Processing of Polyvinyl Acetate Phthalate in Hot-Melt Extrusion—Preparation of Amorphous Solid Dispersions


Abstract
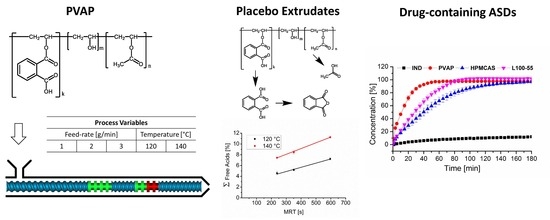
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Thermogravimetric Analysis (TGA)
2.3. Differential Scanning Calorimetry (DSC)
2.4. X-Ray Powder Diffraction (XRPD)
2.5. Screening for Plasticizers
2.6. Melt Rheology
2.7. Hot-Melt Extrusion (HME)
2.8. High Performance Liquid Chromatography (HPLC)
2.9. Dissolution Time
2.10. Non-Sink Dissolution
2.11. Stability Studies
3. Results and Discussion
3.1. Thermal Analysis of Neat PVAP
3.2. Screening for Plasticizers
3.3. Characterization of Placebo Extrudates
3.3.1. Hot-Melt Extrusion and Mean Residence Time
3.3.2. Influence of Process Parameters on Content of Free Acids
3.3.3. Solid-State
3.3.4. Dissolution Time
3.4. Characterization of Drug-Containing ASDs
3.4.1. Drug Content
3.4.2. Solid-State
3.4.3. Non-Sink Dissolution
3.4.4. Stability
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Medium | Saturation Solubility (µg/mL) |
|---|---|
| pH 5.5 | 42.6 ± 0.8 |
| pH 6.8 | 388.5 ± 14.5 |
Appendix B
| Drug | d10 (µm) | d50 (µm) | d90 (µm) |
|---|---|---|---|
| Indomethacin | 23.6 | 75.1 | 265.1 |
| Dipyridamole | 31.4 | 76.4 | 211.0 |
References
- Pham-The, H.; Garrigues, T.; Bermejo, M.; González-Álvarez, I.; Monteagudo, M.C.; Cabrera-Pérez, M.Á. Provisional Classification and in Silico Study of Biopharmaceutical System Based on Caco-2 Cell Permeability and Dose Number. Mol. Pharm. 2013, 10, 2445–2461. [Google Scholar] [CrossRef] [PubMed]
- Repka, M.A.; Majumdar, S.; Kumar Battu, S.; Srirangam, R.; Upadhye, S.B. Applications of hot-melt extrusion for drug delivery. Expert Opin. Drug Deliv. 2008, 5, 1357–1376. [Google Scholar] [CrossRef] [PubMed]
- Breitenbach, J. Melt extrusion: From process to drug delivery technology. Eur. J. Pharm. Biopharm. 2002, 54, 107–117. [Google Scholar] [CrossRef]
- Chiou, W.L.; Riegelman, S. Pharmaceutical Applications of Solid Dispersion Systems. J. Pharm. Sci. 1971, 60, 1281–1302. [Google Scholar] [CrossRef]
- Okonogi, S.; Oguchi, T.; Yonemochi, E.; Puttipipatkhachorn, S.; Yamamoto, K. Improved dissolution of ofloxacin via solid dispersion. Int. J. Pharm. 1997, 156, 175–180. [Google Scholar] [CrossRef]
- Van den Mooter, G. The use of amorphous solid dispersions: A formulation strategy to overcome poor solubility and dissolution rate. Drug Discov. Today Technol. 2012, 9, e79–e85. [Google Scholar] [CrossRef]
- Yoshioka, M.; Hancock, B.C.; Zografi, G. Crystallization of Indomethacin from the Amorphous State below and above Its Glass Transition Temperature. J. Pharm. Sci. 1994, 83, 1700–1705. [Google Scholar] [CrossRef]
- Lin, X.; Hu, Y.; Liu, L.; Su, L.; Li, N.; Yu, J.; Tang, B.; Yang, Z. Physical Stability of Amorphous Solid Dispersions: A Physicochemical Perspective with Thermodynamic, Kinetic and Environmental Aspects. Pharm. Res. 2018, 35. [Google Scholar] [CrossRef]
- Bochmann, E.S.; Neumann, D.; Gryczke, A.; Wagner, K.G. Micro-Scale prediction method for API-Solubility in polymeric matrices and process model for forming amorphous solid dispersion by hot-Melt extrusion. Eur. J. Pharm. Biopharm. 2016, 107, 40–48. [Google Scholar] [CrossRef]
- Hancock, B.C.; Shamblin, S.L.; Zografi, G. Molecular Mobility of Amorphous Pharmaceutical Solids Below Their Glass Transition Temperatures. Pharm. Res. 1995, 12, 799–806. [Google Scholar] [CrossRef]
- Yuan, X.; Sperger, D.; Munson, E.J. Investigating Miscibility and Molecular Mobility of Nifedipine-PVP Amorphous Solid Dispersions Using Solid-State NMR Spectroscopy. Mol. Pharm. 2014, 11, 329–337. [Google Scholar] [CrossRef] [PubMed]
- Vasconcelos, T.; Sarmento, B.; Costa, P. Solid dispersions as strategy to improve oral bioavailability of poor water soluble drugs. Drug Discov. Today 2007, 12, 1068–1075. [Google Scholar] [CrossRef] [PubMed]
- Ilevbare, G.A.; Liu, H.; Edgar, K.J.; Taylor, L.S. Understanding Polymer Properties Important for Crystal Growth Inhibition—Impact of Chemically Diverse Polymers on Solution Crystal Growth of Ritonavir. Cryst. Growth Des. 2012, 12, 3133–3143. [Google Scholar] [CrossRef]
- Vo, C.L.-N.; Park, C.; Lee, B.-J. Current trends and future perspectives of solid dispersions containing poorly water-Soluble drugs. Eur. J. Pharm. Biopharm. 2013, 85, 799–813. [Google Scholar] [CrossRef] [PubMed]
- Van Duong, T.; Van den Mooter, G. The role of the carrier in the formulation of pharmaceutical solid dispersions. Part II: Amorphous carriers. Expert Opin. Drug Deliv. 2016, 13, 1681–1694. [Google Scholar] [CrossRef]
- Miller, D.A.; DiNunzio, J.C.; Yang, W.; McGinity, J.W.; Williams, R.O. Enhanced In Vivo Absorption of Itraconazole via Stabilization of Supersaturation Following Acidic-to-Neutral pH Transition. Drug Dev. Ind. Pharm. 2008, 34, 890–902. [Google Scholar] [CrossRef]
- Sarode, A.L.; Sandhu, H.; Shah, N.; Malick, W.; Zia, H. Hot melt extrusion (HME) for amorphous solid dispersions: Predictive tools for processing and impact of drug–Polymer interactions on supersaturation. Eur. J. Pharm. Sci. 2013, 48, 371–384. [Google Scholar] [CrossRef]
- Zhang, M.; Li, H.; Lang, B.; O′Donnell, K.; Zhang, H.; Wang, Z.; Dong, Y.; Wu, C.; Williams, R.O. Formulation and delivery of improved amorphous fenofibrate solid dispersions prepared by thin film freezing. Eur. J. Pharm. Biopharm. 2012, 82, 534–544. [Google Scholar] [CrossRef]
- Sarabu, S.; Kallakunta, V.R.; Bandari, S.; Batra, A.; Bi, V.; Durig, T.; Zhang, F.; Repka, M.A. Hypromellose acetate succinate based amorphous solid dispersions via hot melt extrusion: Effect of drug physicochemical properties. Carbohydr. Polym. 2020, 233, 115828. [Google Scholar] [CrossRef]
- Balamurali, V.; Pramodkuma, T.M.; Srujana, N.; Venkatesh, M.P.; Gupta, N.V.; Krishna, K.L.; Gangadhara, H.V. pH Sensitive Drug Delivery Systems: A Review. Am. J. Drug Discov. Dev. 2011, 1, 24–48. [Google Scholar] [CrossRef] [Green Version]
- Monschke, M.; Wagner, K.G. Amorphous solid dispersions of weak bases with pH-Dependent soluble polymers to overcome limited bioavailability due to gastric pH variability–An in-vitro approach. Int. J. Pharm. 2019, 564, 162–170. [Google Scholar] [CrossRef] [PubMed]
- Zecevic, D.E.; Meier, R.; Daniels, R.; Wagner, K.-G. Site specific solubility improvement using solid dispersions of HPMC-AS/HPC SSL–Mixtures. Eur. J. Pharm. Biopharm. 2014, 87, 264–270. [Google Scholar] [CrossRef] [PubMed]
- Albadarin, A.B.; Potter, C.B.; Davis, M.T.; Iqbal, J.; Korde, S.; Pagire, S.; Paradkar, A.; Walker, G. Development of stability-Enhanced ternary solid dispersions via combinations of HPMCP and Soluplus® processed by hot melt extrusion. Int. J. Pharm. 2017, 532, 603–611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schilling, S.U.; Lirola, H.L.; Shah, N.H.; Waseem Malick, A.; McGinity, J.W. Influence of plasticizer type and level on the properties of Eudragit® S100 matrix pellets prepared by hot-Melt extrusion. J. Microencapsul. 2010, 27, 521–532. [Google Scholar] [CrossRef]
- Repka, M.A.; Bandari, S.; Kallakunta, V.R.; Vo, A.Q.; McFall, H.; Pimparade, M.B.; Bhagurkar, A.M. Melt extrusion with poorly soluble drugs–An integrated review. Int. J. Pharm. 2018, 535, 68–85. [Google Scholar] [CrossRef]
- Crowley, M.M.; Zhang, F.; Repka, M.A.; Thumma, S.; Upadhye, S.B.; Kumar Battu, S.; McGinity, J.W.; Martin, C. Pharmaceutical Applications of Hot-Melt Extrusion: Part I. Drug Dev. Ind. Pharm. 2007, 33, 909–926. [Google Scholar] [CrossRef]
- Zhang, D.; Lee, Y.-C.; Shabani, Z.; Frankenfeld Lamm, C.; Zhu, W.; Li, Y.; Templeton, A. Processing Impact on Performance of Solid Dispersions. Pharmaceutics 2018, 10, 142. [Google Scholar] [CrossRef] [Green Version]
- Zheng, K.; Lin, Z.; Capece, M.; Kunnath, K.; Chen, L.; Davé, R.N. Effect of Particle Size and Polymer Loading on Dissolution Behavior of Amorphous Griseofulvin Powder. J. Pharm. Sci. 2019, 108, 234–242. [Google Scholar] [CrossRef] [Green Version]
- LaFountaine, J.S.; McGinity, J.W.; Williams, R.O. Challenges and Strategies in Thermal Processing of Amorphous Solid Dispersions: A Review. AAPS PharmSciTech 2016, 17, 43–55. [Google Scholar] [CrossRef] [Green Version]
- Sarode, A.L.; Obara, S.; Tanno, F.K.; Sandhu, H.; Iyer, R.; Shah, N. Stability assessment of hypromellose acetate succinate (HPMCAS) NF for application in hot melt extrusion (HME). Carbohydr. Polym. 2014, 101, 146–153. [Google Scholar] [CrossRef]
- Karandikar, H.; Ambardekar, R.; Kelly, A.; Gough, T.; Paradkar, A. Systematic identification of thermal degradation products of HPMCP during hot melt extrusion process. Int. J. Pharm. 2015, 486, 252–258. [Google Scholar] [CrossRef] [PubMed]
- Meena, A.; Parikh, T.; Gupta, S.S.; Serajuddin, A.T.M. Investigation of thermal and viscoelastic properties of polymers relevant to hot melt extrusion-II: Cellulosic polymers. J. Excip. Food Chem. 2014, 5, 46–55. [Google Scholar]
- Parikh, T.; Gupta, S.S.; Meena, A.; Serajuddin, A.T.M. Investigation of thermal and viscoelastic properties of polymers relevant to hot melt extrusion-III: Polymethacrylates and polymethacrylic acid based polymers. J. Excip. Food Chem. 2014, 5, 56–64. [Google Scholar]
- Mehuys, E.; Remon, J.-P.; Vervaet, C. Production of enteric capsules by means of hot-Melt extrusion. Eur. J. Pharm. Sci. 2005, 24, 207–212. [Google Scholar] [CrossRef] [PubMed]
- DiNunzio, J.C.; Miller, D.A.; Yang, W.; McGinity, J.W.; Williams, R.O. Amorphous Compositions Using Concentration Enhancing Polymers for Improved Bioavailability of Itraconazole. Mol. Pharm. 2008, 5, 968–980. [Google Scholar] [CrossRef]
- Mahmoudi, Z.N.; Upadhye, S.B.; Ferrizzi, D.; Rajabi-Siahboomi, A.R. In Vitro Characterization of a Novel Polymeric System for Preparation of Amorphous Solid Drug Dispersions. AAPS J. 2014, 16, 685–697. [Google Scholar] [CrossRef] [Green Version]
- Peng, T.; She, Y.; Zhu, C.; Shi, Y.; Huang, Y.; Niu, B.; Bai, X.; Pan, X.; Wu, C. Influence of Polymers on the Physical and Chemical Stability of Spray-Dried Amorphous Solid Dispersion: Dipyridamole Degradation Induced by Enteric Polymers. AAPS PharmSciTech 2018, 19, 2620–2628. [Google Scholar] [CrossRef]
- Puri, V.; Dantuluri, A.K.; Bansal, A.K. Barrier Coated Drug Layered Particles for Enhanced Performance of Amorphous Solid Dispersion Dosage Form. J. Pharm. Sci. 2012, 101, 342–353. [Google Scholar] [CrossRef]
- Dong, Z.; Choi, D.S. Hydroxypropyl Methylcellulose Acetate Succinate: Potential Drug–Excipient Incompatibility. AAPS PharmSciTech 2008, 9, 991–997. [Google Scholar] [CrossRef] [Green Version]
- Zecevic, D.E.; Wagner, K.G. Rational Development of Solid Dispersions via Hot-Melt Extrusion Using Screening, Material Characterization, and Numeric Simulation Tools. J. Pharm. Sci. 2013, 102, 2297–2310. [Google Scholar] [CrossRef]
- Gupta, S.S.; Solanki, N.; Serajuddin, A.T.M. Investigation of Thermal and Viscoelastic Properties of Polymers Relevant to Hot Melt Extrusion, IV: AffinisolTM HPMC HME Polymers. AAPS PharmSciTech 2016, 17, 148–157. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Obara, S.; Ioannidis, N.; Suwardie, J.; Gogos, C.; Kikuchi, S. Understanding the Processing Window of Hypromellose Acetate Succinate for Hot-Melt Extrusion, Part I: Polymer Characterization and Hot-Melt Extrusion. Adv. Polym. Technol. 2018, 37, 154–166. [Google Scholar] [CrossRef]
- O′Brien, M.; McCauley, J.; Cohen, E. Indomethacin. In Analytical Profiles of Drug Substances; Elsevier: Amsterdam, The Netherlands, 1984; Volume 13, pp. 211–238. ISBN 978-0-12-260813-1. [Google Scholar]
- Abuhelwa, A.Y.; Foster, D.J.R.; Upton, R.N. A Quantitative Review and Meta-Models of the Variability and Factors Affecting Oral Drug Absorption—Part I: Gastrointestinal pH. AAPS J. 2016, 18, 1309–1321. [Google Scholar] [CrossRef] [PubMed]
- Vertzoni, M.; Augustijns, P.; Grimm, M.; Koziolek, M.; Lemmens, G.; Parrott, N.; Pentafragka, C.; Reppas, C.; Rubbens, J.; Van Den Abeele, J.; et al. Impact of regional differences along the gastrointestinal tract of healthy adults on oral drug absorption: An UNGAP review. Eur. J. Pharm. Sci. 2019, 134, 153–175. [Google Scholar] [CrossRef]










| HME Conditions | Free Acids (%) | ||||||
|---|---|---|---|---|---|---|---|
| Temperature (°C) | Feed Rate (g/min) | Torque (%) | SME (kWh/t) | PA | PAH | AA | Sum |
| Physical blend | - | - | - | 0.70 ± 0.02 | 0 | 0.66 ± 0.02 | 1.36 |
| 120 | 1 | 63.3 | 1447.1 | 5.67 ± 0.16 | 0.49 ± 0.04 | 1.06 ± 0.01 | 7.22 |
| 120 | 2 | 70.0 | 811.2 | 4.04 ± 0.15 | 0.16 ± 0.01 | 0.99 ± 0.03 | 5.19 |
| 120 | 3 | 83.3 | 656.8 | 3.65 ± 0.26 | 0.16 ± 0.01 | 0.77 ± 0.05 | 4.58 |
| 140 | 1 | 56.6 | 1271.7 | 9.18 ± 0.11 | 0.98 ± 0.02 | 1.07 ± 0.01 | 11.23 |
| 140 | 2 | 52.0 | 575.7 | 6.70 ± 0.12 | 0.75 ± 0.17 | 1.01 ± 0.02 | 8.46 |
| 140 | 3 | 53.4 | 396.0 | 6.09 ± 0.08 | 0.42 ± 0.02 | 0.93 ± 0.03 | 7.44 |
| Process Condition | pH 6.8 | pH 5.5 |
|---|---|---|
| Dissolution Time (min) | Dissolution Time (min) | |
| Raw material | 7.5 | 10 |
| 120 °C, 1 g/min | 20 | 15 |
| 120 °C, 2 g/min | 10 | 15 |
| 120 °C, 3 g/min | 5 | 15 |
| 140 °C, 1 g/min | incomplete | incomplete |
| 140 °C, 2 g/min | 10 | 20 |
| 140 °C, 3 g/min | 10 | 15 |
| Composition | HME Conditions | Content (Assay) (%) | ||
|---|---|---|---|---|
| Temperature (°C) | Feed Rate (%) | Torque (%) | ||
| IND/PEG3000/PVAP 10/10/80 | 120 | 1 | 49.3 | 99.5 ± 3.21 |
| 120 | 2 | 60.0 | 99.3 ± 1.13 | |
| 120 | 3 | 66.7 | 104.2 ± 0.04 | |
| DPD/PEG3000/PVAP 10/10/80 | 120 | 1 | 50.0 | 0.85 ± 0.01 |
| 120 | 2 | 56.6 | 4.3 ± 0.01 | |
| 120 | 3 | 66.7 | 7.5 ± 0.12 | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Monschke, M.; Kayser, K.; Wagner, K.G. Processing of Polyvinyl Acetate Phthalate in Hot-Melt Extrusion—Preparation of Amorphous Solid Dispersions. Pharmaceutics 2020, 12, 337. https://doi.org/10.3390/pharmaceutics12040337
Monschke M, Kayser K, Wagner KG. Processing of Polyvinyl Acetate Phthalate in Hot-Melt Extrusion—Preparation of Amorphous Solid Dispersions. Pharmaceutics. 2020; 12(4):337. https://doi.org/10.3390/pharmaceutics12040337
Chicago/Turabian StyleMonschke, Marius, Kevin Kayser, and Karl G. Wagner. 2020. "Processing of Polyvinyl Acetate Phthalate in Hot-Melt Extrusion—Preparation of Amorphous Solid Dispersions" Pharmaceutics 12, no. 4: 337. https://doi.org/10.3390/pharmaceutics12040337