Melt Amorphisation of Orlistat with Mesoporous Silica Using a Supercritical Carbon Dioxide: Effects of Pressure, Temperature, and Drug Loading Ratio and Comparison with Other Conventional Amorphisation Methods


Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
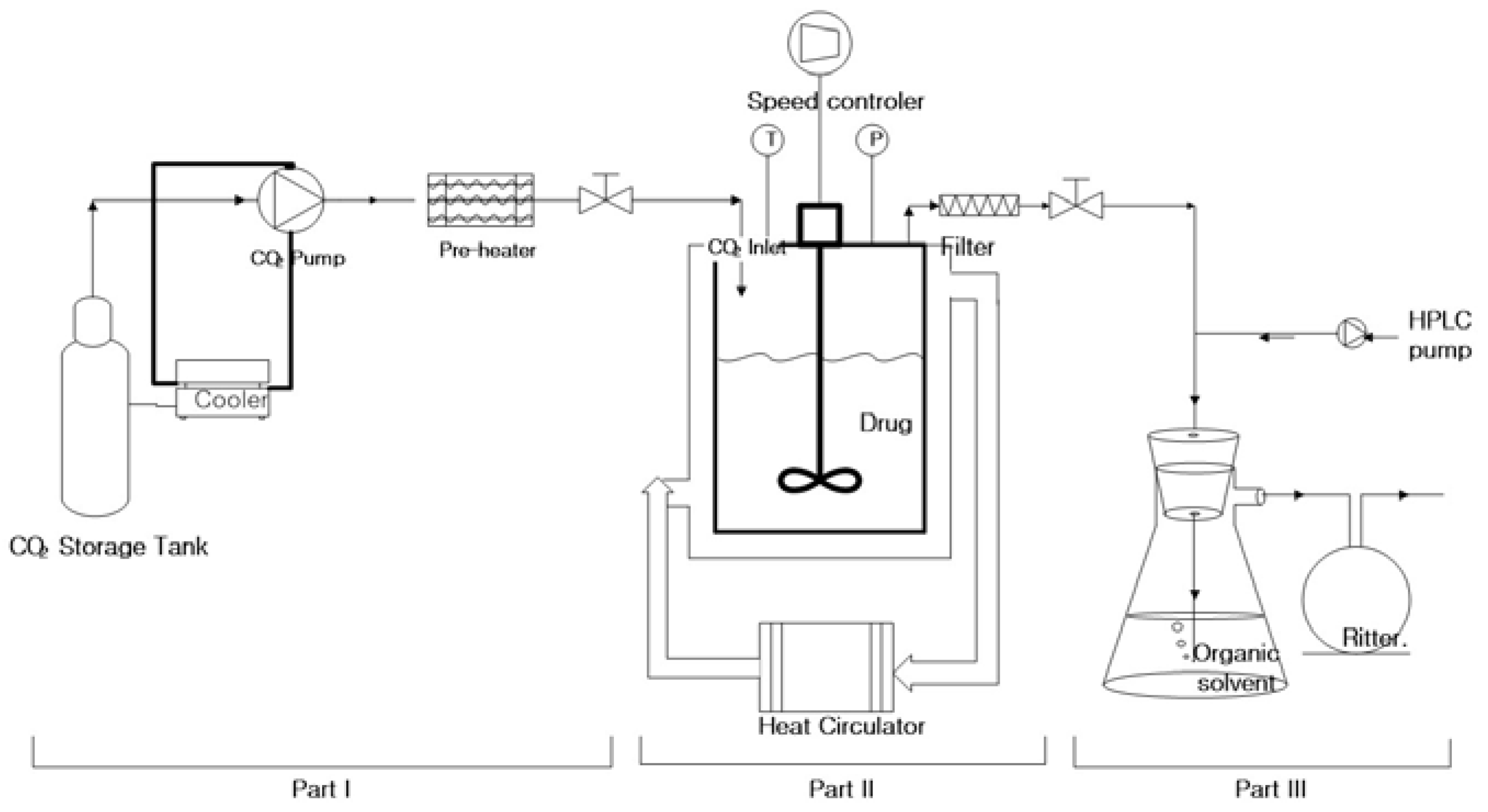
2.2. Measurement of Orlistat Solubility in SC-CO2
2.3. Correlation of Density of SC-CO2 and Orlistat Solubility in SC-CO2 by Semi-Empirical Models
2.4. Determination of Melting Point Depression by Solid–Liquid (S-L) Phase Behavior Experiment
2.5. Drug Quantification Method
2.6. Preparation of Amorphous Orlistat-Loaded Neusilin® UFL2 Using MA-SCF Process
2.6.1. Effect of Pressure and Temperature
2.6.2. Effect of Mesoporous Silica:Drug Mass Ratio
2.7. SE Method
2.8. HM Method
2.9. Specific Surface Area and Total Pore Volume
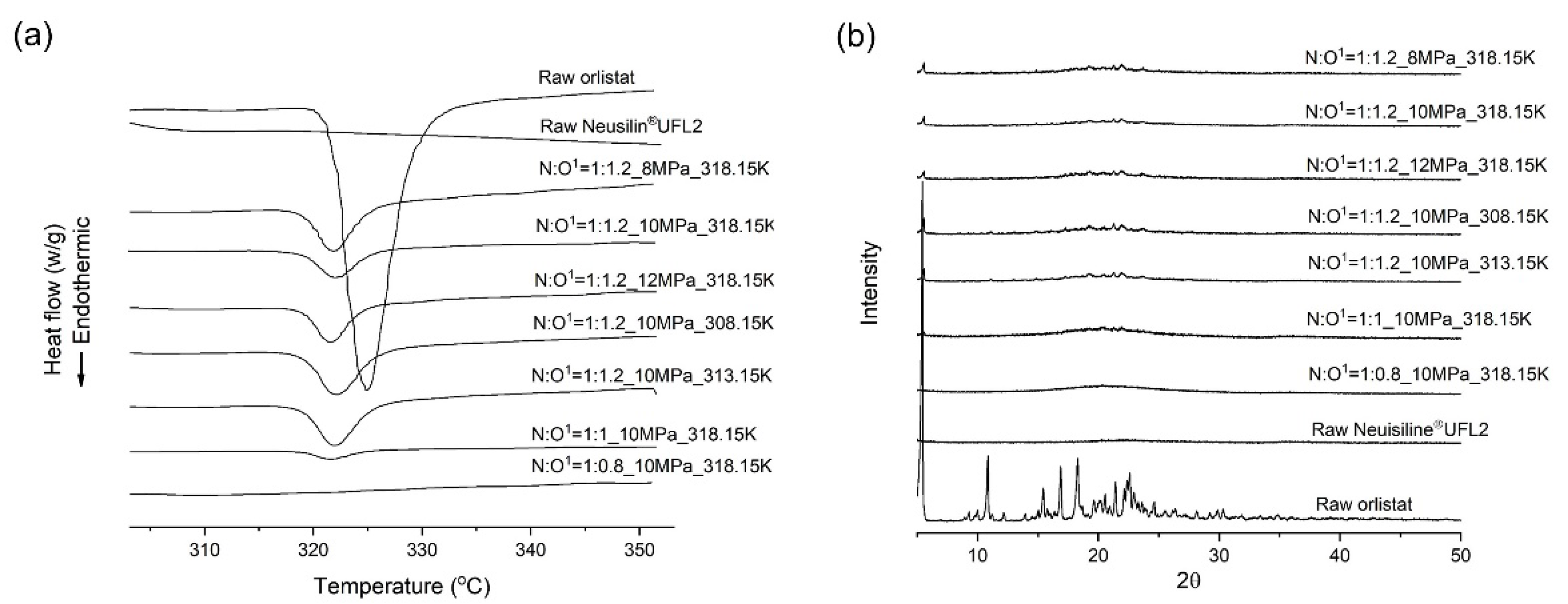
2.10. Differential Scanning Calorimetry (DSC)
2.11. PXRD
2.12. SEM
2.13. Analyses of Drug Content (%) and Uniformity
2.14. In-Vitro Dissolution Study
2.15. Statistical Analysis
3. Results and Discussions
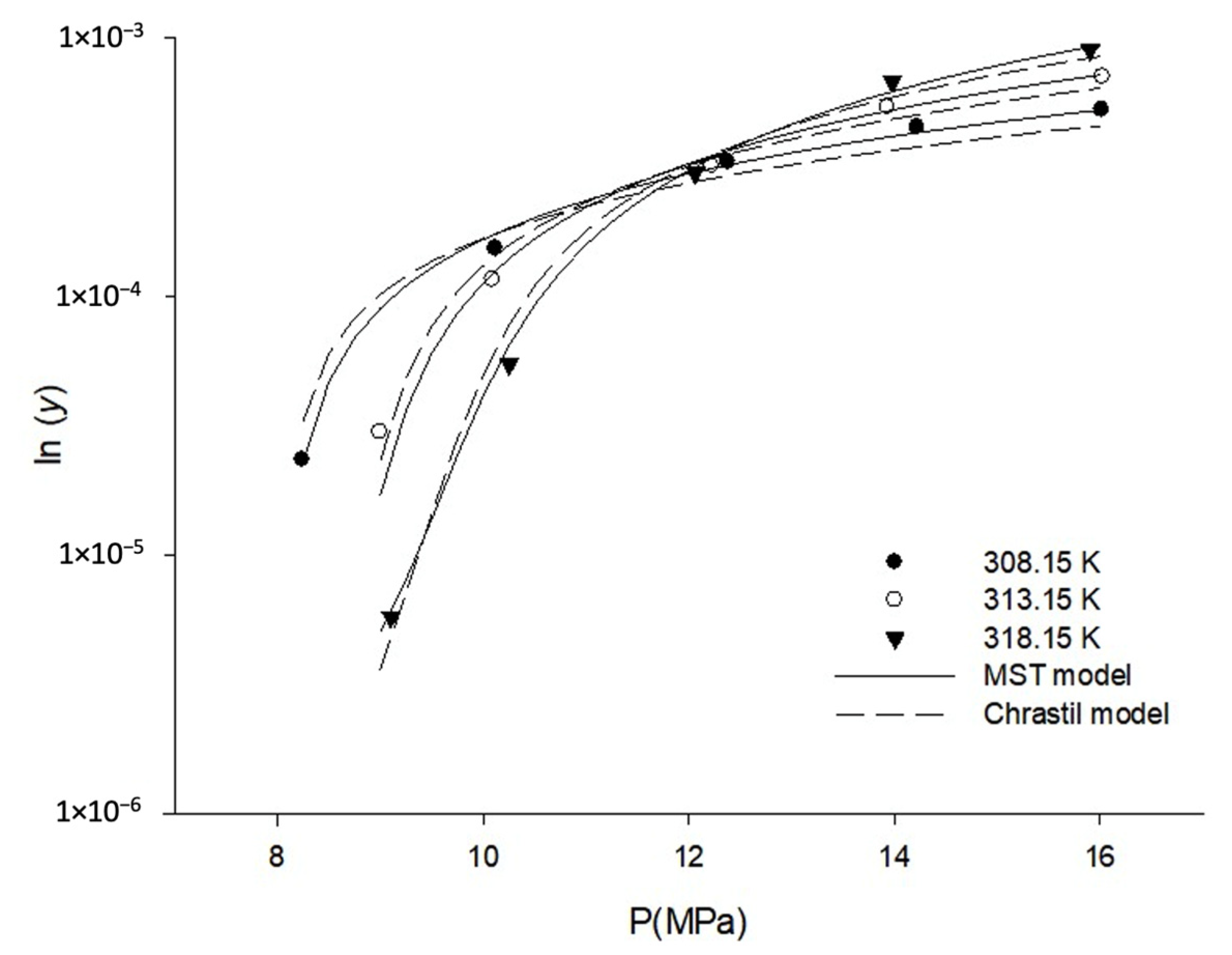
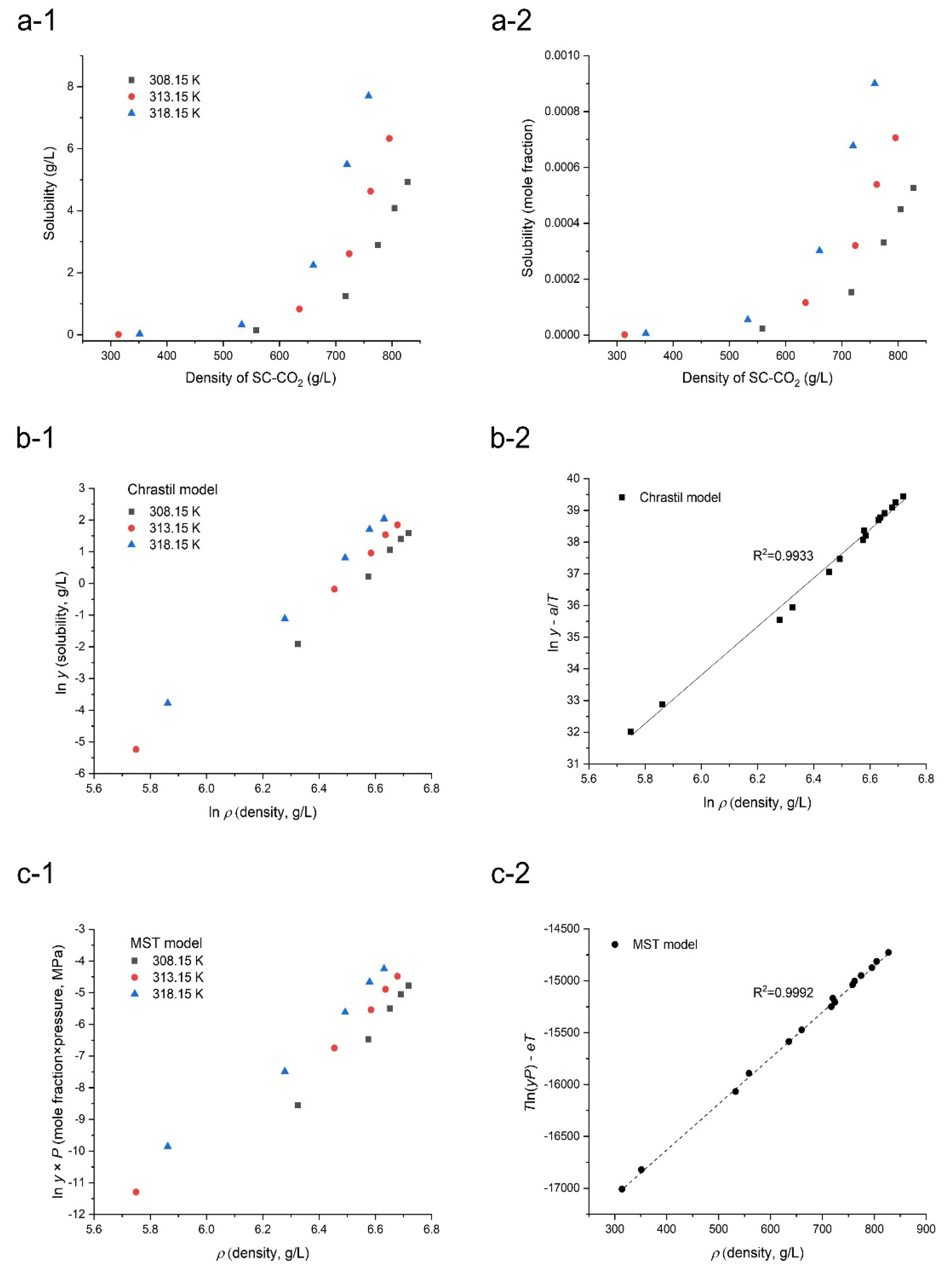
3.1. Solubility of Orlistat in SC-CO2
3.2. Melting Point Depression of Orlistat
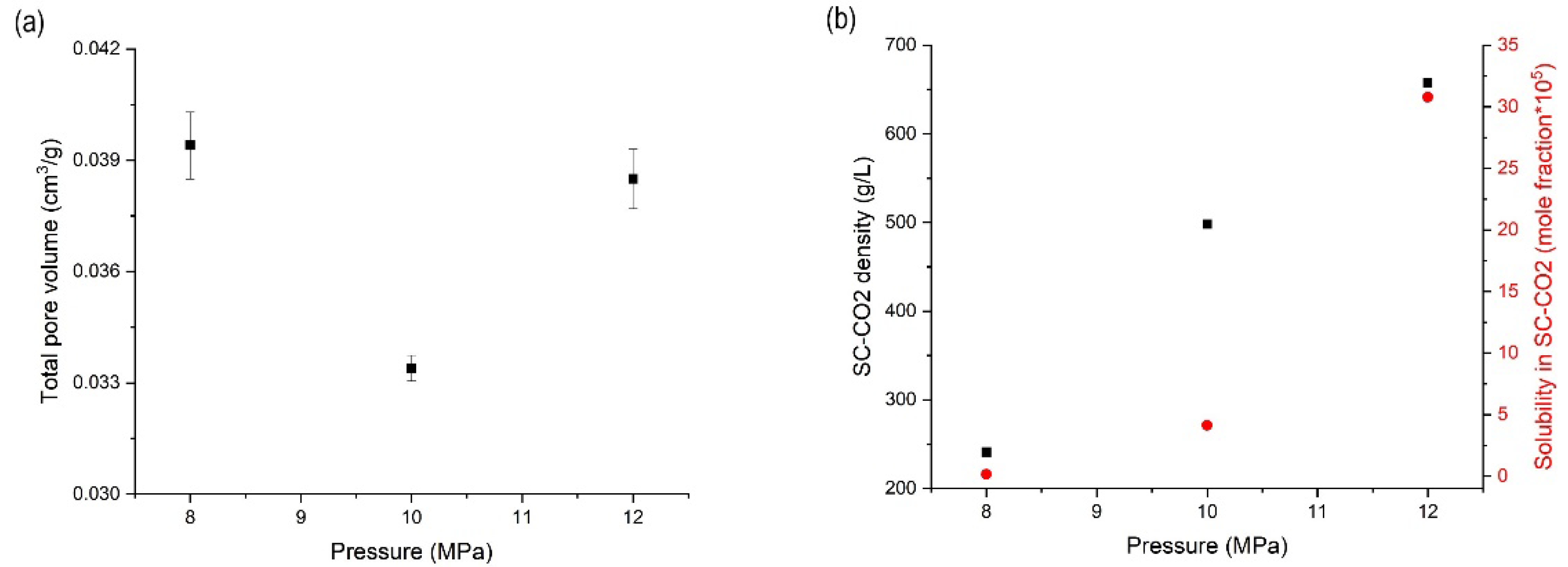
3.3. Effects of Pressure on the Orlistat Loading onto Mesoporous Silica
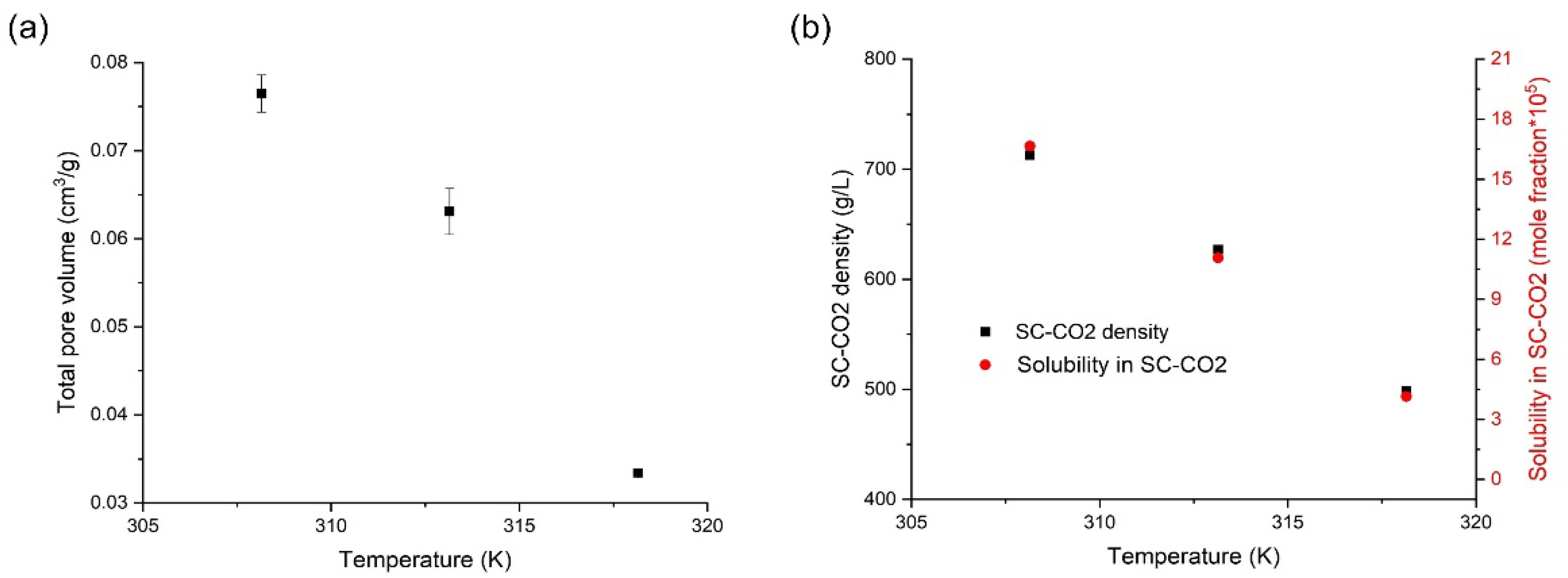
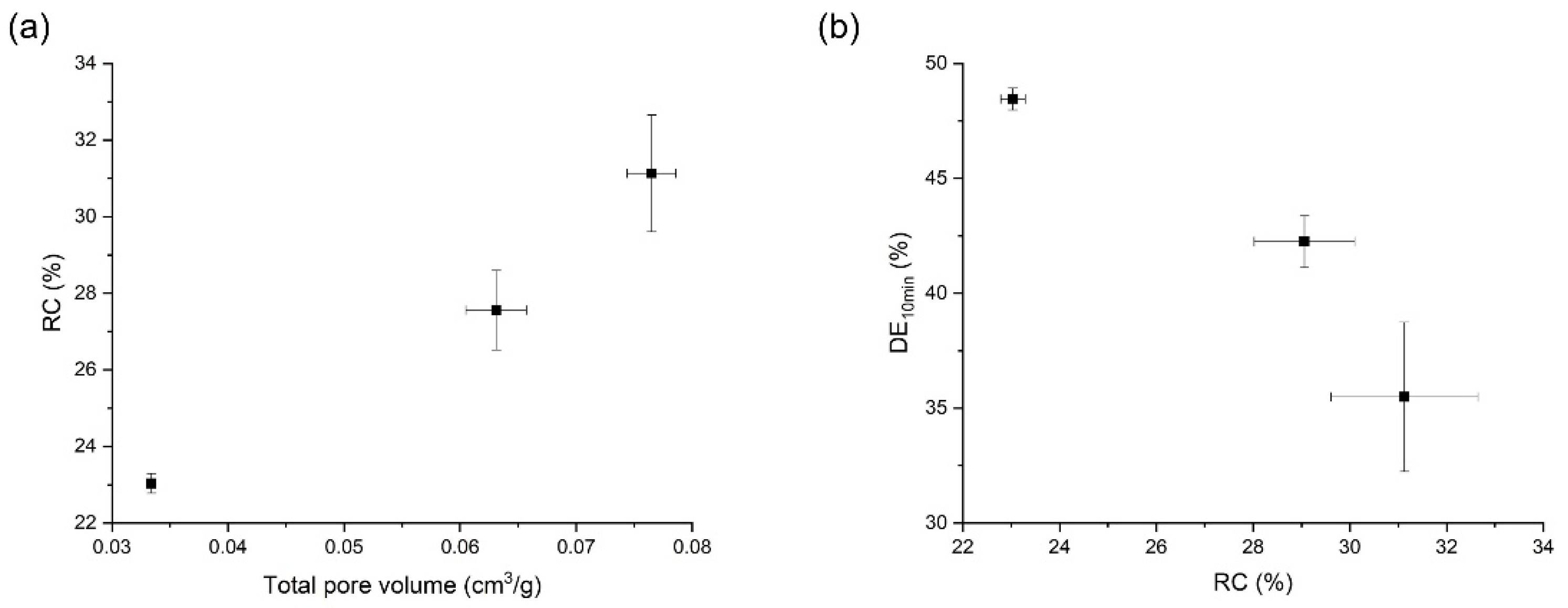
3.4. Effects of Temperature on the Orlistat Loading onto Mesoporous Silica
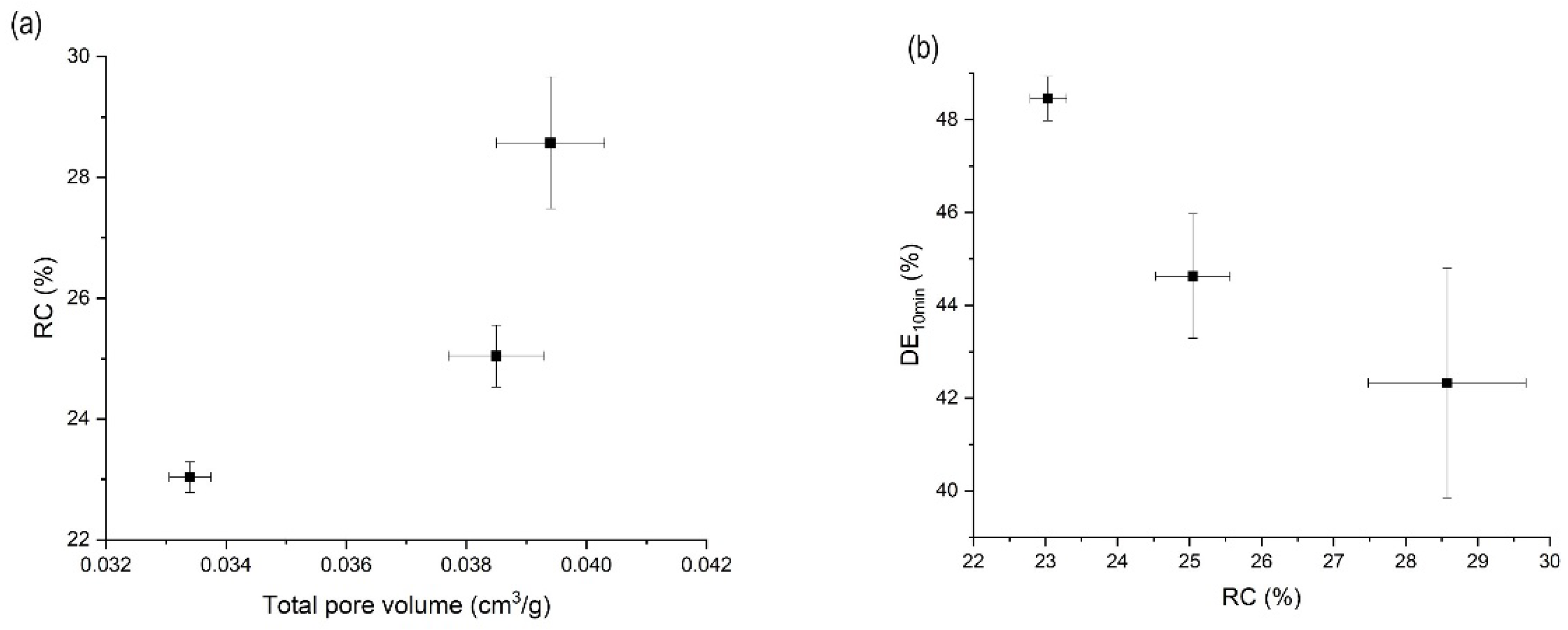
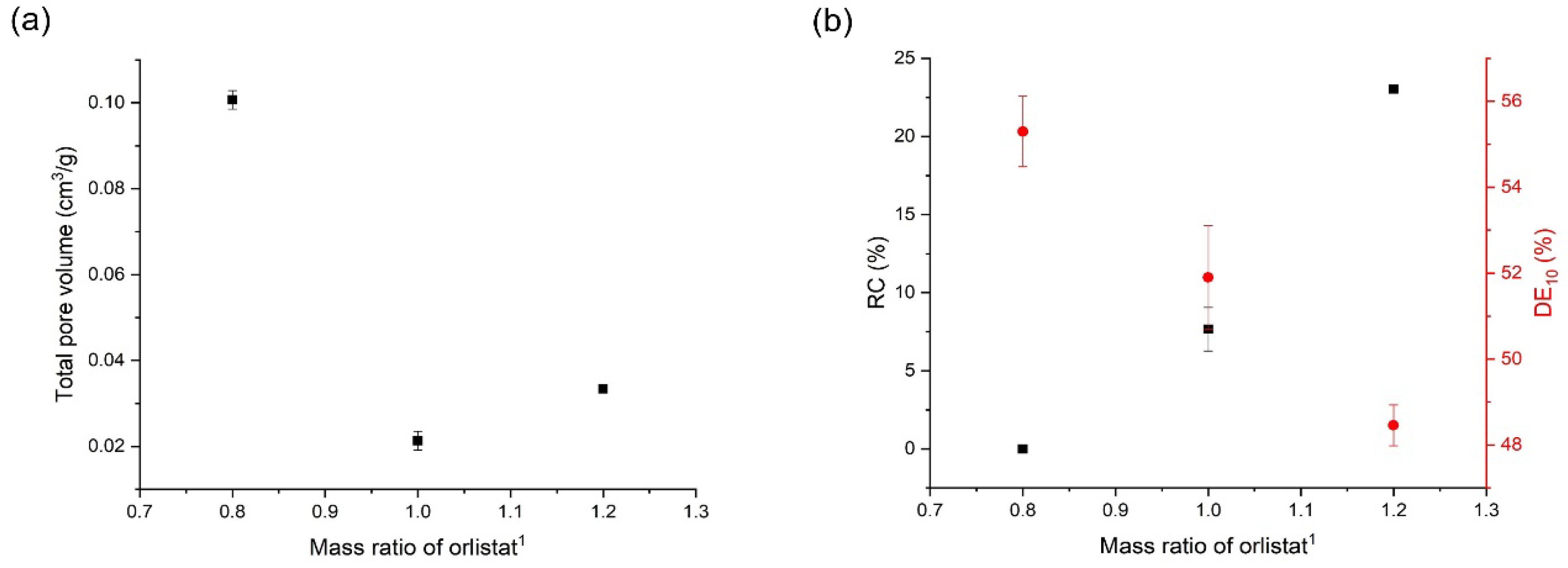
3.5. Effects of Mesoporous Silica:Drug Mass Ratio on the Orlistat Loading Behavior onto Mesoporous Silica
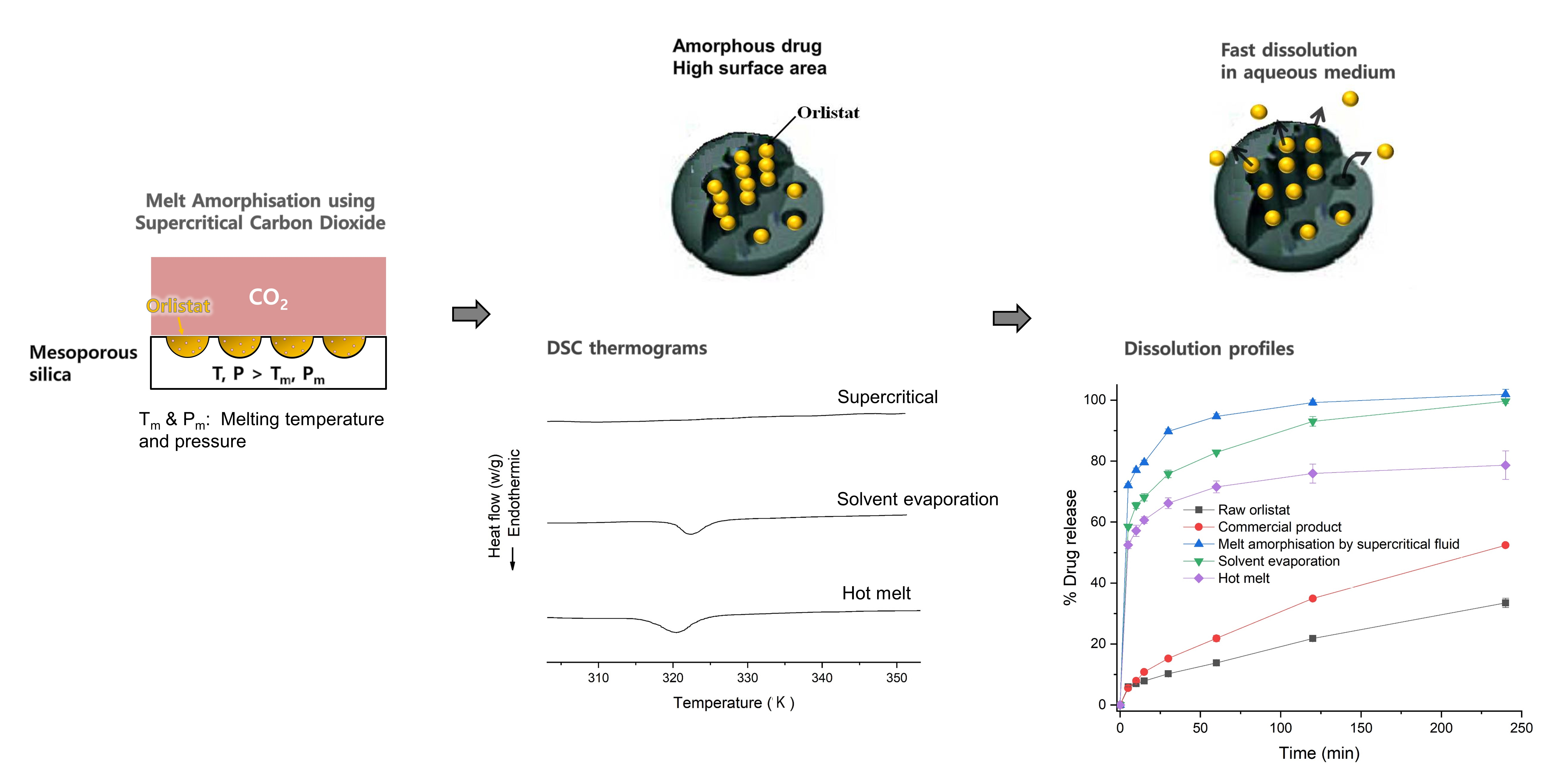
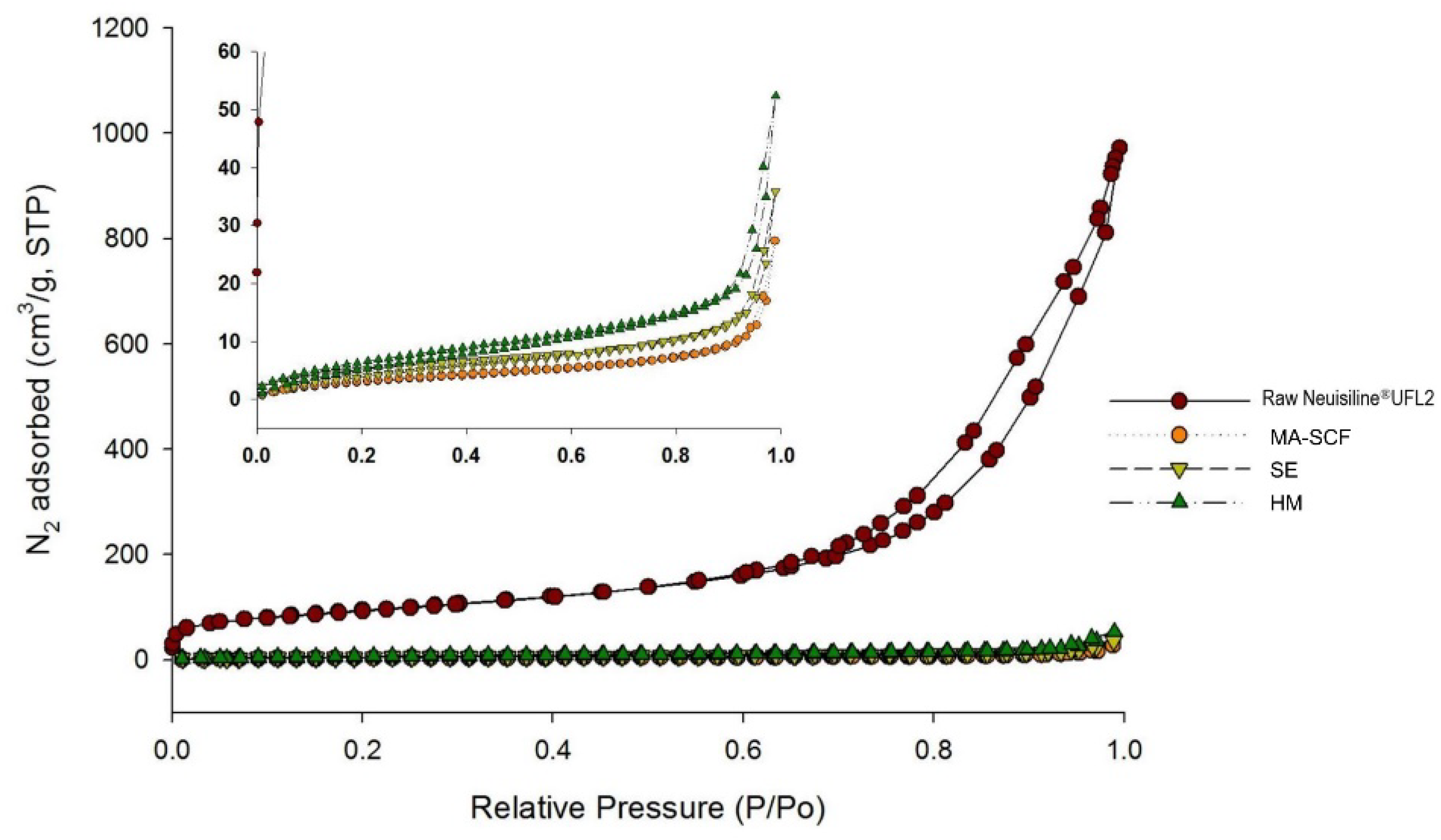
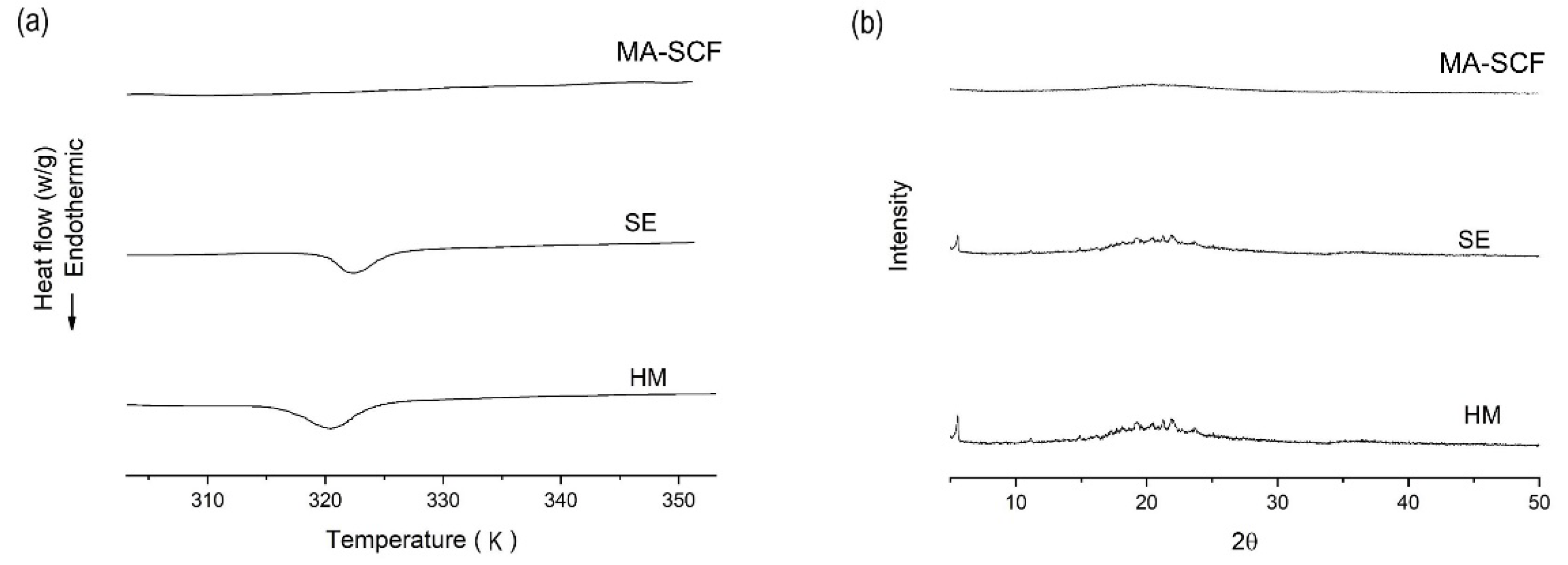
3.6. Comparison of Amorphisation Method
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Ballinger, A.; Peikin, S.R. Orlistat: Its current status as an anti-obesity drug. Eur. J. Pharmacol. 2002, 440, 109–117. [Google Scholar] [CrossRef]
- Harp, J.B. An assessment of the efficacy and safety of orlistat for the long-term management of obesity. J. Nutr. Biochem. 1998, 9, 516–521. [Google Scholar] [CrossRef]
- Hsieh, C.-J.; Wang, P.-W.; Liu, R.-T.; Tung, S.-C.; Chien, W.-Y.; Chen, J.-F.; Chen, C.-H.; Kuo, M.-C.; Hu, Y.-H. Orlistat for obesity: Benefits beyond weight loss. Diabetes Res. Clin. Pract. 2005, 67, 78–83. [Google Scholar] [CrossRef] [PubMed]
- Zhi, J.; Melia, A.T.; Eggers, H.; Joly, R.; Patel, I.H. Review of limited systemic absorption of orlistat, a lipase inhibitor, in healthy human volunteers. J. Clin. Pharmacol. 1995, 35, 1103–1108. [Google Scholar] [CrossRef]
- Davidson, M.H.; Hauptman, J.; DiGirolamo, M.; Foreyt, J.P.; Halsted, C.H.; Heber, D.; Heimburger, D.C.; Lucas, C.P.; Robbins, D.C.; Chung, J. Weight control and risk factor reduction in obese subjects treated for 2 years with orlistat: A randomized controlled trial. Jama 1999, 281, 235–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clarysse, S.; Psachoulias, D.; Brouwers, J.; Tack, J.; Annaert, P.; Duchateau, G.; Reppas, C.; Augustijns, P. Postprandial changes in solubilizing capacity of human intestinal fluids for BCS class II drugs. Pharm. Res. 2009, 26, 1456–1466. [Google Scholar] [CrossRef]
- Dolenc, A.; Govedarica, B.; Kocbek, P.; Srčič, S.; Kristl, J. Nanosized particles of orlistat with enhanced in vitro dissolution rate and lipase inhibition. Int. J. Pharm. 2010, 396, 149–155. [Google Scholar] [CrossRef]
- Choi, Y.H.; Han, H.-K. Nanomedicines: Current status and future perspectives in aspect of drug delivery and pharmacokinetics. J. Pharm. Investig. 2018, 48, 43–60. [Google Scholar] [CrossRef] [Green Version]
- Bhardwaj, K.; Sharma, R. Formulation and evaluation of solid lipid nanoparticles loaded with orlistat for oral drug delivery. Master’s Thesis, Lovely Professional University, Punjab, India, 2017. [Google Scholar]
- Singh, A. Evaluation of orlistat solid dispersion using poloxomer 188 as hydrophilic carrier. Sch. Res. J. 2011, 1, 48–51. [Google Scholar] [CrossRef]
- Payghan, S.A. Potential Screening of Spray Dried Solid Dispersion of Orlistat using Three Dimensional Solubility Parameter. Asian J. Pharm. 2018, 11, 760–772. [Google Scholar]
- Liu, X.; Feng, X.; Williams, R.O.; Zhang, F. Characterization of amorphous solid dispersions. J. Pharm. Investig. 2018, 48, 19–41. [Google Scholar] [CrossRef]
- Kim, D.H.; Kim, J.Y.; Kim, R.M.; Maharjan, P.; Ji, Y.-G.; Jang, D.-J.; Min, K.A.; Koo, T.-S.; Cho, K.H. Orlistat-loaded solid sNeDDs for the enhanced solubility, dissolution, and in vivo performance. Int. J. Nanomedicine 2018, 13, 7095. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.H.; Maharjan, P.; Kim, J.Y.; Jang, D.-J.; Koo, T.-S.; Min, K.A.; Cho, K.H. Enhanced solubility, in-vitro dissolution and lipase inhibition of a self-nanoemulsifying drug delivery system containing orlistat. J. Nanosci. Nanotechnol. 2019, 19, 634–639. [Google Scholar] [CrossRef] [PubMed]
- Ahsan, M.N.; Verma, P.R.P. Enhancement of in vitro dissolution and pharmacodynamic potential of olanzapine using solid SNEDDS. J. Pharm. Investig. 2018, 48, 269–278. [Google Scholar] [CrossRef]
- Gaikwad, S.; Lonare, M.; Lonare, D.; Tajne, M.; Narnaware, P. Formulation development, characterization and evaluation of liquisolid tablet contatining orlistat. IJCRT 2018, 6, 142–152. [Google Scholar]
- Dischinger, A.; Page, S.; Kleinebudde, P. Fast dissolving fillers in dry foam formulation. Pow. Technol. 2015, 270, 494–501. [Google Scholar] [CrossRef]
- Singh, D.; Bedi, N.; Tiwary, A.K. Enhancing solubility of poorly aqueous soluble drugs: Critical appraisal of techniques. J. Pharm. Investig. 2018, 48, 509–526. [Google Scholar] [CrossRef]
- Friedrich, H.; Fussnegger, B.; Kolter, K.; Bodmeier, R. Dissolution rate improvement of poorly water-soluble drugs obtained by adsorbing solutions of drugs in hydrophilic solvents onto high surface area carriers. Eur. J. Pharm. Biopharm. 2006, 62, 171–177. [Google Scholar] [CrossRef]
- Sharma, S.; Sher, P.; Badve, S.; Pawar, A.P. Adsorption of meloxicam on porous calcium silicate: Characterization and tablet formulation. AAPS PharmSciTech 2005, 6, E618–E625. [Google Scholar] [CrossRef] [Green Version]
- Sher, P.; Ingavle, G.; Ponrathnam, S.; Pawar, A.P. Low density porous carrier: Drug adsorption and release study by response surface methodology using different solvents. Int. J. Pharm. 2007, 331, 72–83. [Google Scholar] [CrossRef]
- Witschi, C.; Doelker, E. Residual solvents in pharmaceutical products: Acceptable limits, influences on physicochemical properties, analytical methods and documented values. Eur. J. Pharm. Biopharm. 1997, 43, 215–242. [Google Scholar] [CrossRef]
- Hong, S.H.; Choi, Y. Mesoporous silica-based nanoplatforms for the delivery of photodynamic therapy agents. J. Pharm. Investig. 2018, 48, 3–17. [Google Scholar] [CrossRef] [Green Version]
- Park, H.J.; Kim, M.-S.; Lee, S.; Kim, J.-S.; Woo, J.-S.; Park, J.-S.; Hwang, S.-J. Recrystallization of fluconazole using the supercritical antisolvent (SAS) process. Int. J. Pharm. 2007, 328, 152–160. [Google Scholar] [CrossRef]
- Abuzar, S.M.; Hyun, S.-M.; Kim, J.-H.; Park, H.J.; Kim, M.-S.; Park, J.-S.; Hwang, S.-J. Enhancing the solubility and bioavailability of poorly water-soluble drugs using supercritical antisolvent (SAS) process. Int. J. Pharm. 2018, 538, 1–13. [Google Scholar] [CrossRef]
- Kim, M.-S.; Jin, S.-J.; Kim, J.-S.; Park, H.J.; Song, H.-S.; Neubert, R.H.; Hwang, S.-J. Preparation, characterization and in vivo evaluation of amorphous atorvastatin calcium nanoparticles using supercritical antisolvent (SAS) process. Eur. J. Pharm. Biopharm. 2008, 69, 454–465. [Google Scholar] [CrossRef]
- Pasquali, I.; Bettini, R. Are pharmaceutics really going supercritical? Int. J. Pharm. 2008, 364, 176–187. [Google Scholar] [CrossRef]
- Pasquali, I.; Bettini, R.; Giordano, F. Supercritical fluid technologies: An innovative approach for manipulating the solid-state of pharmaceuticals. Adv. Drug Deliv. Rev. 2008, 60, 399–410. [Google Scholar] [CrossRef]
- Yeo, S.-D.; Kiran, E. Formation of polymer particles with supercritical fluids: A review. J. Supercrit. Fluids 2005, 34, 287–308. [Google Scholar] [CrossRef]
- Pasquali, I.; Bettini, R.; Giordano, F. Solid-state chemistry and particle engineering with supercritical fluids in pharmaceutics. Eur. J. Pharm. Sci. 2006, 27, 299–310. [Google Scholar] [CrossRef]
- Sanganwar, G.P.; Gupta, R.B. Dissolution-rate enhancement of fenofibrate by adsorption onto silica using supercritical carbon dioxide. Int. J. Pharm. 2008, 360, 213–218. [Google Scholar] [CrossRef]
- Cha, K.-H.; Cho, K.-J.; Kim, M.-S.; Kim, J.-S.; Park, H.J.; Park, J.; Cho, W.; Park, J.-S.; Hwang, S.-J. Enhancement of the dissolution rate and bioavailability of fenofibrate by a melt-adsorption method using supercritical carbon dioxide. Int. J. Nanomedicine 2012, 7, 5565. [Google Scholar] [PubMed] [Green Version]
- Knez, Ž.; Škerget, M.; Hrnčič, M.K.; Čuček, D. Particle formation using sub-and supercritical fluids. In Supercritical Fluid Technology for Energy and Environmental Applications; Elsevier: Amsterdam, The Netherlands, 2014; pp. 31–67. [Google Scholar]
- Pasquali, I.; Comi, L.; Pucciarelli, F.; Bettini, R. Swelling, melting point reduction and solubility of PEG 1500 in supercritical CO2. Int. J. Pharm. 2008, 356, 76–81. [Google Scholar] [CrossRef] [PubMed]
- Pasquali, I.; Andanson, J.-M.; Kazarian, S.G.; Bettini, R. Measurement of CO2 sorption and PEG 1500 swelling by ATR-IR spectroscopy. J. Supercrit. Fluids 2008, 45, 384–390. [Google Scholar] [CrossRef]
- Stassi, A.; Bettini, R.; Gazzaniga, A.; Giordano, F.; Schiraldi, A. Assessment of solubility of ketoprofen and vanillic acid in supercritical CO2 under dynamic conditions. J. Chem. Eng. 2000, 45, 161–165. [Google Scholar]
- Bettini, R.; Bonassi, L.; Castoro, V.; Rossi, A.; Zema, L.; Gazzaniga, A.; Giordano, F. Solubility and conversion of carbamazepine polymorphs in supercritical carbon dioxide. Eur. J. Pharm. Sci. 2001, 13, 281–286. [Google Scholar] [CrossRef]
- Fischer, K.; Wilken, M.; Gmehling, J. The effect of gas pressure on the melting behavior of compounds. Fluid Ph. Equilibria 2003, 210, 199–214. [Google Scholar] [CrossRef]
- Su, C.-S.; Chen, Y.-P. Measurement and correlation for the solid solubility of non-steroidal anti-inflammatory drugs (NSAIDs) in supercritical carbon dioxide. J. Supercrit. Fluids 2008, 43, 438–446. [Google Scholar] [CrossRef]
- Chen, Y.-M.; Lin, P.-C.; Tang, M.; Chen, Y.-P. Solid solubility of antilipemic agents and micronization of gemfibrozil in supercritical carbon dioxide. J. Supercrit. Fluids 2010, 52, 175–182. [Google Scholar] [CrossRef]
- Lemmon, E.W.; Huber, M.L.; McLinden, M.O. NIST Standard Reference Database: Reference Fluid Thermodynamic and Transport Properties-REFPROP, Version 8.0; National Institute of Standards and Technology: Gaithersburg, MD, USA, 2018.
- Chrastil, J. Solubility of solids and liquids in supercritical gases. J. Phys. Chem. 1982, 86, 3016–3021. [Google Scholar] [CrossRef]
- Méndez-Santiago, J.; Teja, A.S. The solubility of solids in supercritical fluids. Fluid Ph. Equilibria 1999, 158, 501–510. [Google Scholar] [CrossRef]
- Hussein, K.; Türk, M.; Wahl, M.A. Drug loading into β-cyclodextrin granules using a supercritical fluid process for improved drug dissolution. Eur. J. Pharm. Sci. 2008, 33, 306–312. [Google Scholar] [CrossRef]
- Hudson-Curtis, B.; Novick, S. Assessing content uniformity. In Nonclinical Statistics for Pharmaceutical and Biotechnology Industries; Springer: Cham, Switzerland, 2016; pp. 631–651. [Google Scholar]
- Huang, Z.; Chiew, Y.C.; Lu, W.-D.; Kawi, S. Solubility of aspirin in supercritical carbon dioxide/alcohol mixtures. Fluid Ph. Eequilibria 2005, 237, 9–15. [Google Scholar] [CrossRef]
- Hezave, A.Z.; Khademi, M.H.; Esmaeilzadeh, F. Measurement and modeling of mefenamic acid solubility in supercritical carbon dioxide. Fluid Ph. Equilibria 2012, 313, 140–147. [Google Scholar] [CrossRef]
- Huang, Z.; Kawi, S.; Chiew, Y. Solubility of cholesterol and its esters in supercritical carbon dioxide with and without cosolvents. J. Supercrit. Fluids 2004, 30, 25–39. [Google Scholar] [CrossRef]
- Sabegh, M.A.; Rajaei, H.; Esmaeilzadeh, F.; Lashkarbolooki, M. Solubility of ketoprofen in supercritical carbon dioxide. J. Supercrit. Fluids 2012, 72, 191–197. [Google Scholar] [CrossRef]
- Sauceau, M.; Letourneau, J.-J.; Freiss, B.; Richon, D.; Fages, J. Solubility of eflucimibe in supercritical carbon dioxide with or without a co-solvent. J. Supercrit. Fluids 2004, 31, 133–140. [Google Scholar] [CrossRef] [Green Version]
- Huang, Z.; Guo, Y.-H.; Miao, H.; Teng, L.-J. Solubility of progesterone in supercritical carbon dioxide and its micronization through RESS. Pow. Technol. 2014, 258, 66–77. [Google Scholar] [CrossRef]
- Yamini, Y.; Moradi, M. Measurement and correlation of antifungal drugs solubility in pure supercritical CO2 using semiempirical models. J. Chem. Thermodyn. 2011, 43, 1091–1096. [Google Scholar] [CrossRef]
- Vatanara, A.; Najafabadi, A.R.; Khajeh, M.; Yamini, Y. Solubility of some inhaled glucocorticoids in supercritical carbon dioxide. J. Supercrit. Fluids 2005, 33, 21–25. [Google Scholar] [CrossRef]
- Keri, V.; Csorvasi, A.; Aronhime, J. Preparation of orlistat and orlistat crystalline forms. International PCT WO2003/047531 A2, 12 June 2003. [Google Scholar]
- Mellaerts, R.; Mols, R.; Jammaer, J.A.; Aerts, C.A.; Annaert, P.; Van Humbeeck, J.; Van den Mooter, G.; Augustijns, P.; Martens, J.A. Increasing the oral bioavailability of the poorly water soluble drug itraconazole with ordered mesoporous silica. Eur. J. Pharm. Biopharm. 2008, 69, 223–230. [Google Scholar] [CrossRef]
- Salonen, J.; Laitinen, L.; Kaukonen, A.M.; Tuura, J.; Björkqvist, M.; Heikkilä, T.; Vähä-Heikkilä, K.; Hirvonen, J.; Lehto, V.-P. Mesoporous silicon microparticles for oral drug delivery: Loading and release of five model drugs. J. Control. Release 2005, 108, 362–374. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhi, Z.; Jiang, T.; Zhang, J.; Wang, Z.; Wang, S. Spherical mesoporous silica nanoparticles for loading and release of the poorly water-soluble drug telmisartan. J. Control. Release 2010, 145, 257–263. [Google Scholar] [CrossRef]
- Sliwinska-Bartkowiak, M.; Dudziak, G.; Gras, R.; Sikorski, R.; Radhakrishnan, R.; Gubbins, K.E. Freezing behavior in porous glasses and MCM-41. Colloids Surf. A, Physicochem. Eng. Asp. 2001, 187, 523–529. [Google Scholar] [CrossRef]
- Healy, A.M.; Worku, Z.A.; Kumar, D.; Madi, A.M. Pharmaceutical solvates, hydrates and amorphous forms: A special emphasis on cocrystals. Adv. Drug Deliv. Rev. 2017, 117, 25–46. [Google Scholar] [CrossRef] [Green Version]
- Lee, T.; Wang, Y.W. Initial salt screening procedures for manufacturing ibuprofen. Drug Dev. Ind. Pharm. 2009, 35, 555–567. [Google Scholar] [CrossRef]
- Zůza, D.; Šoltys, M.; Mužík, J.; Lizoňová, D.; Lhotka, M.; Ulbrich, P.; Kašpar, O.; Štěpánek, F. Silica particles with three levels of porosity for efficient melt amorphisation of drugs. Microporous Mesoporous Mater. 2019, 274, 61–69. [Google Scholar] [CrossRef]
- Wakayama, H.; Goto, Y.; Fukushima, Y. A novel method for tailoring porous structures of nanoporous materials using supercritical solvents. Phys. Chem. Chem. Phys. 2003, 5, 3784–3788. [Google Scholar] [CrossRef]
- Takeuchi, H.; Nagira, S.; Yamamoto, H.; Kawashima, Y. Solid dispersion particles of amorphous indomethacin with fine porous silica particles by using spray-drying method. Int. J. Pharm. 2005, 293, 155–164. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Temperature (K) | Pressure (MPa) | CO2 Density (g/L) [41] | Solubility of Orlistat ± S.D. y (Mole Fraction) × 105 | s (Mean Value, g/L = kg/m3) |
|---|---|---|---|---|
| 308.15 | 8.25 | 558.62 | 2.3 ± 0.1 | 0.15 |
| 10.12 | 717.2 | 15.3 ± 0.2 | 1.24 | |
| 12.38 | 774.62 | 33.1 ± 1.1 | 2.89 | |
| 14.23 | 804.64 | 45.0 ± 1.4 | 4.08 | |
| 16.02 | 827.4 | 52.7 ± 1.2 | 4.92 | |
| 313.15 | 8.28 | 314.02 | 0.2 ± 0.0 | 0.01 |
| 10.10 | 635.51 | 11.6 ± 0.2 | 0.83 | |
| 12.23 | 724.12 | 32.0 ± 0.9 | 2.61 | |
| 13.94 | 762.06 | 53.9 ± 1.5 | 4.63 | |
| 16.03 | 795.28 | 70.6 ± 2.6 | 6.33 | |
| 318.15 | 9.10 | 351.49 | 0.6 ± 0.0 | 0.02 |
| 10.25 | 532.91 | 5.5 ± 0.1 | 0.33 | |
| 12.06 | 660.27 | 30.1 ± 0.6 | 2.24 | |
| 13.98 | 719.99 | 67.7 ± 2.7 | 5.50 | |
| 15.90 | 758.35 | 90.1 ± 3.5 | 7.70 |
| Binary System (CO2 + Orliastat) | ||
|---|---|---|
| Pressure (MPa) 1 ± S.D. | Tm_p (K) 2 | ΔTm (K) 4 |
| Ambient | 321.05 3 | - |
| 2.3 ± 0.1 | 313.15 | −8 |
| 3.2 ± 0.1 | 308.15 | −13 |
| 3.7 ± 0.1 | 303.15 | −18 |
| 4.0 ± 0.1 | 298.15 | −23 |
| 4.3 ± 0.1 | 293.15 | −28 |
| 4.8 ± 0.1 | 288.15 | −33 |
| Factors | Results (n = 3, mean ± S.D.) | |||||
|---|---|---|---|---|---|---|
| Mass Ratio (N:O) 1 | T (K) 2 | P (MPa) 3 | Total Pore Volume (cm3/g) | Drug Content (%) | RC (%) 4 | DE10 (%)5 |
| 1:1.2 | 318.15 | 8 | 0.0394 ± 0.0009 | 97.5 ± 1.2 | 29.6 ± 1.1 | 42.3 ± 2.5 |
| 1:1.2 | 318.15 | 10 | 0.0334 ± 0.0004 | 97.7 ± 1.1 | 23.0 ± 0.3 | 48.5 ± 0.5 |
| 1:1.2 | 318.15 | 12 | 0.0385 ± 0.0008 | 93.4 ± 2.1 | 25.0 ± 0.5 | 44.6 ± 1.3 |
| 1:1.2 | 308.15 | 10 | 0.0765 ± 0.0021 | 94.4 ± 0.9 | 31.1 ± 1.5 | 35.5 ± 3.3 |
| 1:1.2 | 313.15 | 10 | 0.0631 ± 0.0026 | 97.2 ± 2.5 | 29.1 ± 1.0 | 42.3 ± 1.1 |
| 1:1 | 318.15 | 10 | 0.0213 ± 0.0021 | 98.6 ± 1.3 | 7.7 ± 1.4 | 51.9 ± 1.2 |
| 1:0.8 | 318.15 | 10 | 0.1007 ± 0.0022 | 99.8 ± 1.0 | 0.00 ± 0.0 | 55.3 ± 0.8 |
| Model | Parameters | AARD (%) |
|---|---|---|
| Chrastil | k = 7.727, a = −11,663.6, b = −12.61 | 16.67 |
| Méndez -Santiago and Teja (MST) | c = −18,341.6, d = 4.369, e = 43.018 | 6.37 |
| Method | RC (%) 1 | % Drug Dissolved 2 | DE60 (%) 3 | Content (%) 4 | AV (%) 5 | |
|---|---|---|---|---|---|---|
| 10 min | 30 min | |||||
| Raw Orlistat | 100 | 7.0 ± 1.1 | 10.2 ± 0.9 | 9.7 ± 1.1 | - | - |
| Commercial Product | - | 7.9 ± 2.1 | 15.3 ± 1.4 | 14.1 ± 1.3 | - | - |
| MA-SCF | 0 | 77.0 ± 0.7 | 89.8 ± 0.8 | 83.0 ± 1.1 | 99.8 ± 1.0 | 2.4 |
| SE | 17.4 | 65.5 ± 1.0 | 75.8 ± 1.2 | 70.8 ± 1.9 | 100.5 ± 3.6 | 8.7 |
| HM | 24.8 | 57.2 ± 1.9 | 66.2 ± 1.8 | 61.9 ± 3.3 | 98.5 ± 7.1 | 17.0 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, H.; Cha, K.-H.; Hong, S.H.; Abuzar, S.M.; Ha, E.-S.; Kim, J.-S.; Kim, M.-S.; Hwang, S.-J. Melt Amorphisation of Orlistat with Mesoporous Silica Using a Supercritical Carbon Dioxide: Effects of Pressure, Temperature, and Drug Loading Ratio and Comparison with Other Conventional Amorphisation Methods. Pharmaceutics 2020, 12, 377. https://doi.org/10.3390/pharmaceutics12040377
Park H, Cha K-H, Hong SH, Abuzar SM, Ha E-S, Kim J-S, Kim M-S, Hwang S-J. Melt Amorphisation of Orlistat with Mesoporous Silica Using a Supercritical Carbon Dioxide: Effects of Pressure, Temperature, and Drug Loading Ratio and Comparison with Other Conventional Amorphisation Methods. Pharmaceutics. 2020; 12(4):377. https://doi.org/10.3390/pharmaceutics12040377
Chicago/Turabian StylePark, Heejun, Kwang-Ho Cha, Seung Hyeon Hong, Sharif Md Abuzar, Eun-Sol Ha, Jeong-Soo Kim, Min-Soo Kim, and Sung-Joo Hwang. 2020. "Melt Amorphisation of Orlistat with Mesoporous Silica Using a Supercritical Carbon Dioxide: Effects of Pressure, Temperature, and Drug Loading Ratio and Comparison with Other Conventional Amorphisation Methods" Pharmaceutics 12, no. 4: 377. https://doi.org/10.3390/pharmaceutics12040377