. ACS Symposium Series, 2nd ed.; Polymers for Biomedical Applications; American Chemical Society: Washington, DC, USA, 2008; pp. 1–7. [Google Scholar]
- Maitz, F.M. Applications of synthetic polymers in clinical medicine. Biosurf. Biotribol. 2015, 1, 161–176. [Google Scholar] [CrossRef] [Green Version]
- Mohamed, F.; van der Walle, C.F. Engineering biodegradable polyester particles with specific drug targeting and drug release properties. J. Pharm. Sci. 2008, 97, 71–87. [Google Scholar] [CrossRef]
- Thomas, C.M. Stereocontrolled ring-opening polymerization of cyclic esters: Synthesis of new polyester microstructures. Chem. Soc. Rev. 2010, 39, 165–173. [Google Scholar] [CrossRef]
- Van der Mee, L.; Helmich, F.; de Bruijn, R.; Vekemans, J.A.J.M.; Palmans, A.R.A.; Meijer, E.W. Investigation of lipase-catalyzed ring-opening polymerizations of lactones with various ring sizes: Kinetic evaluation. Macromolecules 2006, 39, 5021–5027. [Google Scholar] [CrossRef]
- Oledzka, E.; Sokolowski, K.; Sobczak, M.; Kolodziejski, W. α-Amino acids as initiators of ε-caprolactone and l,l-lactide polymerization. Polym. Int. 2011, 60, 787–793. [Google Scholar] [CrossRef]
- Oledzka, E.; Narine, S.S. Organic acids catalyzed polymerization of ε-caprolactone: Synthesis and characterization. J. Appl. Polym. Sci. 2011, 119, 1873–1882. [Google Scholar] [CrossRef]
- Davis, M.E.; Brewster, M.E. Cyclodextrin-based pharmaceutics: Past, present and future. Nat. Rev. Drug. Discov. 2004, 3, 1023–1035. [Google Scholar] [CrossRef]
- Rajewski, R.A.; Stella, V.J. Pharmaceutical applications of cyclodextrins. 2. In vivo drug delivery. J. Pharm. Sci. 1996, 85, 1142–1169. [Google Scholar] [CrossRef] [PubMed]
- Loftsson, T.; Brewster, M.E. Pharmaceutical applications of cyclodextrins. 1. Drug solubilization and stabilization. J. Pharm. Sci. 1996, 85, 1017–1025. [Google Scholar] [CrossRef] [PubMed]
- Dodziuk, H. Cyclodextrins and Their Complexes: Chemistry, Analytical Methods, Applications; Dodziuk, H., Ed.; Wiley-VCH: Weinheim, Germany, 2006; pp. 1–30. [Google Scholar]
- Takashima, Y.; Kawaguchi, Y.; Nakagawa, S.; Harada, A. Inclusion Complex Formation and Hydrolysis of Lactones by Cyclodextrins. Chem. Lett. 2003, 32, 1122–1123. [Google Scholar] [CrossRef]
- Takayanagi, M.; Ito, S.; Matsumoto, K.; Nagaoka, M. Formation of reactant complex structure for initiation reaction of lactone ring-opening polymerization by cooperation of multiple cyclodextrin. J. Phys. Chem. B 2016, 120, 7174–7181. [Google Scholar] [CrossRef]
- Takashima, Y.; Osaki, M.; Harada, A. Cyclodextrin-initiated polymerization of cyclic esters in bulk: Formation of polyester-tethered cyclodextrins. J. Am. Chem. Soc. 2004, 126, 13588–13589. [Google Scholar] [CrossRef]
- Osaki, M.; Takashima, Y.; Yamaguchi, H.; Harada, A. An artificial molecular chaperone: Poly-pseudo-rotaxane with an extensible axle. J. Am. Chem. Soc. 2007, 129, 14452–14457. [Google Scholar] [CrossRef]
- Osaki, M.; Takashima, Y.; Yamaguchi, H.; Harada, A. Polymerization of lactones initiated by cyclodextrins: Effects of cyclodextrins on the initiation and propagation reactions. Macromolecules 2007, 40, 3154–3158. [Google Scholar] [CrossRef]
- Harada, A.; Osaki, M.; Takashima, Y.; Yamaguchi, H. Ring-opening polymerization of cyclic esters by cyclodextrins. Acc. Chem. Res. 2008, 41, 1143–1152. [Google Scholar] [CrossRef]
- Nogueira, G.; Favrelle, A.; Bria, M.; Prates Ramalho, J.P.; Mendes, P.J.; Valente, A.; Zinck, P. Adenine as an organocatalyst for the ring-opening polymerization of lactide: Scope, mechanism and access to adenine-functionalized polylactide. React. Chem. Eng. 2016, 1, 508–520. [Google Scholar] [CrossRef] [Green Version]
- Pujeri, S.S.; Khader, A.M.A.; Seetharamappa, J. Validated Stability-Indicating HPLC Method for the Separation of Pindolol Enantiomers and Its Related Substances. J. Food Drug Anal. 2011, 19, 73–84. [Google Scholar]
- Ferreira Almeida, P.; Almeida, A.J. Cross-linked alginate-gelatine beads: A new matrix for controlled release of pindolol. J. Control. Release 2004, 97, 431–439. [Google Scholar] [CrossRef] [PubMed]
- Knapp, S.; Keipert, S. Interactions between Cyclodextrins and Antiglaucoma Agents and Their Influence on the Transcorneal Permeability In Vitro. In Cyclodextrin: From Basic Research to Market, International Cyclodextrin Symposium, 10th ed.; Ann Arbor, M.I., Ed.; Wacker Biochem Corp.: Adrian, MI, USA, 2000; pp. 399–406. [Google Scholar]
- Gazpio, C.; Sánchez, M.; Zornoza, A.; Martín, C.; Martínez-Ohárriz, C.; Vélaz, I. A fluorimetric study of pindolol and its complexes with cyclodextrins. Talanta 2003, 60, 477–482. [Google Scholar] [CrossRef]
- Gazpio, C.; Sánchez, M.; Isasi, J.R.; Vélaz, I.; Martín, C.; Martínez-Ohárriz, C.; Zornoza, A. Sorption of pindolol and related compounds by a β-cyclodextrin polymer: Isosteric heat of sorption. Carbohydr. Polym. 2008, 71, 140–146. [Google Scholar] [CrossRef]
- International Organization for Standardization. Available online: https://www.iso.org/standard/73342.html (accessed on 5 March 2020).
- Nałęcz-Jawecki, G. Spirotox—Spirostomum ambiguum acute toxicity test—10 years of experience. Environ. Toxicol. 2004, 19, 359–364. [Google Scholar] [CrossRef] [PubMed]
- Oda, Y.; Nakamura, S.; Oki, I.; Kato, T.; Shinagawa, H. Evaluation of the new system (umu-test) for the detection of environmental mutagens and carcinogens. Mutat. Res. Mutagen. Relat. Subj. 1985, 147, 219–229. [Google Scholar] [CrossRef]
- European Pharmacopoeia. European Directorate for the Quality of Medicines & HealthCare (EDQM); European Pharmacopoeia Commission: Strasbourg, France, 2014. [Google Scholar]
- Veregin, R.P.; Fyfe, C.A.; Marchessault, R.H.; Taylor, M.G. Correlation of C-13 chemical-shifts with torsional angles from high-resolution, C-13 CP-MAS NMR-studies of crystalline cyclomalto-oligosaccharide complexes, and their relation to the structures of the starch polymorphs. Carbohyd. Res. 1987, 160, 41–56. [Google Scholar] [CrossRef]
- Jarvis, M.C. Relationship of chemical shift to glycosidic conformation in the solid-state 13C NMR spectra of (1-->4)-linked glucose polymers and oligomers: Anomeric and related effects. Carbohyd. Res. 1994, 259, 311–318. [Google Scholar] [CrossRef]
- Price, D.M.; Hourston, D.J.; Dumont, F. Thermogravimetry of Polymers. In Encyclopedia of Analytical Chemistry; Meyers, R.A., Ed.; John Wiley & Sons Ltd.: Chichester, UK, 2006; pp. 8094–8105. [Google Scholar]
- Abdolmohammadi, S.; Siyamak, S.; Ibrahim, N.A.; Yunus, W.M.; Ab Rahman, M.Z.; Azizi, S.; Fatehi, A. Enhancement of mechanical and thermal properties of polycaprolactone/chitosan blend by calcium carbonate nanoparticles. Int. J. Mol. Sci. 2012, 13, 4508–4522. [Google Scholar] [CrossRef]
- Spassky, N.; Simic, V.; Montaudo, M.S.; Hubert-Pfalzgraf, L.G. Inter- and intramolecular ester exchange reactions in the ring-opening polymerization of (D,L)-lactide using lanthanide alkoxide initiators. Macromol. Chem. Phys. 2000, 201, 2432–2440. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | [I]/[M] | Temperature (°C) | DP a | DS b | Mn(NMR) c (Da) | Mn(SEC-MALLS) d (Da) | ĐM d | Yield (%) |
|---|---|---|---|---|---|---|---|---|
| β-CD/PCL50 | 1/50 | 140 | 5.6 | 3.3 | 3200 | 3800 | 1.23 | 85 |
| β-CD/PLLA50 | 1/50 | 140 | 8.1 | 11.3 | 7700 | 8800 | 1.35 | 73 |
| β-CD/PCL100 | 1/100 | 140 | 11.2 | 13.0 | 17600 | 18700 | 1.26 | 91 |
| β-CD/PLA100 | 1/100 | 140 | 5.6 | 19.6 | 9000 | 10100 | 1.25 | 69 |
| β-CD/PLLA100 | 1/100 | 140 | 6.4 | 19.3 | 10000 | 10800 | 1.41 | 73 |
| β-CD/PCL100 | 1/100 | 100 | 6.9 | 8.0 | 7400 | 7300 | 1.23 | 47 |
| β-CD/PLA100 | 1/100 | 100 | 2.9 | 8.7 | 4800 | 5200 | 1.47 | 34 |
| β-CD/PLLA100 | 1/100 | 100 | 3.3 | 8.3 | 5100 | 6300 | 1.36 | 51 |
| Substitution Position of β-CD | DP = 20 DS = 1 | DP = 5 DS = 4 Type A | DP = 5 DS = 4 Type B | DP = 5 DS = 4 Type C | DP = 5 DS = 4 Type D |
|---|---|---|---|---|---|
| OH-2 | −232.028 | −266.359 | −313.119 | −302.218 | −267.362 |
| OH-3 | −226.762 | −258.627 | −310.287 | −275.790 | −265.445 |
| Entry | Spirotox 24 h-PE 1 | Microtox 15 min-PE 1 |
|---|---|---|
| β-CD/PLA100 | 0 | 18 ± 2 |
| β-CD/PLLA100 | 0 | 16 ± 6 |
| β-CD/PCL100 | 0 | 10 ± 3 |
| Entry | −S9 a | +S9 b | ||
|---|---|---|---|---|
| G c ± SD | IR d ± SD | Gc ± SD | IR d ± SD | |
| β-CD/PLA100 | 1.00 ± 0.0 | 0.75 ± 0.09 | 0.80 ± 0.02 | 1.04 ± 0.09 |
| β-CD/PLLA100 | 0.93 ± 0.1 | 0.91 ± 0.13 | 1.01 ± 0.09 | 0.69 ± 0.12 |
| β-CD/PCL100 | 1.12 ± 0.0 | 0.87 ± 0.21 | 1.02 ± 0.02 | 1.01 ± 0.06 |
| Negative Control | 1.01 ± 0.0 | 0.91 ± 0.17 | 1.00 ± 0.10 | 0.99 ± 0.14 |
| Solvent Control | 0.92 ± 0.1 | 0.83 ± 0.12 | 0.93 ± 0.07 | 0.84 ± 0.07 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lis-Cieplak, A.; Charuk, F.; Sobczak, M.; Zgadzaj, A.; Drobniewska, A.; Szeleszczuk, Ł.; Oledzka, E. Development and Evaluation of Matrices Composed of β-cyclodextrin and Biodegradable Polyesters in the Controlled Delivery of Pindolol. Pharmaceutics 2020, 12, 500. https://doi.org/10.3390/pharmaceutics12060500
Lis-Cieplak A, Charuk F, Sobczak M, Zgadzaj A, Drobniewska A, Szeleszczuk Ł, Oledzka E. Development and Evaluation of Matrices Composed of β-cyclodextrin and Biodegradable Polyesters in the Controlled Delivery of Pindolol. Pharmaceutics. 2020; 12(6):500. https://doi.org/10.3390/pharmaceutics12060500
Chicago/Turabian StyleLis-Cieplak, Agnieszka, Filip Charuk, Marcin Sobczak, Anna Zgadzaj, Agata Drobniewska, Łukasz Szeleszczuk, and Ewa Oledzka. 2020. "Development and Evaluation of Matrices Composed of β-cyclodextrin and Biodegradable Polyesters in the Controlled Delivery of Pindolol" Pharmaceutics 12, no. 6: 500. https://doi.org/10.3390/pharmaceutics12060500