Improved Dissolution and Pharmacokinetics of Abiraterone through KinetiSol® Enabled Amorphous Solid Dispersions


Abstract
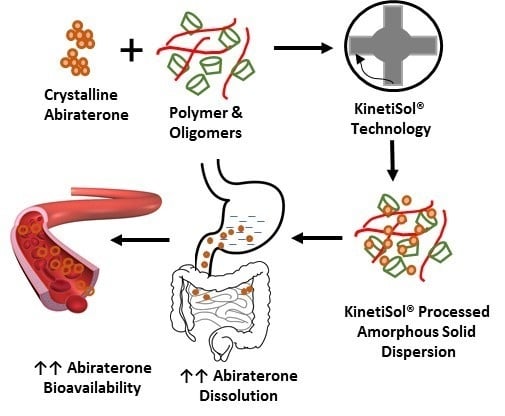
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
2.2.1. Development of KSDs
KinetiSol® Processing
Milling
2.2.2. Physicochemical Characterization of KSDs
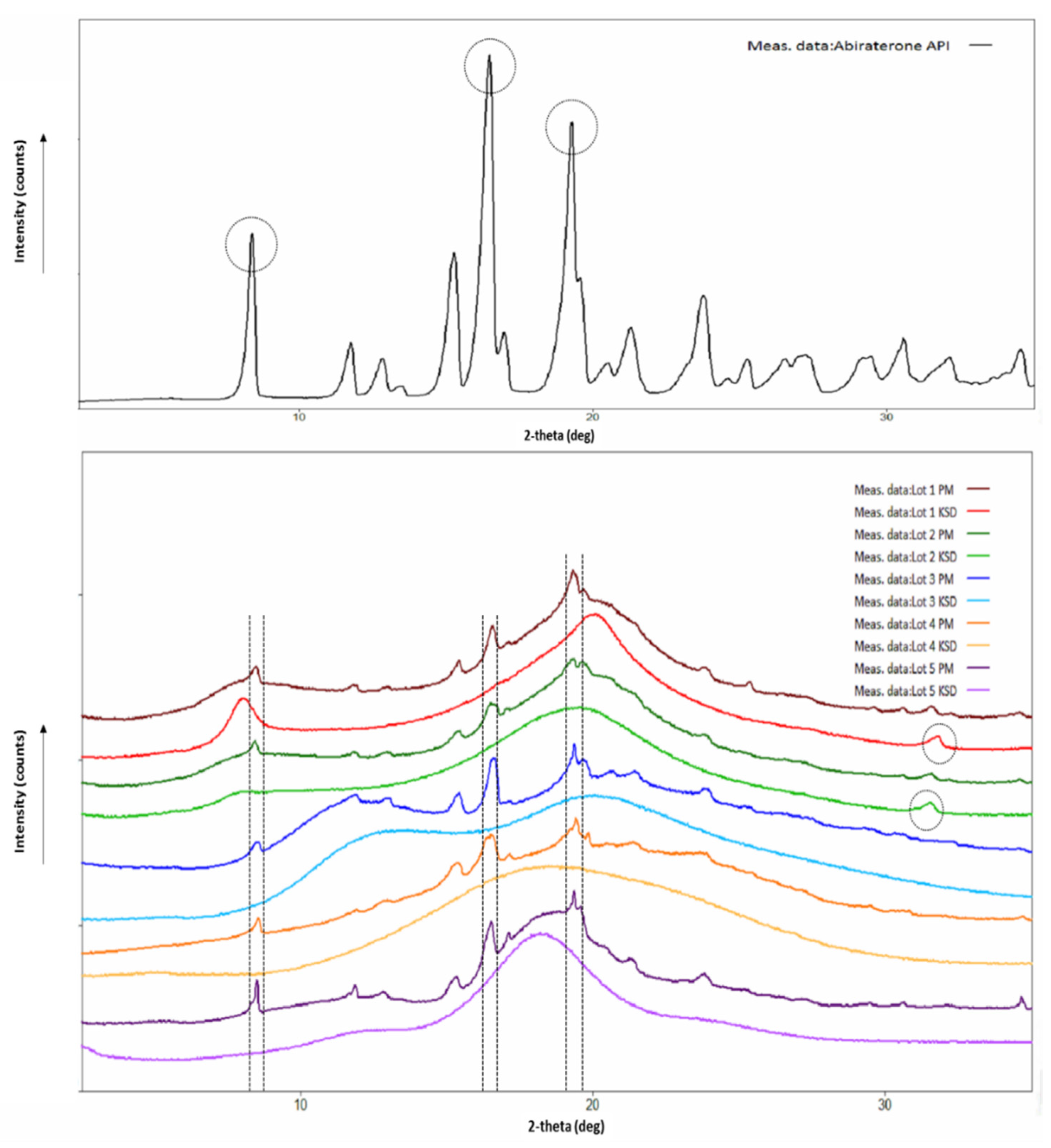
X-Ray Powder Diffraction
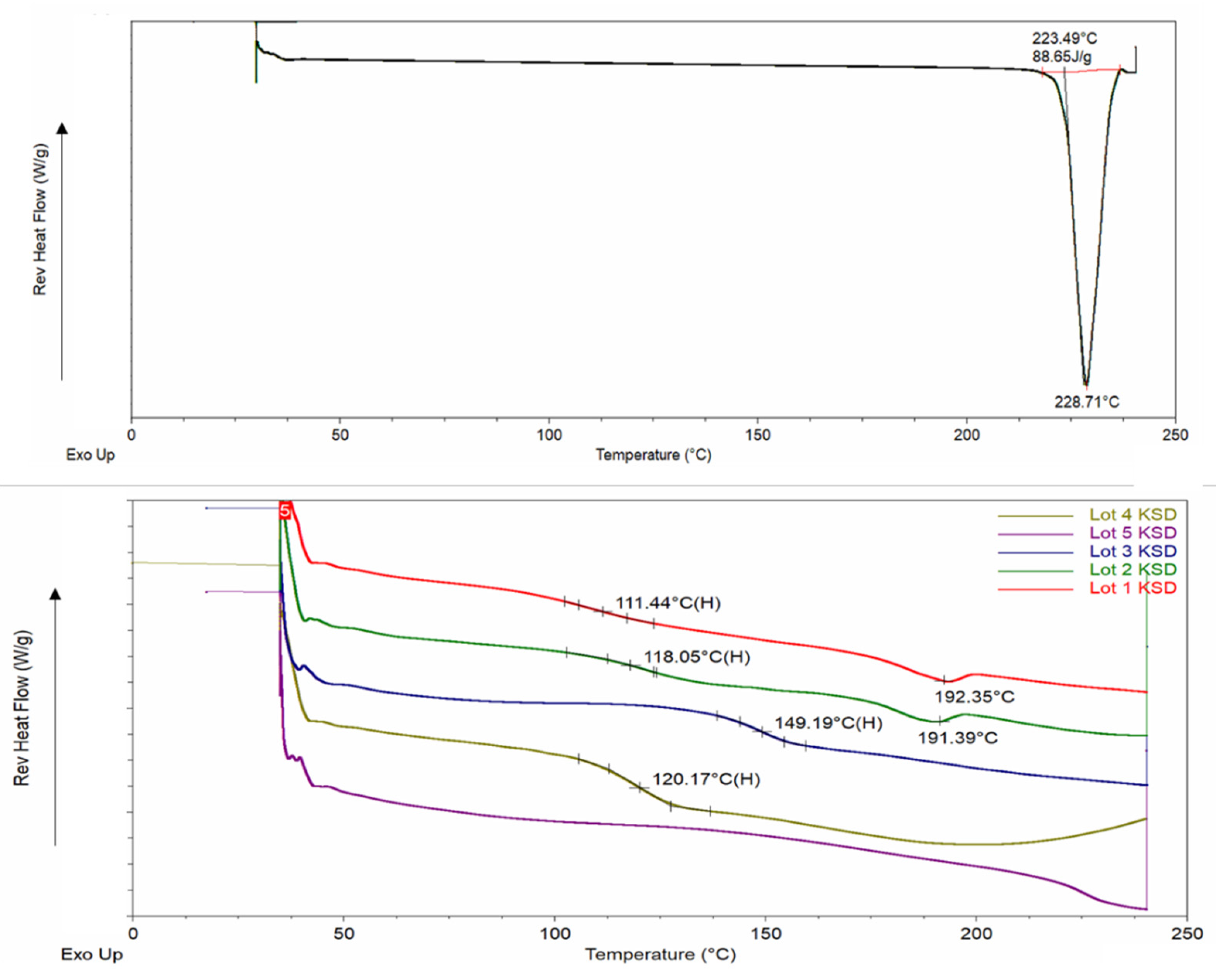
Modulated Differential Scanning Calorimetry
HPLC Analysis
Dissolution
Tableting
Pharmacokinetic Study in Beagle Dogs
Pharmacokinetic Analysis
3. Results and Discussion
3.1. Development of Binary KSDs
3.2. Physicochemical Characterization of Binary KSDs
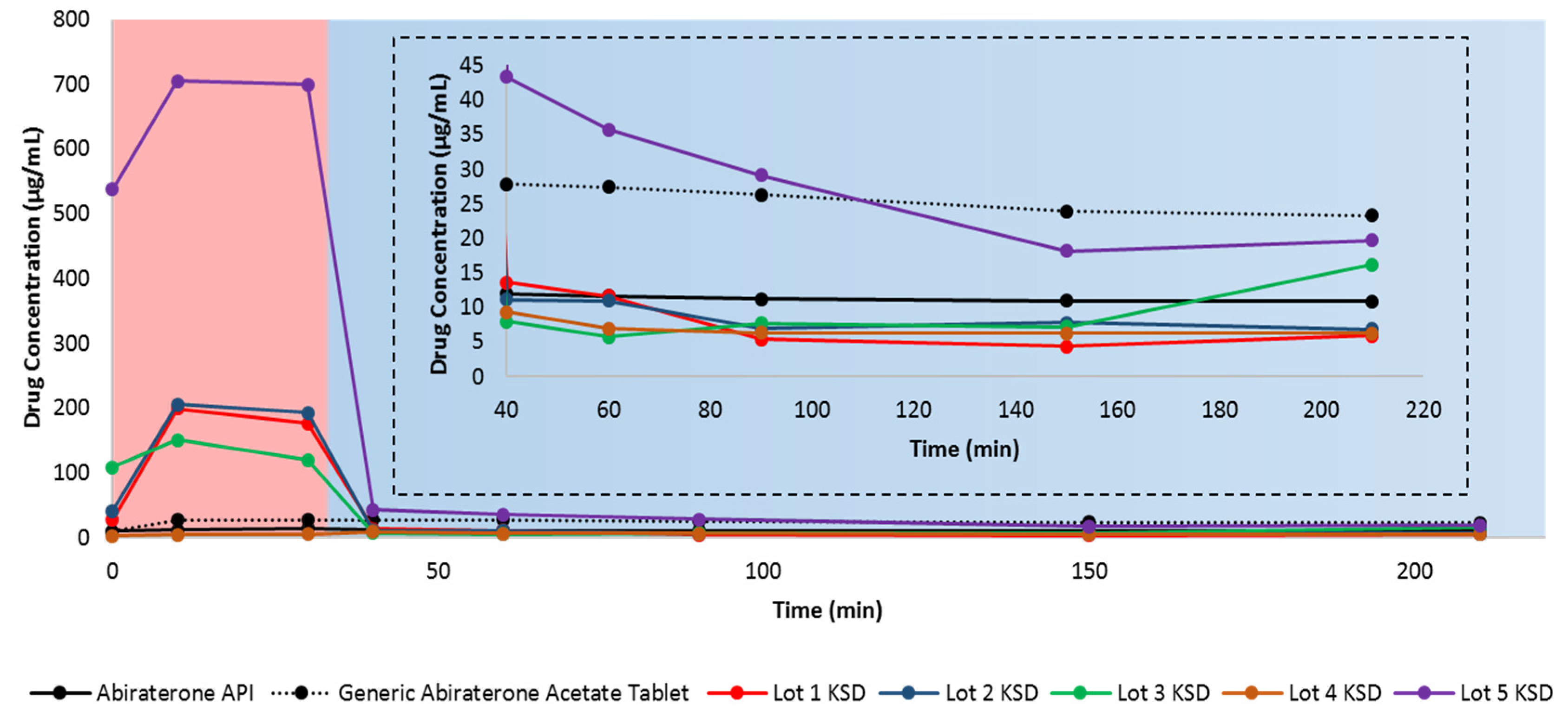
3.3. Dissolution of Binary KSDs
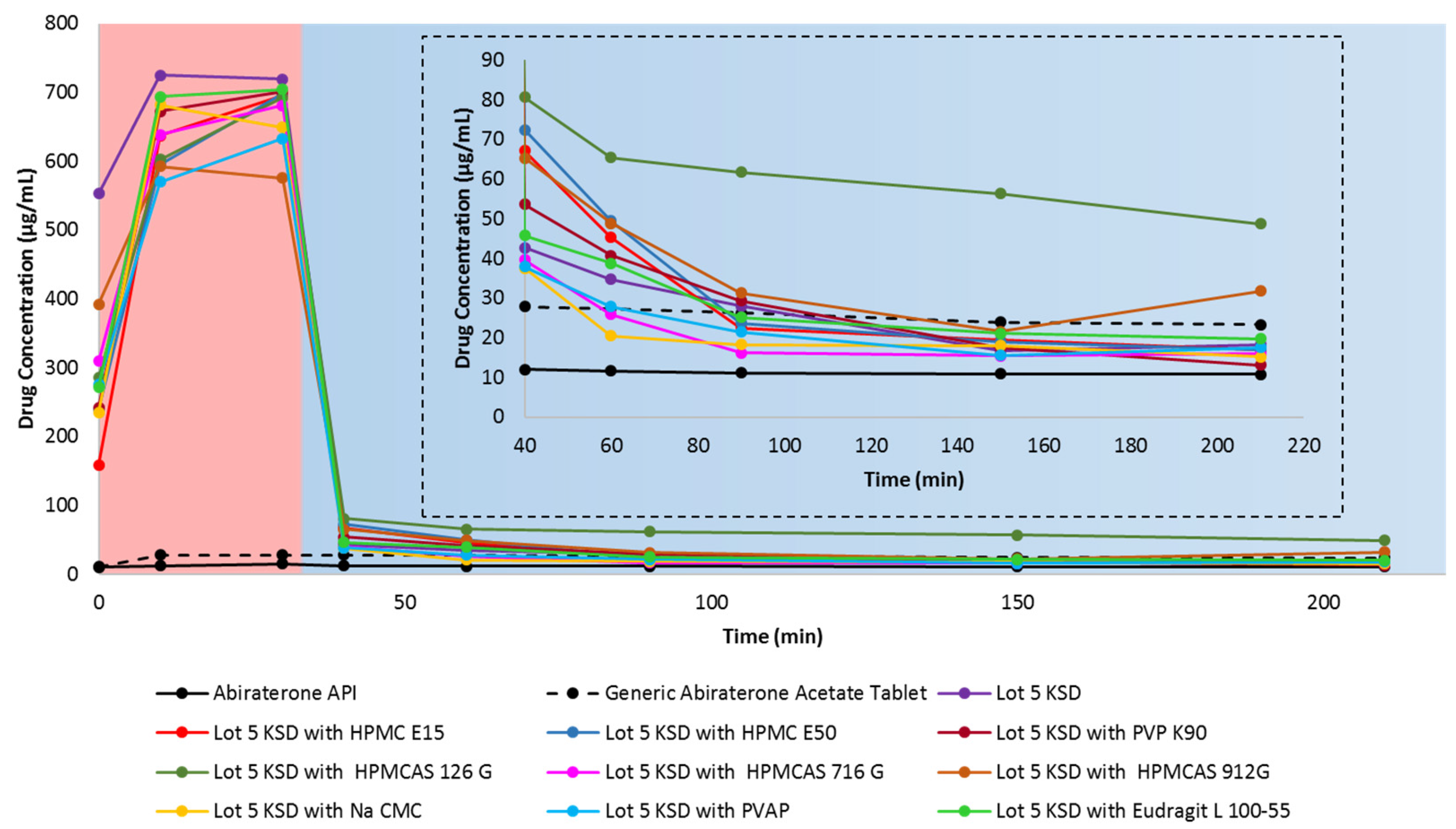
3.4. Selection of a Suitable Ternary Component for KSDs
3.5. Development of Ternary KSDs
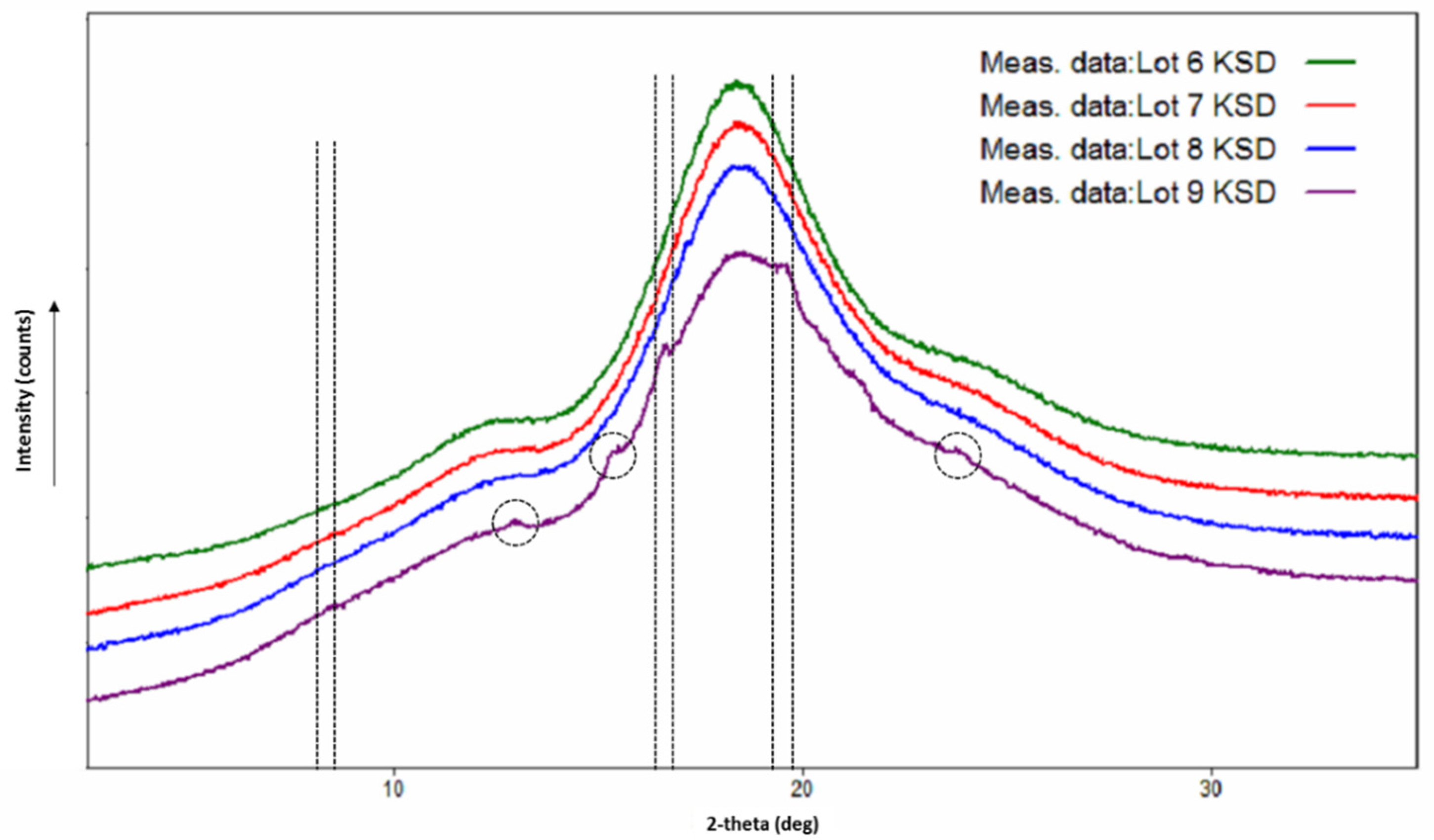
3.6. Physicochemical Characterization of Ternary KSDs
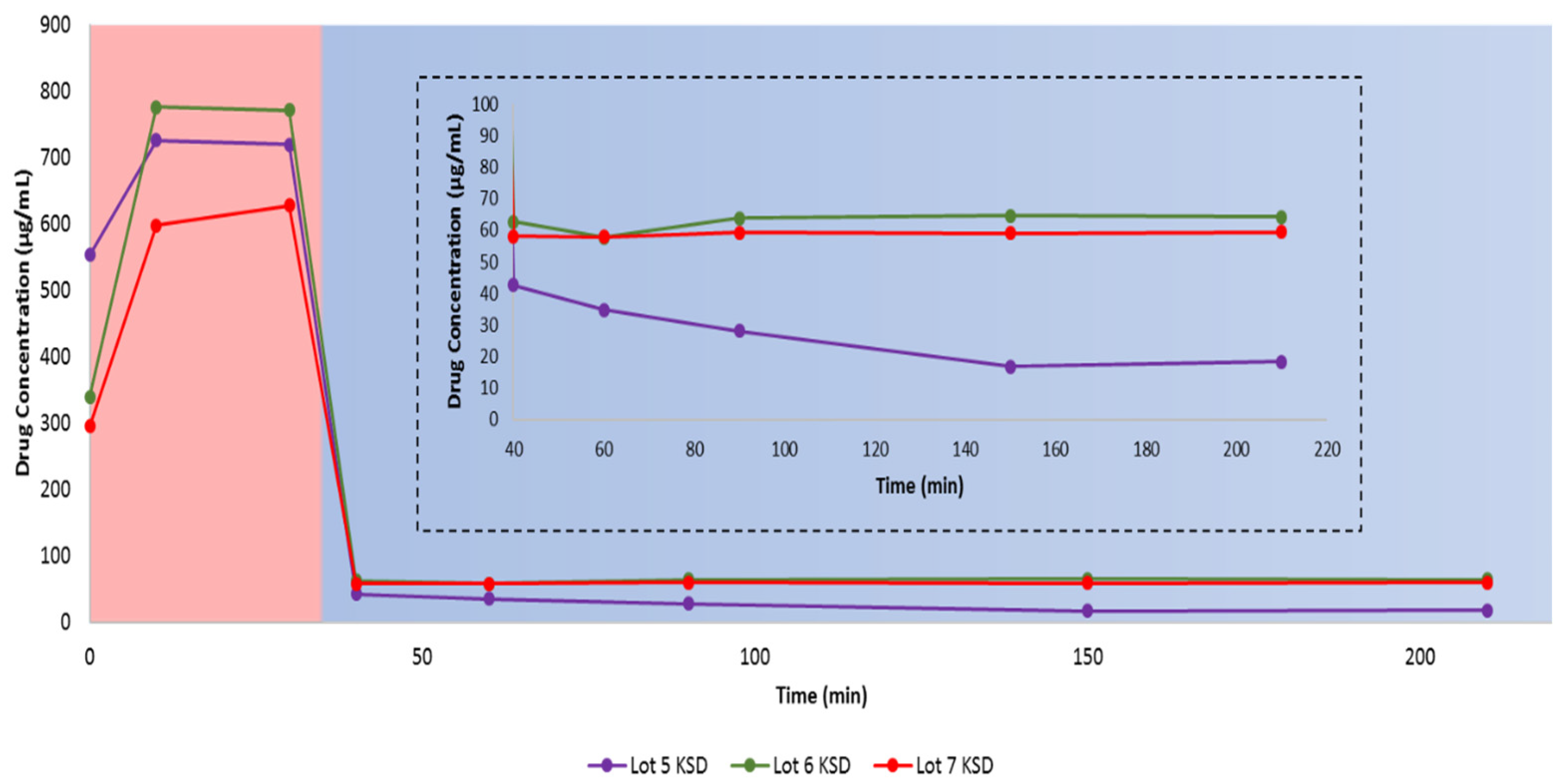
3.7. Dissolution of Ternary KSDs
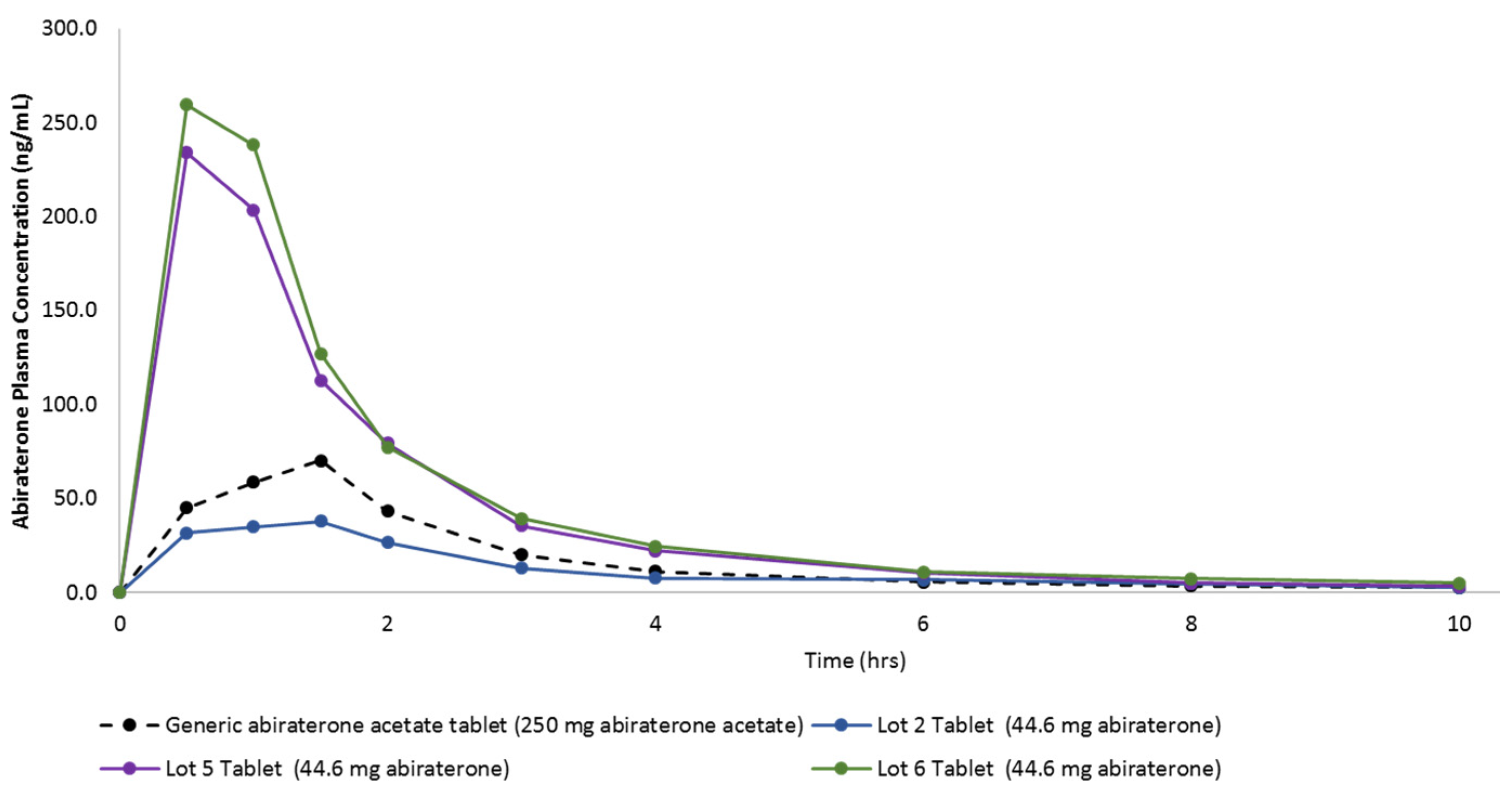
3.8. Pharmacokinetic Study in Beagle Dogs
4. Conclusions
5. Patents
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Fizazi, K.; Tran, N.; Fein, L.; Matsubara, N.; Rodriguez-Antolin, A.; Alekseev, B.Y.; Özgüroğlu, M.; Ye, D.; Feyerabend, S.; Protheroe, A.; et al. Abiraterone plus prednisone in metastatic, castration-sensitive prostate cancer. N. Engl. J. Med. 2017, 377, 352–360. [Google Scholar] [CrossRef] [PubMed]
- Stappaerts, J.; Geboers, S.; Snoeys, J.; Brouwers, J.; Tack, J.; Annaert, P.; Augustijns, P. Rapid conversion of the ester prodrug abiraterone acetate results in intestinal supersaturation and enhanced absorption of abiraterone: In vitro, rat in situ and human in vivo studies. Eur. J. Pharm. Biopharm. 2015, 90, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Rehman, Y.; Rosenberg, J.E. Abiraterone acetate: Oral androgen biosynthesis inhibitor for treatment of castration-resistant prostate cancer. Drug Des. Devel. Ther. 2012, 6, 13–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Attard, G.; Reid, A.H.; Yap, T.A. Phase I clinical trial of a selective inhibitor of CYP17, abiraterone acetate, confirms that castration-resistant prostate cancer commonly remains hormone driven. J. Clin. Oncol. 2008, 26, 4563–4571. [Google Scholar] [CrossRef]
- US FDA. Highlights of Prescribing Information-Zytiga®. 2011-19. Available online: http://www.janssenlabels.com/package-insert/product-monograph/prescribing-information/ZYTIGA-pi.pdf (accessed on 13 April 2020).
- Solymosi, T.; Tóth, F.; Orosz, J.; Basa-Dénes, O.; Angi, R.; Jordán, T.; Ötvös, Z.; Glavinas, H. Solubility measurements at 296 and 310 K and physicochemical characterization of abiraterone and abiraterone acetate. J. Chem. Eng. Data 2018, 63, 4453–4458. [Google Scholar] [CrossRef]
- US FDA. Clinical Pharmacology and Biopharmaceutics Review(s)-Zytiga®. 2010. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2011/202379orig1s000clinpharmr.pdf (accessed on 13 April 2020).
- Chi, K.N.; Spratlin, J.; Kollmannsberger, C.; North, S.; Pankras, C.; Gonzalez, M.; Bernard, A.; Stieltjes, H.; Peng, L.; Jiao, J.; et al. Food effects on abiraterone pharmacokinetics in healthy subjects and patients with metastatic castration-resistant prostate cancer. J. Clin. Pharm. 2015, 55, 1406–1414. [Google Scholar] [CrossRef]
- Xu, X.S.; Ryan, C.J.; Stuyckens, K.; Smith, M.R.; Saad, F.; Griffin, T.W.; Park, Y.C.; Yu, M.K.; De Porre, P.; Vermeulen, A.; et al. Modeling the relationship between exposure to abiraterone and prostate-specific antigen dynamics in patients with metastatic castration-resistant prostate cancer. Clin. Pharmacokinet. 2017, 56, 55–63. [Google Scholar] [CrossRef]
- Li, R.; Evaul, K.; Sharma, K.K.; Chang, K.H.; Yoshimoto, J.; Liu, J.; Auchus, R.J.; Sharifi, N. Abiraterone inhibits 3beta-hydroxysteroid dehydrogenase: A rationale for increasing drug exposure in castration-resistant prostate cancer. Clin. Cancer Res. 2012, 18, 3571–3579. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.S.; Ryan, C.J.; Stuyckens, K.; Smith, M.R.; Saad, F.; Griffin, T.W.; Park, Y.C.; Yu, M.K.; Vermeulen, A.; Poggesi, I.; et al. Correlation between prostate-specific antigen kinetics and overall survival in abiraterone acetate-treated castration-resistant prostate cancer patients. Clin. Cancer Res. 2015, 21, 3170–3177. [Google Scholar] [CrossRef] [Green Version]
- Goldwater, R.; Hussaini, A.; Bosch, B.; Nemeth, P. Comparison of a novel formulation of abiraterone acetate vs. the originator formulation in healthy male subjects: Two randomized, open-label, crossover studies. Clin. Pharmacokinet. 2017, 56, 803–813. [Google Scholar] [CrossRef]
- US FDA. Highlights of Prescribing Information-Yonsa®. 2018. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/210308s000lbl.pdf (accessed on 13 April 2020).
- Stein, C.A.; Levin, R.; Given, R.; Higano, C.S.; Nemeth, P.; Bosch, B.; Chapas-Reed, J.; Dreicer, R. Randomized phase 2 therapeutic equivalence study of abiraterone acetate fine particle formulation vs. originator abiraterone acetate in patients with metastatic castration-resistant prostate cancer: The STAAR study. Urol. Oncol. 2018, 36, 81.e9–81.e16. [Google Scholar] [CrossRef]
- Solymosi, T.; Otvos, Z.; Angi, R.; Ordasi, B.; Jordan, T.; Semsey, S.; Molnar, L.; Ranky, S.; Filipcsei, G.; Heltovics, G.; et al. Development of an abiraterone acetate formulation with improved oral bioavailability guided by absorption modeling based on in vitro dissolution and permeability measurements. Int. J. Pharm. 2017, 532, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Stolarczyk, E.U.; Laszcz, M.; Les, A.; Kubiszewski, M.; Kuziak, K.; Sidoryk, K.; Stolarczyk, K. Design and molecular modeling of abiraterone-functionalized gold nanoparticles. Nanomaterials 2018, 8, 641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sokol, M.B.; Nikolskaya, E.D.; Yabbarov, N.G.; Zenin, V.A.; Faustova, M.R.; Belov, A.V.; Zhunina, O.A.; Mollaev, M.D.; Zabolotsky, A.I.; Tereshchenko, O.G.; et al. Development of novel PLGA nanoparticles with co-encapsulation of docetaxel and abiraterone acetate for a highly efficient delivery into tumor cells. J. Biomed. Mater. Res. B Appl. Biomater. 2019, 107, 1150–1158. [Google Scholar] [CrossRef] [PubMed]
- Williams, H.; Agarwal, P.; Jule, E. Abiraterone Acetate Lipid Formulations. U.S. Patent 15/565,400, 25 March 2016. [Google Scholar]
- Grenier, P.; Vergnault, G. Pharmaceutical Composition Comprising Abiraterone Acetate. U.S. Patent 14/398,841, 3 May 2013. [Google Scholar]
- Legen, I.; Peternel, L.; Novak, S.M.; Homar, M.; Rozman, P.T.; Klancar, U. Self-Microemulsifying Drug Delivery System of Abiraterone or Abiraterone Acetate. Patent WO2014009434, 10 July 2013. [Google Scholar]
- Shah, N.; Sandhu, H.; Choi, D.S.; Chokshi, H.; Malick, A.W. Amorphous Solid Dispersions: Theory and Practice; Springer: New York, NY, USA, 2014. [Google Scholar]
- Jermain, S.V.; Brough, C.; Williams, R.O., III. Amorphous solid dispersions and nanocrystal technologies for poorly water-soluble drug delivery—An update. Int. J. Pharm. 2018, 535, 379–392. [Google Scholar] [CrossRef] [PubMed]
- Van den Mooter, G. The use of amorphous solid dispersions: A formulation strategy to overcome poor solubility and dissolution rate. Drug Discov. Today Technol. 2012, 9, e79–e85. [Google Scholar] [CrossRef] [PubMed]
- Sawicki, E.; Schellens, J.H.M.; Beijnen, J.H.; Nuijen, B. Inventory of oral anticancer agents: Pharmaceutical formulation aspects with focus on the solid dispersion technique. Cancer Treat. Rev. 2016, 50, 247–263. [Google Scholar] [CrossRef] [Green Version]
- Tran, P.; Pyo, Y.-C.; Kim, D.-H.; Lee, S.-E.; Kim, J.-K.; Park, J.-S. Overview of the manufacturing methods of solid dispersion technology for improving the solubility of poorly water-soluble drugs and application to anticancer drugs. Pharmaceutics 2019, 11, 132. [Google Scholar] [CrossRef] [Green Version]
- Gala, U.H.; Miller, D.A.; Williams, R.O. Harnessing the therapeutic potential of anticancer drugs through amorphous solid dispersions. Biochim. Biophys. Acta Rev. Cancer 2020, 1873, 188319. [Google Scholar] [CrossRef]
- Godugu, C.; Doddapaneni, R.; Patel, A.R.; Singh, R.; Mercer, R.; Singh, M. Novel gefitinib formulation with improved oral bioavailability in treatment of A431 skin carcinoma. Pharm. Res. 2016, 33, 137–154. [Google Scholar] [CrossRef] [Green Version]
- Shah, N.; Iyer, R.M.; Mair, H.J.; Choi, D.S.; Tian, H.; Diodone, R.; Fähnrich, K.; Pabst-Ravot, A.; Tang, K.; Scheubel, E. Improved human bioavailability of vemurafenib, a practically insoluble drug, using an amorphous polymer—Stabilized solid dispersion prepared by a solvent—Controlled coprecipitation process. J. Pharm. Sci. 2013, 102, 967–981. [Google Scholar] [CrossRef] [PubMed]
- Meng, F.; Gala, U.; Chauhan, H. Classification of solid dispersions: Correlation to (i) stability and solubility (ii) preparation and characterization techniques. Drug Dev. Ind. Pharm. 2015, 41, 1401–1415. [Google Scholar] [CrossRef] [PubMed]
- Nagy, Z.K.; Balogh, A.; Demuth, B.; Pataki, H.; Vigh, T.; Szabo, B.; Molnar, K.; Schmidt, B.T.; Horak, P.; Marosi, G.; et al. High speed electrospinning for scaled-up production of amorphous solid dispersion of itraconazole. Int. J. Pharm. 2015, 480, 137–142. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Li, H.; Lang, B.; O’Donnell, K.; Zhang, H.; Wang, Z.; Dong, Y.; Wu, C.; Williams, R.O. Formulation and delivery of improved amorphous fenofibrate solid dispersions prepared by thin film freezing. Eur. J. Pharm. Biopharm. 2012, 82, 534–544. [Google Scholar] [CrossRef] [PubMed]
- LaFountaine, J.S.; McGinity, J.W.; Williams, R.O. Challenges and strategies in thermal processing of amorphous solid dispersions: A review. AAPS Pharmscitech. 2016, 17, 43–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haser, A.; Huang, S.; Listro, T.; White, D.; Zhang, F. An approach for chemical stability during melt extrusion of a drug substance with a high melting point. Int. J. Pharm. 2017, 524, 55–64. [Google Scholar] [CrossRef]
- Singh, A.; Van den Mooter, G. Spray drying formulation of amorphous solid dispersions. Adv. Drug Deliv. Rev. 2016, 100, 27–50. [Google Scholar] [CrossRef]
- Ellenberger, D.J.; Miller, D.A.; Williams, R.O. Expanding the application and formulation space of amorphous solid dispersions with KinetiSol®: A review. AAPS Pharmscitech. 2018, 19, 1933–1956. [Google Scholar] [CrossRef]
- Miller, D.A.; DiNunzio, J.C.; Hughey, J.R.; Williams, R.O.; McGinity, J.W. KinetiSol: A New Processing Paradigm for Amorphous Solid Dispersion Systems; Drug development and Delivery: Montville, NJ, USA, 2012. [Google Scholar]
- Brouwers, J.; Brewster, M.E.; Augustijns, P. Supersaturating drug delivery systems: The answer to solubility-limited oral bioavailability? J. Pharm. Sci. 2009, 98, 2549–2572. [Google Scholar] [CrossRef]
- Lim, S.M.; Pang, Z.W.; Tan, H.Y.; Shaikh, M.; Adinarayana, G.; Garg, S. Enhancement of docetaxel solubility using binary and ternary solid dispersion systems. Drug Dev. Ind. Pharm. 2015, 41, 1847–1855. [Google Scholar] [CrossRef]
- Prasad, D.; Chauhan, H.; Atef, E. Amorphous stabilization and dissolution enhancement of amorphous ternary solid dispersions: Combination of polymers showing drug–polymer interaction for synergistic effects. J. Pharm. Sci. 2014, 103, 3511–3523. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Qian, X.; Yang, R. Thermal conductivity of polymers and polymer nanocomposites. Mater. Sci. Eng. R Rep. 2018, 132, 1–22. [Google Scholar] [CrossRef] [Green Version]
- Gopakumar, T.G.; Pagé, D.J.Y.S. Compounding of nanocomposites by thermokinetic mixing. J. Appl. Polym. Sci. 2005, 96, 1557–1563. [Google Scholar] [CrossRef]
- Hughey, J.R.; Keen, J.M.; Miller, D.A.; Brough, C.; McGinity, J.W. Preparation and characterization of fusion processed solid dispersions containing a viscous thermally labile polymeric carrier. Int. J. Pharm. 2012, 438, 11–19. [Google Scholar] [CrossRef]
- LaFountaine, J.S.; Prasad, L.K.; Brough, C.; Miller, D.A.; McGinity, J.W.; Williams, R.O. Thermal processing of PVP- and HPMC-based amorphous solid dispersions. AAPS Pharmscitech. 2016, 17, 120–132. [Google Scholar] [CrossRef] [Green Version]
- Kumar, V.; Yang, T.; Yang, Y. Interpolymer complexation. I. Preparation and characterization of a polyvinyl acetate phthalate-polyvinylpyrrolidone (PVAP-PVP) complex. Int. J. Pharm. 1999, 188, 221–232. [Google Scholar] [CrossRef]
- Rowe, R.C.; Sheskey, P.; Quinn, M. Handbook of Pharmaceutical Excipients, 7th ed.; The Pharmaceutical Press: London, UK; The American Pharmacists Association: Washington, DC, USA, 2012. [Google Scholar]
- Sá Couto, A.R.; Ryzhakov, A.; Loftsson, T. 2-Hydroxypropyl-β-Cyclodextrin aggregates: Identification and development of analytical techniques. Materials 2018, 11, 1971. [Google Scholar] [CrossRef] [Green Version]
- Guo, M.; Hu, X.; Wang, C.; Ai, L. Polysaccharides: Structure and Solubility; IntechOpen Ltd.: London, UK, 2017. [Google Scholar]
- Burke, D.F.; Laughton, C.A.; Snook, C.F.; Neidle, S.; Potter, G.A.; Jarman, M. Active-site conformation of 17-(3-pyridyl)androsta-5,16-dien-3β-ol, a potent inhibitor of the P450 enzyme C17α-hydroxylase/C17-20 lyase. Bioorganic Med. Chem. Lett. 1995, 5, 1125–1130. [Google Scholar] [CrossRef]
- Dow. METHOCEL Cellulose Ethers in Aqueous Systems for Tablet Coating; 2002. Available online: http://msdssearch.dow.com/PublishedLiteratureDOWCOM/dh_004a/0901b8038004ab56.pdf?filepath=/198-00755.pdf?filepath=/198-00755.pdf&fromPage=GetDoc (accessed on 13 April 2020).
- Lee, Y.-E.; Jo, J.; Kim, I.; Yoo, Y.-S. Influence of NaCl concentration on food-waste biochar structure and templating effects. Energies 2018, 11, 2341. [Google Scholar] [CrossRef] [Green Version]
- DeVore, N.M.; Scott, E.E. Structures of cytochrome P450 17A1 with prostate cancer drugs abiraterone and TOK-001. Nature 2012, 482, 116–119. [Google Scholar] [CrossRef] [Green Version]
- Khedr, A.; Darwish, I.; Bamane, F. Analysis of abiraterone stress degradation behavior using liquid chromatography coupled to ultraviolet detection and electrospray ionization mass spectrometry. J. Pharm. Biomed. Anal. 2013, 74, 77–82. [Google Scholar] [CrossRef] [PubMed]
- Roxin, P.; Karlsson, A.; Singh, S.K. Characterization of cellulose acetate phthalate (CAP). Drug Dev. Ind. Pharm. 1998, 24, 1025–1041. [Google Scholar] [CrossRef] [PubMed]
- Saokham, P.; Muankaew, C.; Jansook, P.; Loftsson, T. Solubility of cyclodextrins and drug/cyclodextrin complexes. Molecules 2018, 23, 1161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loftsson, T.; Brewster, M.E. Cyclodextrins as functional excipients: Methods to enhance complexation efficiency. J. Pharm. Sci. 2012, 101, 3019–3032. [Google Scholar] [CrossRef] [PubMed]
- Varma, M.M.; Kumar, P.S. Formulation and evaluation of GLZ tablets containing PVP K30 and hydroxyl propyl beta cyclodextrin solid dispersion. Int. J. Pharm. Sci. Nanotechnol. 2012, 5, 1706–1719. [Google Scholar]
- Wilson, V.; Lou, X.; Osterling, D.J.; Stolarik, D.F.; Jenkins, G.; Gao, W.; Zhang, G.G.Z.; Taylor, L.S. Relationship between amorphous solid dispersion in vivo absorption and in vitro dissolution: Phase behavior during dissolution, speciation, and membrane mass transport. J. Control. Release 2018, 292, 172–182. [Google Scholar] [CrossRef]
- Xu, S.; Dai, W.-G. Drug precipitation inhibitors in supersaturable formulations. Int. J. Pharm. 2013, 453, 36–43. [Google Scholar] [CrossRef]
- Mosquera, L.; Taylor, L.; Santiago, D. Molecular mobility as a tool for understanding the impact of Polyvinylpyrrolidone (polymer) and tpgs (surfactant) in crystallization kinetics of amorphous celecoxib. In Proceedings of the AIChE Annual Meeting, Salt Lake City, UT, USA, 8–13 November 2015. [Google Scholar]
- Sarode, A.L.; Obara, S.; Tanno, F.K.; Sandhu, H.; Iyer, R.; Shah, N. Stability assessment of hypromellose acetate succinate (HPMCAS) NF for application in hot melt extrusion (HME). Carbohydr. Polym. 2014, 101, 146–153. [Google Scholar] [CrossRef]
- Loftsson, T.; Masson, M. The effects of water-soluble polymers on cyclodextrins and cyclodextrin solubilization of drugs. J. Drug Deliv. Sci. Technol. 2004, 14, 35–43. [Google Scholar] [CrossRef]
- Curatolo, W.; Nightingale, J.A.; Herbig, S.M. Utility of hydroxypropylmethylcellulose acetate succinate (HPMCAS) for initiation and maintenance of drug supersaturation in the GI Milieu. Pharm. Res. 2009, 26, 1419–1431. [Google Scholar] [CrossRef]
- Friesen, D.T.; Shanker, R.; Crew, M.; Smithey, D.T.; Curatolo, W.J.; Nightingale, J.A. Hydroxypropyl methylcellulose acetate succinate-based spray-dried dispersions: An overview. Mol. Pharm. 2008, 5, 1003–1019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinto, J.M.O.; Leão, A.F.; Riekes, M.K.; França, M.T.; Stulzer, H.K. HPMCAS as an effective precipitation inhibitor in amorphous solid dispersions of the poorly soluble drug candesartan cilexetil. Carbohydr. Polym. 2018, 184, 199–206. [Google Scholar] [CrossRef] [PubMed]
- Ueda, K.; Higashi, K.; Yamamoto, K.; Moribe, K. The effect of HPMCAS functional groups on drug crystallization from the supersaturated state and dissolution improvement. Int. J. Pharm. 2014, 464, 205–213. [Google Scholar] [CrossRef] [PubMed]
- Ueda, K.; Higashi, K.; Yamamoto, K.; Moribe, K. Equilibrium state at supersaturated drug concentration achieved by hydroxypropyl methylcellulose acetate succinate: Molecular characterization using 1H NMR technique. Mol. Pharm. 2015, 12, 1096–1104. [Google Scholar] [CrossRef] [PubMed]
- Loftsson, T.; Moya-Ortega, M.D.; Alvarez-Lorenzo, C.; Concheiro, A. Pharmacokinetics of cyclodextrins and drugs after oral and parenteral administration of drug/cyclodextrin complexes. J. Pharm. Pharmcol. 2016, 68, 544–555. [Google Scholar] [CrossRef] [PubMed]










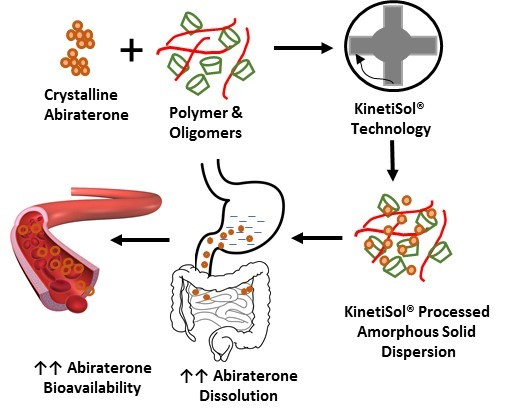{kind=link}
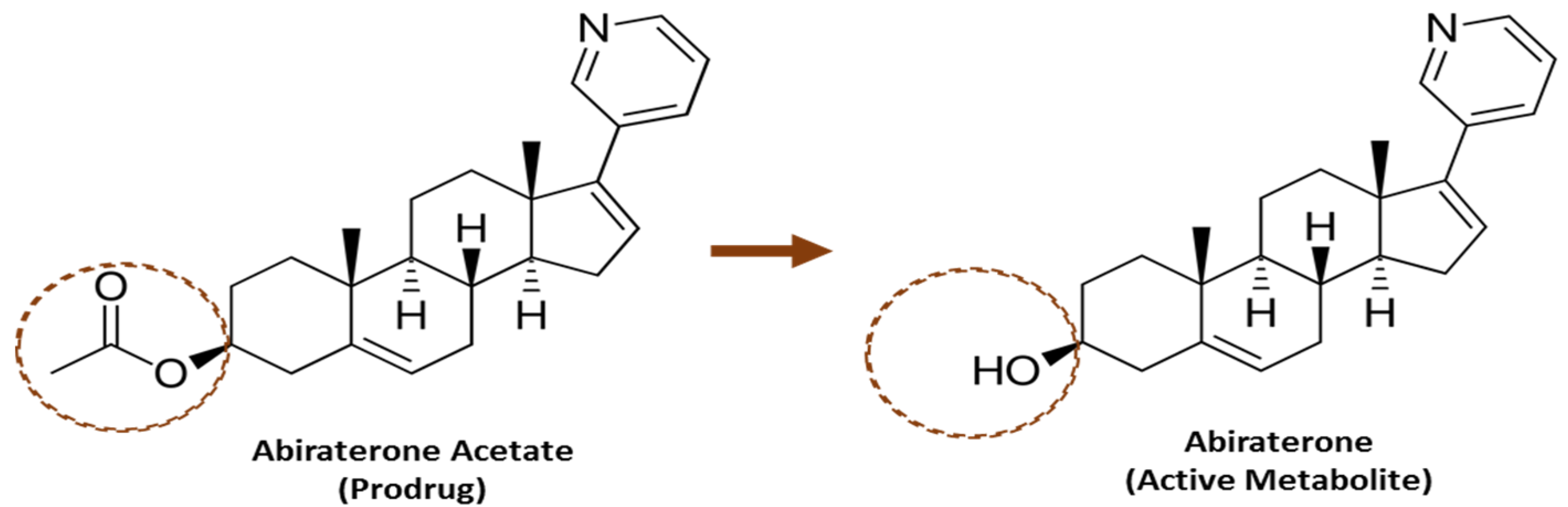{kind=link}
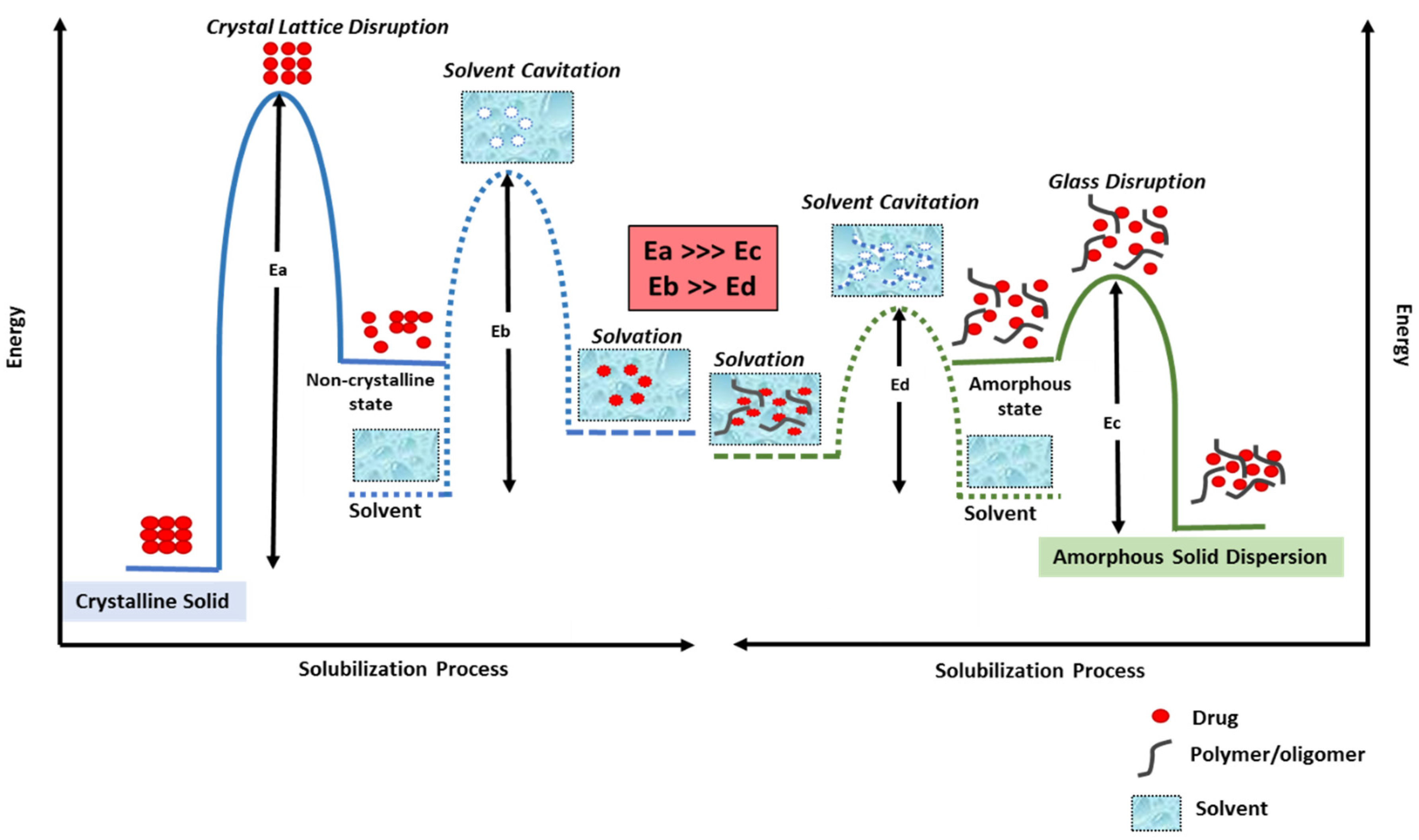{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Polymer/Oligomer | Hydroxy Propyl Methyl Cellulose (HPMC E3) | Hydroxy Propyl Methyl Cellulose (HPMC E5) | Polyvinyl Pyrrolidone (PVP 30) | Polyvinyl Acetate Phthalate (PVAP) | Hydroxy Propyl β Cyclodextrin (HPBCD) |
|---|---|---|---|---|---|
| Commercial Product used | Methocel™ E3 Premium LV | Methocel™ E5 Premium LV | Kollidon® 30 | Phthalavin® | Kleptose® HPB |
| Chemistry | Cellulose based | Cellulose based | Pyrrolidone based | Phthalate based | Glucose based |
| Monomer Structure |  |  |  |  | |
| Architecture | Long-chain Linear Polymer | Long-chain Linear Polymer | Long-chain Linear Polymer | Long-chain Linear Polymer | Short-chain Cyclic Oligomer |
| Molecular Weight | ~20,000 | ~28,700 | ~50,000 | ~60,700 | 1399 |
| Viscosity | 2.4–3.6 (mPa.s: 2% w/v in water at 20 °C) | 4.0–6.0 (mPa.s: 2% w/v in water at 20 °C) | 5.5–8.5 (mPa.s: 10% w/v in water at 20 °C) | 7–11 (mPa.s: in water at 25 °C) | <1.5 (mPa.s: 10% w/v in water at 25 °C) |
| Lot No. | Composition | Batch Size (g) | Processing Temperature (°C) | Shear Stress (Rotational Speed (rpm)) | * Processing Time (Seconds) | Appearance | |
|---|---|---|---|---|---|---|---|
| Drug (% wt) | Primary Polymer/Oligomer (% wt) | ||||||
| 1 | Abiraterone (10) | HPMC E3 (90) | 10 | 160 | 4000, 5000, 6000 | 10 + 10 + 6.9 |  |
| 2 | Abiraterone (10) | HPMC E5 (90) | 10 | 160 | 4000, 5000, 6000 | 10 + 10 + 7.1 |  |
| 3 | Abiraterone (10) | PVP K30 (90) | 10 | 160 | 4000, 5000, 6000 | 10 + 10 + 3.7 |  |
| 4 | Abiraterone (10) | PVAP (90) | 10 | 160 | 4000, 5000, 6000 | 10 + 10 +23 |  |
| 5 | Abiraterone (10) | HPBCD (90) | 10 | 160 | 4000, 5000, 6000 | 10+10+6.3 |  |
| Sample | Percent Rel. AUDCTotal (Relative to Lot 5 KSD) | Percent Rel. AUDC0.01 N HCl (Relative to Lot 5 KSD) | Percent Rel. AUDCFaSSIF (Relative to Lot 5 KSD) |
|---|---|---|---|
| Lot 5 KSD | 100.0 | 100.0 | 100.0 |
| Abiraterone API | 8.4 | 1.8 | 46.2 |
| Generic Abiraterone Acetate Tablet | 18.4 | 3.6 | 104.1 |
| Lot 5 KSD with HPMC E15 | 89.0 | 83.1 | 109.1 |
| Lot 5 KSD with HPMC E50 | 89.2 | 82.1 | 113.8 |
| Lot 5 KSD with PVP K90 | 91.8 | 87.9 | 105.0 |
| Lot 5 KSD with HPMCAS 126 G | 108.9 | 83.5 | 244.8 |
| Lot 5 KSD with HPMCAS 716 G | 85.9 | 86.1 | 77.4 |
| Lot 5 KSD with HPMCAS 912G | 88.1 | 79.7 | 134.6 |
| Lot 5 KSD with Na CMC | 85.4 | 85.8 | 79.1 |
| Lot 5 KSD with PVAP | 80.4 | 78.0 | 85.3 |
| Lot 5 KSD with Eudragit L-100 55 | 93.8 | 90.3 | 107.5 |
| Lot No. | Composition | Batch Size (g) | Processing Temperature (°C) | Shear Stress (Rotational Speed-Rpm) | * Processing Time (Seconds) | Appearance | ||
|---|---|---|---|---|---|---|---|---|
| API (% Wt) | Primary Cyclic Oligomer (% Wt) | Secondary Polymer (% Wt) | ||||||
| 6 | Abiraterone (10) | HPBCD (80) | HPMCAS 126 G (10) | 10 | 160 | 4000, 5000 | 10 + 3.6 |  |
| 7 | Abiraterone (10) | HPBCD (70) | HPMCAS 126 G (20) | 10 | 160 | 4000, 5000 | 10 + 6.3 |  |
| 8 | Abiraterone (10) | HPBCD (60) | HPMCAS 126 G (30) | 10 | 160 | 4000, 5000 | 10 + 5.2 |  |
| 9 | Abiraterone (10) | HPBCD (50) | HPMCAS 126 G (40) | 10 | 160 | 4000 | 9.6 |  |
| Generic Abiraterone Acetate Tablet (250 mg Abiraterone Acetate) | Lot 2 Tablet (44.6 mg Abiraterone) | Lot 5 Tablet (44.6 mg Abiraterone) | Lot 6 Tablet (44.6 mg Abiraterone) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| PK parameters | Units | Average | %CV | Average | %CV | Average | %CV | Average | %CV |
| Cmax | ng/mL | 86.32 | 66.57% | 53.58 | 58.84% | 280.00 | 33.51% | 305.00 | 20.72% |
| Tmax | hr | 1.20 | 37.27% | 1.20 | 55.90% | 0.80 | 34.23% | 0.70 | 39.12% |
| T1/2 | hr | 4.78 | 50.70% | 3.12 | 42.20% | 2.30 | 32.30% | 3.47 | 28.90% |
| AUC0–10 h | ng*hr/mL | 177.29 | 77.53% | 122.21 | 25.64% | 438.01 | 34.29% | 487.58 | 14.11% |
| F Value (Dose Adjusted) | unitless | 1.0 | 3.4 | 12.4 | 13.8 | ||||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gala, U.; Miller, D.; Williams, R.O., III. Improved Dissolution and Pharmacokinetics of Abiraterone through KinetiSol® Enabled Amorphous Solid Dispersions. Pharmaceutics 2020, 12, 357. https://doi.org/10.3390/pharmaceutics12040357
Gala U, Miller D, Williams RO III. Improved Dissolution and Pharmacokinetics of Abiraterone through KinetiSol® Enabled Amorphous Solid Dispersions. Pharmaceutics. 2020; 12(4):357. https://doi.org/10.3390/pharmaceutics12040357
Chicago/Turabian StyleGala, Urvi, Dave Miller, and Robert O. Williams, III. 2020. "Improved Dissolution and Pharmacokinetics of Abiraterone through KinetiSol® Enabled Amorphous Solid Dispersions" Pharmaceutics 12, no. 4: 357. https://doi.org/10.3390/pharmaceutics12040357