MHC Class I Stability is Modulated by Cell Surface Sialylation in Human Dendritic Cells

 , , , and
, , , and
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Generation of Human DCs
2.2. Cell Lines
2.3. Sialidase Treatment
2.4. Anti-MHC-I antibodies
2.5. Flow Cytometry
2.6. Confocal Laser Scanning Microscopy
2.7. Protein Extraction, Immunoprecipitation, and Western Blotting
2.8. Mass Spectrometry and Bioinformatics
2.9. Peptide Stability Assays
2.10. Surface MHC-I Stability Assays
2.11. Gene Expression Assays
2.12. DC: T Cell Interaction/ Doublet Analysis
2.13. Cytokine Production Evaluation
2.14. Statistical Analysis
3. Results
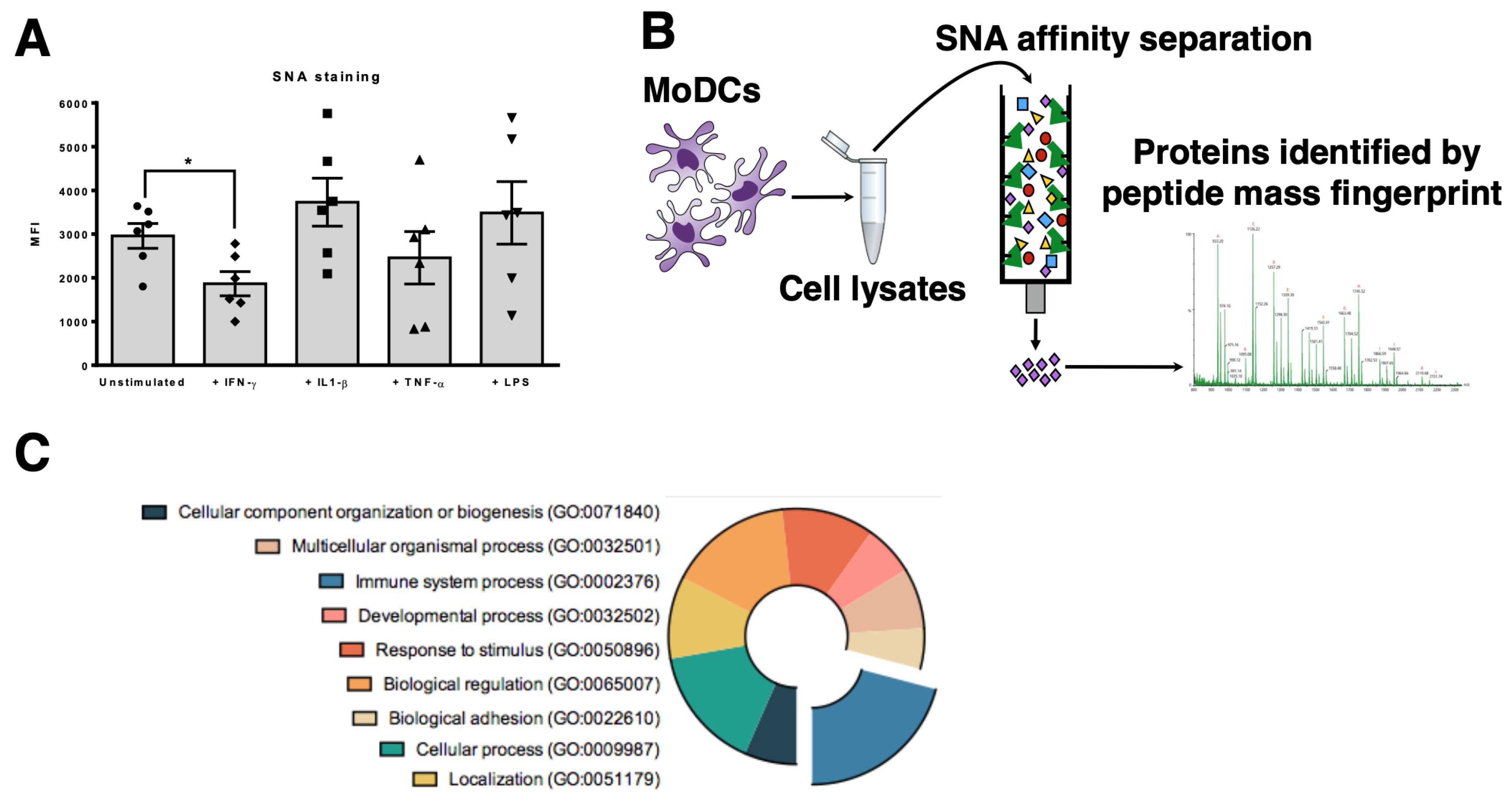
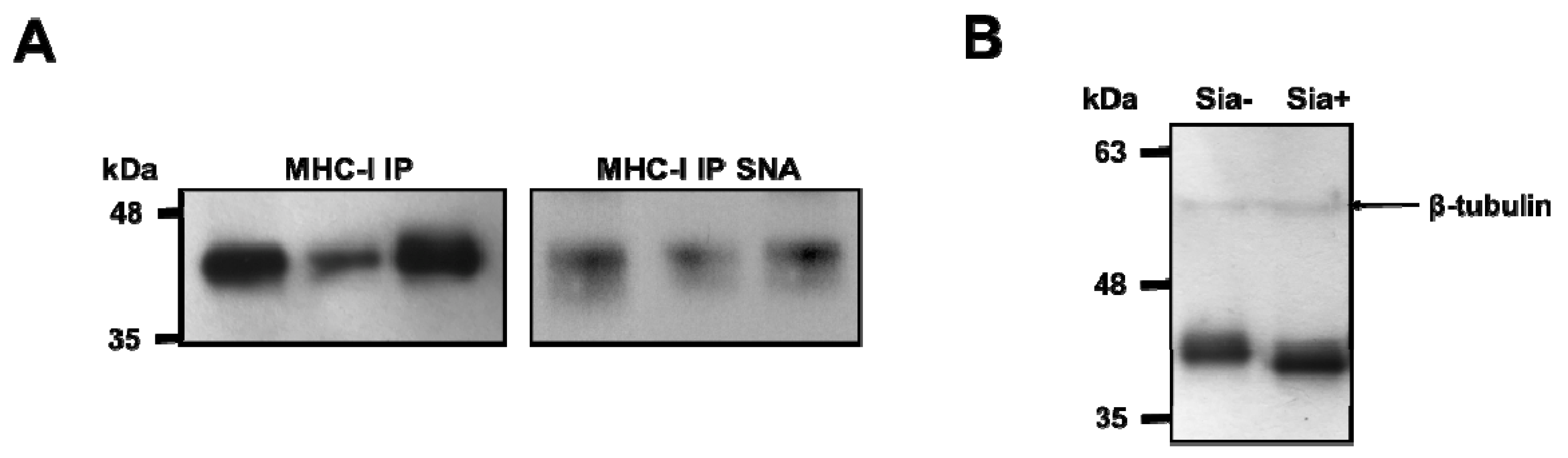
3.1. MHC Class I Molecules on Human Dcs are Sialylated
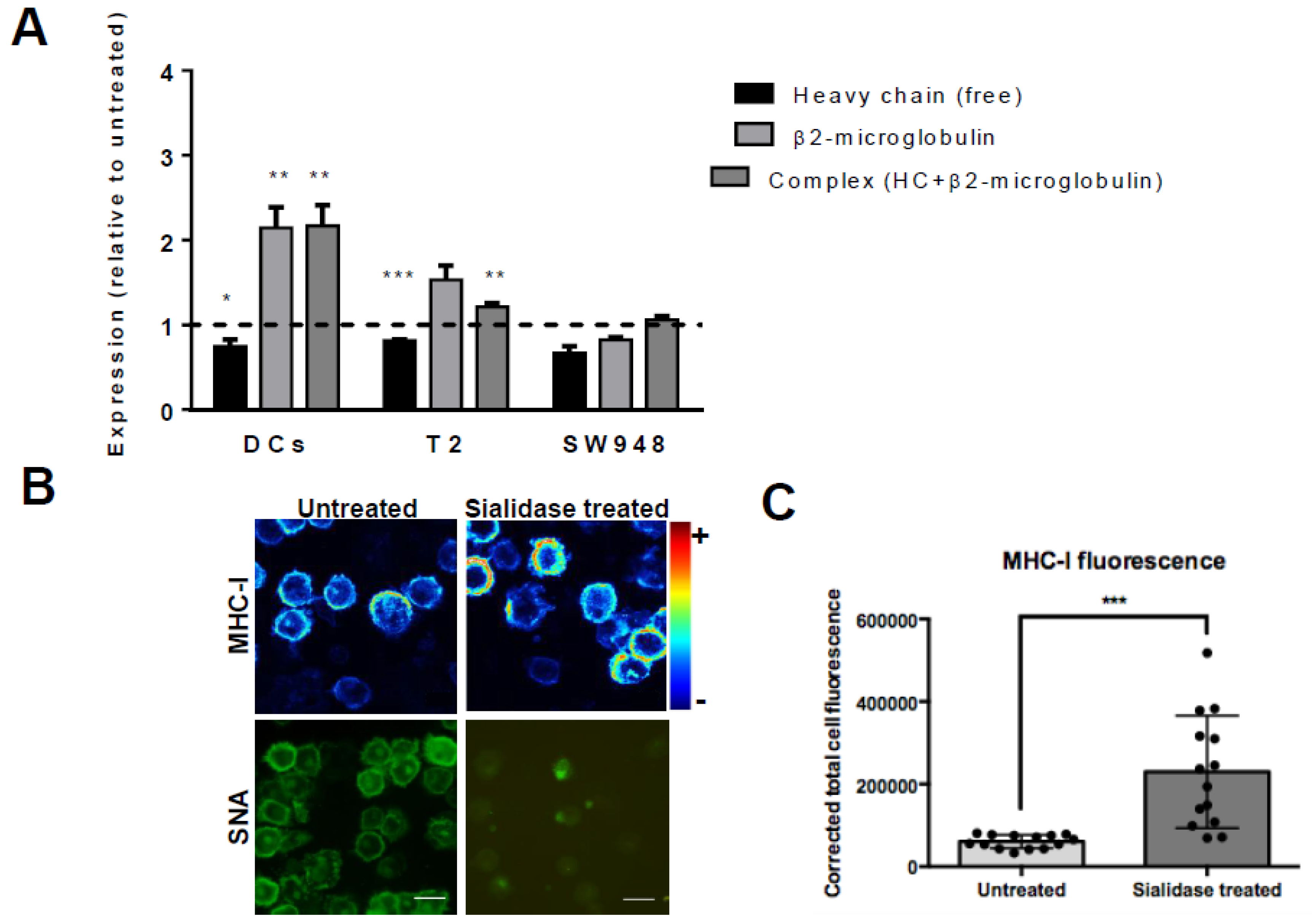
3.2. Desialylation Increases the Number of Mhc Class I-Β2m Dimer Complex in Dcs and T2 Cell Line
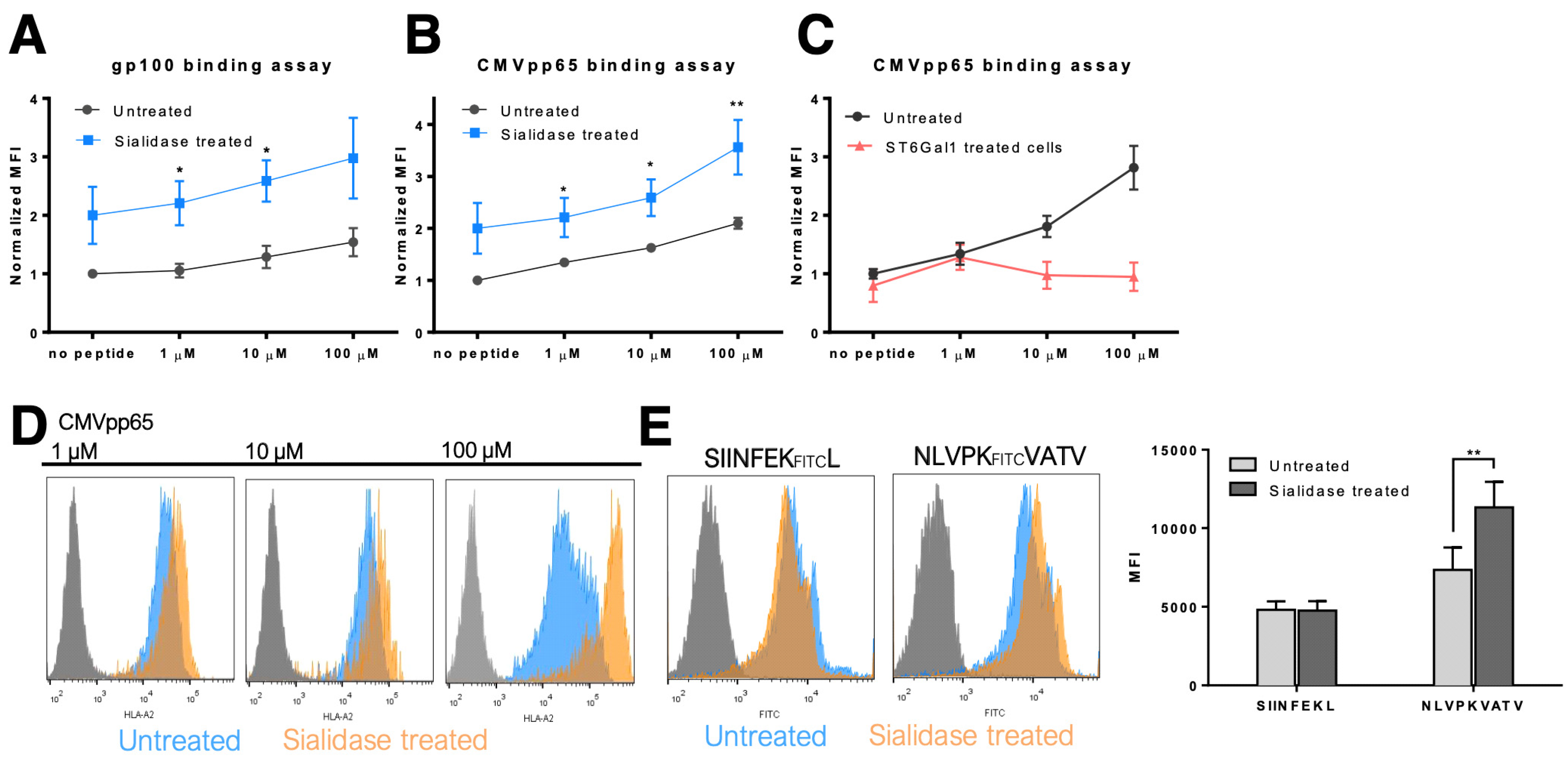
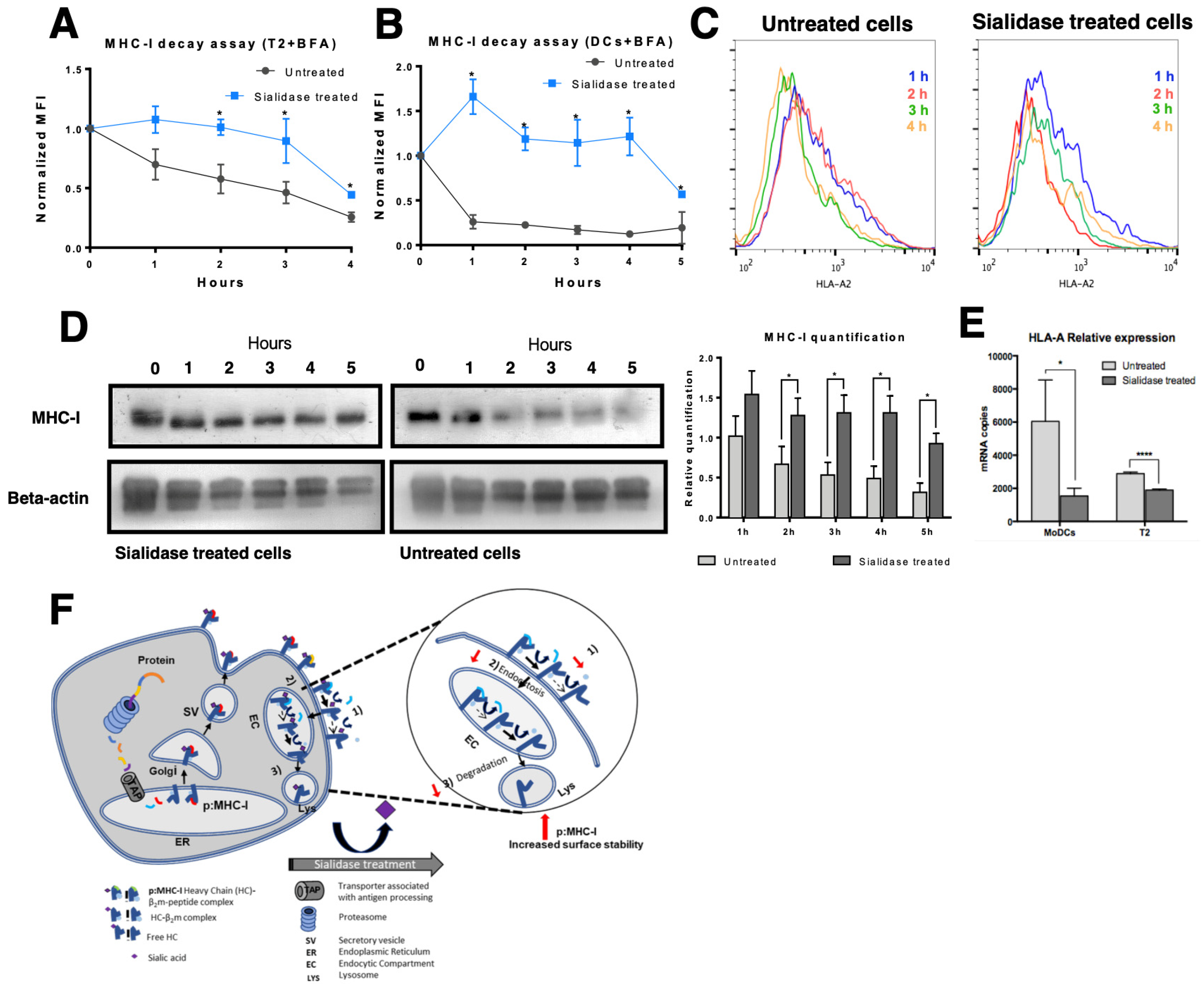
3.3. MHC Class I Molecules Show Increased Peptide Stability at the Cell Surface after Desialylation
3.4. MHC Class I Molecules Show Slower Turnover after Desialylation
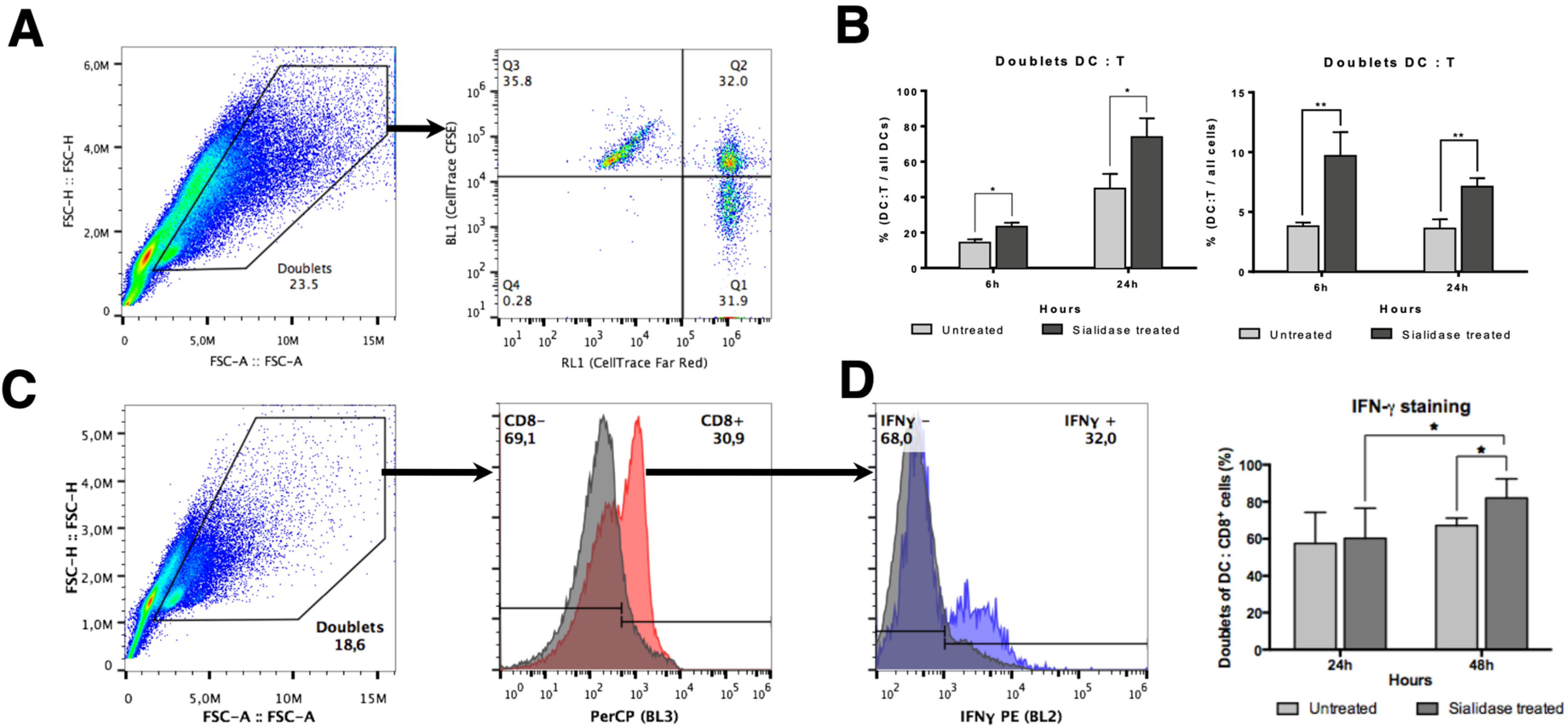
3.5. DC:T Cell Doublet Formation and T Cell Activation are Improved by Desialylated DCs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Rossi, M.; Young, J.W. Human Dendritic Cells: Potent Antigen-Presenting Cells at the Crossroads of Innate and Adaptive Immunity. J. Immunol. 2005, 175, 1373–1381. [Google Scholar] [CrossRef]
- Palucka, K.; Banchereau, J. Dendritic Cells: A Link Between Innate and Adaptive Immunity. J. Clin. Immunol. 1999, 19, 12–25. [Google Scholar] [CrossRef]
- Anguille, S.; Smits, E.L.; Lion, E.; Van Tendeloo, V.F.; Berneman, Z.N. Clinical use of dendritic cells for cancer therapy. Lancet Oncol. 2014, 15, e257–e267. [Google Scholar] [CrossRef]
- Garg, A.D.; Coulie, P.G.; Van den Eynde, B.J.; Agostinis, P. Integrating Next-Generation Dendritic Cell Vaccines into the Current Cancer Immunotherapy Landscape. Trends Immunol. 2017, 38, 577–593. [Google Scholar] [CrossRef] [PubMed]
- Bol, K.F.; Schreibelt, G.; Gerritsen, W.R.; de Vries, I.J.M.; Figdor, C.G. Dendritic Cell-Based Immunotherapy: State of the Art and Beyond. Clin. Cancer Res. 2016, 22, 1897–1906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Förster, R.; Schubel, A.; Breitfeld, D.; Kremmer, E.; Renner-Müller, I.; Wolf, E.; Lipp, M. CCR7 coordinates the primary immune response by establishing functional microenvironments in secondary lymphoid organs. Cell 1999, 99, 23–33. [Google Scholar] [CrossRef] [Green Version]
- Valitutti, S.; Miller, S.; Cella, M.; Padovan, E.; Lanzavecchia, A. Serial triggering of many T-cell receptors by a few peptide-MHC complexes. Nature 1995, 375, 148–151. [Google Scholar] [CrossRef] [PubMed]
- Matsui, K.; Boniface, J.; Reay, P.; Schild, H.; Fazekas de St. Groth, B.; Davis, M. Low affinity interaction of peptide-MHC complexes with T cell receptors. Science 1991, 254, 1788–1791. [Google Scholar] [CrossRef]
- Hansen, T.H.; Leet, D.R. Mechanism of Class I Assembly with β2 Microglobulin and Loading with Peptide. Adv. Immunol. 1997, 64, 105–137. [Google Scholar]
- Banchereau, J.; Briere, F.; Caux, C.; Davoust, J.; Lebecque, S.; Liu, Y.J.; Pulendran, B.; Palucka, K. Immunobiology of dendritic cells. Annu. Rev. Immunol. 2000, 18, 767–811. [Google Scholar] [CrossRef]
- Mellman, I.; Steinman, R.M. Dendritic cells: Specialized and regulated antigen processing machines. Cell 2001, 106, 255–258. [Google Scholar] [CrossRef] [Green Version]
- Varki, A.; Gagneux, P. Multifarious roles of sialic acids in immunity. Ann. N. Y. Acad. Sci. 2012, 1253, 16–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bax, M.; García-Vallejo, J.J.; Jang-Lee, J.; North, S.J.; Gilmartin, T.J.; Hernández, G.; Crocker, P.R.; Leffler, H.; Head, S.R.; Haslam, S.M.; et al. Dendritic cell maturation results in pronounced changes in glycan expression affecting recognition by siglecs and galectins. J. Immunol. 2007, 179, 8216–8224. [Google Scholar] [CrossRef] [PubMed]
- Videira, P.A.; Amado, I.F.; Crespo, H.J.; Algueró, M.C.; Dall’Olio, F.; Cabral, M.G.; Trindade, H. Surface alpha 2–3–- and alpha 2–6–sialylation of human monocytes and derived dendritic cells and its influence on endocytosis. Glycoconj. J. 2008, 25, 259–268. [Google Scholar] [CrossRef]
- Cabral, M.G.; Piteira, A.R.; Silva, Z.; Ligeiro, D.; Brossmer, R.; Videira, P.A. Human dendritic cells contain cell surface sialyltransferase activity. Immunol. Lett. 2010, 131, 89–96. [Google Scholar] [CrossRef]
- Crespo, H.J.; Guadalupe Cabral, M.; Teixeira, A.V.; Lau, J.T.Y.; Trindade, H.; Videira, P.A. Effect of sialic acid loss on dendritic cell maturation. Immunology 2009, 128, e621–e631. [Google Scholar] [CrossRef]
- Silva, M.; Silva, Z.; Marques, G.; Ferro, T.; Gonçalves, M.; Monteiro, M.; van Vliet, S.J.; Mohr, E.; Lino, A.C.; Fernandes, A.R.; et al. Sialic acid removal from dendritic cells improves antigen cross-presentation and boosts anti-tumor immune responses. Oncotarget 2016, 7, 41053–41066. [Google Scholar] [CrossRef]
- Carrascal, M.A.; Severino, P.F.; Guadalupe Cabral, M.; Silva, M.; Ferreira, J.A.; Calais, F.; Quinto, H.; Pen, C.; Ligeiro, D.; Santos, L.L.; et al. Sialyl Tn-expressing bladder cancer cells induce a tolerogenic phenotype in innate and adaptive immune cells. Mol. Oncol. 2014, 8, 753–765. [Google Scholar] [CrossRef]
- Barnstable, C.J.; Bodmer, W.F.; Brown, G.; Galfre, G.; Milstein, C.; Williams, A.F.; Ziegler, A. Production of monoclonal antibodies to group A erythrocytes, HLA and other human cell surface antigens-new tools for genetic analysis. Cell 1978, 14, 9–20. [Google Scholar] [CrossRef]
- Stam, N.J.; Spits, H.; Ploegh, H.L. Monoclonal antibodies raised against denatured HLA-B locus heavy chains permit biochemical characterization of certain HLA-C locus products. J. Immunol. 1986, 137, 2299–2306. [Google Scholar]
- Brodsky, F.M.; Bodmer, W.F.; Parham, P. Characterization of a monoclonal anti-β2-microglobulin antibody and its use in the genetic and biochemical analysis of major histocompatibility antigens. Eur. J. Immunol. 1979, 9, 536–545. [Google Scholar] [CrossRef] [PubMed]
- Brodsky, F.M.; Parham, P.; Barnstable, C.J.; Crumpton, M.J.; Bodmer, W.F. Monoclonal antibodies for analysis of the HLA system. Immunol. Rev. 1979, 47, 3–61. [Google Scholar] [CrossRef] [PubMed]
- Pirro, M.; Schoof, E.; van Vliet, S.J.; Rombouts, Y.; Stella, A.; de Ru, A.; Mohammed, Y.; Wuhrer, M.; van Veelen, P.A.; Hensbergen, P.J. Glycoproteomic Analysis of MGL-Binding Proteins on Acute T-Cell Leukemia Cells. J. Proteome Res. 2019, 18, 1125–1132. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, J.A.; Daniel-Da-Silva, A.L.; Alves, R.M.P.; Duarte, D.; Vieira, I.; Santos, L.L.; Vitorino, R.; Amado, F. Synthesis and optimization of lectin functionalized nanoprobes for the selective recovery of glycoproteins from human body fluids. Anal. Chem. 2011, 83, 7035–7043. [Google Scholar] [CrossRef]
- Azevedo, R.; Silva, A.M.N.; Reis, C.A.; Santos, L.L.; Ferreira, J.A. In silico approaches for unveiling novel glycobiomarkers in cancer. J. Proteomics 2018, 171, 95–106. [Google Scholar] [CrossRef]
- Lam, P.V.N.; Goldman, R.; Karagiannis, K.; Narsule, T.; Simonyan, V.; Soika, V.; Mazumder, R. Structure-based Comparative Analysis and Prediction of N-linked Glycosylation Sites in Evolutionarily Distant Eukaryotes. Genom. Proteom. Bioinform. 2013, 11, 96–104. [Google Scholar] [CrossRef] [Green Version]
- Steentoft, C.; Vakhrushev, S.Y.; Joshi, H.J.; Kong, Y.; Vester-Christensen, M.B.; Schjoldager, K.T.; Lavrsen, K.; Dabelsteen, S.; Pedersen, N.B.; Marcos-Silva, L.; et al. Precision mapping of the human O-GalNAc glycoproteome through SimpleCell technology. EMBO J. 2013, 32, 1478–1488. [Google Scholar] [CrossRef] [Green Version]
- Nasirikenari, M.; Veillon, L.; Collins, C.C.; Azadi, P.; Lau, J.T.Y. Remodeling of marrow hematopoietic stem and progenitor cells by non-self ST6Gal-1 sialyltransferase. J. Biol. Chem. 2014, 289, 7178–7189. [Google Scholar] [CrossRef] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Parham, P.; Barnstable, C.J.; Bodmer, W.F. Use of a monoclonal antibody (W6/32) in structural studies of HLA-A.,B.,C., antigens. J. Immunol. 1979, 123, 342–349. [Google Scholar]
- Smith, K.D.; Lutz, C.T. Peptide-dependent expression of HLA-B7 on antigen processing-deficient T2 cells. J. Immunol. 1996, 156, 3755–3764. [Google Scholar] [PubMed]
- Skipper, J.C.; Gulden, P.H.; Hendrickson, R.C.; Harthun, N.; Caldwell, J.A.; Shabanowitz, J.; Engelhard, V.H.; Hunt, D.F.; Slingluff, C.L., Jr. Mass-spectrometric evaluation of HLA-A*0201-associated peptides identifies dominant naturally processed forms of CTL epitopes from MART-1 and gp100. Int J. Cancer 1999, 82, 669–677. [Google Scholar] [CrossRef]
- Hoppes, R.; Oostvogels, R.; Luimstra, J.J.; Wals, K.; Toebes, M.; Bies, L.; Ekkebus, R.; Rijal, P.; Celie, P.H.N.; Huang, J.H.; et al. Altered Peptide Ligands Revisited: Vaccine Design through Chemically Modified HLA-A2–Restricted T Cell Epitopes. J. Immunol. 2014, 193, 4803–4813. [Google Scholar] [CrossRef] [Green Version]
- Donaldson, J.G.; Lippincott-Schwartz, J.; Klausner, R.D. Guanine nucleotides modulate the effects of brefeldin A in semipermeable cells: Regulation of the association of a 110-kD peripheral membrane protein with the Golgi apparatus. J. Cell Biol. 1991, 112, 579–588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ackerman, A.L.; Cresswell, P. Regulation of MHC Class I Transport in Human Dendritic Cells and the Dendritic-Like Cell Line KG-1. J. Immunol. 2003, 170, 4178–4188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iezzi, G.; Karjalainen, K.; Lanzavecchia, A. The duration of antigenic stimulation determines the fate of naive and effector T cells. Immunity 1998, 8, 89–95. [Google Scholar] [CrossRef] [Green Version]
- Müller, S.; Demotz, S.; Bulliard, C.; Valitutti, S. Kinetics and extent of protein tyrosine kinase activation in individual T cells upon antigenic stimulation. Immunology 1999, 97, 287–293. [Google Scholar] [CrossRef]
- Amith, S.R.; Jayanth, P.; Franchuk, S.; Finlay, T.; Seyrantepe, V.; Beyaert, R.; Pshezhetsky, A.V.; Szewczuk, M.R. Neu1 desialylation of sialyl α-2,3-linked β-galactosyl residues of TOLL-like receptor 4 is essential for receptor activation and cellular signaling. Cell. Signal. 2010, 22, 314–324. [Google Scholar] [CrossRef]
- Amith, S.R.; Jayanth, P.; Franchuk, S.; Siddiqui, S.; Seyrantepe, V.; Gee, K.; Basta, S.; Beyaert, R.; Pshezhetsky, A.V.; Szewczuk, M.R. Dependence of pathogen molecule-induced Toll-like receptor activation and cell function on Neu1 sialidase. Glycoconj. J. 2009, 26, 1197–1212. [Google Scholar] [CrossRef]
- Feng, C.; Zhang, L.; Almulki, L.; Faez, S.; Whitford, M.; Hafezi-Moghadam, A.; Cross, A.S. Endogenous PMN sialidase activity exposes activation epitope on CD11b/CD18 which enhances its binding interaction with ICAM-1. J. Leukoc. Biol. 2011, 90, 313–321. [Google Scholar] [CrossRef] [Green Version]
- Ryan, S.O.; Cobb, B.A. Roles for major histocompatibility complex glycosylation in immune function. In Seminars in Immunopathology; Springer: Berlin, Germany, 2012; Volume 34, pp. 425–441. [Google Scholar]
- Bjorkman, P.J.; Saper, M.A.; Samraoui, B.; Bennett, W.S.; Strominger, J.L.; Wiley, D.C. Structure of the human class I histocompatibility antigen, HLA-A2. Nature 1987, 329, 506–512. [Google Scholar] [CrossRef] [PubMed]
- Ryan, S.O.; Cobb, B.A. Host glycans and antigen presentation. Microbes Infect. 2012, 14, 894–903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cole, K.S.; Steckbeck, J.D.; Rowles, J.L.; Desrosiers, R.C.; Montelaro, R.C. Removal of N-linked glycosylation sites in the V1 region of simian immunodeficiency virus gp120 results in redirection of B-cell responses to V3. J. Virol. 2004, 78, 1525–1539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huso, D.L.; Narayan, O.; Hart, G.W. Sialic acids on the surface of caprine arthritis-encephalitis virus define the biological properties of the virus. J. Virol. 1988, 62, 1974–1980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lundegaard, C.; Lamberth, K.; Harndahl, M.; Buus, S.; Lund, O.; Nielsen, M. NetMHC-3.0: Accurate web accessible predictions of human, mouse and monkey MHC class I affinities for peptides of length 8–11. Nucleic Acids Res. 2008, 36, W509–W512. [Google Scholar] [CrossRef] [Green Version]
- Andreatta, M.; Nielsen, M. Gapped sequence alignment using artificial neural networks: Application to the MHC class I system. Bioinformatics 2015, 32, 511–517. [Google Scholar] [CrossRef] [Green Version]
- Miller, S.G.; Carnell, L.; Moore, H.P.H. Post-Golgi membrane traffic: Brefeldin A inhibits export from distal Golgi compartments to the cell surface but not recycling. J. Cell Biol. 1992, 118, 267–283. [Google Scholar] [CrossRef]
- Adiko, A.C.; Babdor, J.; Gutiérrez-Martínez, E.; Guermonprez, P.; Saveanu, L. Intracellular transport routes for MHC I and their relevance for antigen cross-presentation. Front. Immunol. 2015, 6, 1–11. [Google Scholar] [CrossRef]
- Naslavsky, N.; Weigert, R.; Donaldson, J.G. Characterization of a nonclathrin endocytic pathway: Membrane cargo and lipid requirements. Mol. Biol. Cell 2004, 15, 3542–3552. [Google Scholar] [CrossRef] [Green Version]
- Blander, J.M. The comings and goings of MHC class I molecules herald a new dawn in cross-presentation. Immunol. Rev. 2016, 272, 65–79. [Google Scholar] [CrossRef] [Green Version]
- Zeeh, J.C.; Zeghouf, M.; Grauffel, C.; Guibert, B.; Martin, E.; Dejaegere, A.; Cherfils, J. Dual specificity of the interfacial inhibitor brefeldin A for arf proteins and Sec7 domains. J. Biol. Chem. 2006, 281, 11805–11814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neefjes, J.; Sadaka, C. Into the intracellular logistics of cross-presentation. Front. Immunol. 2012, 3, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reid, P.A.; Watts, C. Cycling of cell-surface MHC glycoproteins through primaquine-sensitive intracellular compartments. Nature 1990, 346, 655–657. [Google Scholar] [CrossRef] [PubMed]
- Pfeifer, J.D.; Wick, M.J.; Roberts, R.L.; Findlay, K.; Normark, S.J.; Harding, C.V. Phagocytic processing of bacterial antigens for class I MHC presentation to T cells. Nature 1993, 361, 359–362. [Google Scholar] [CrossRef]
- Chiu, I.; Davis, D.M.; Strominger, J.L. Trafficking of spontaneously endocytosed MHC proteins. Proc. Natl. Acad. Sci. USA 1999, 96, 13944–13949. [Google Scholar] [CrossRef] [Green Version]
- Li, G.; Abediankenari, S.; Kim, Y.J.; Campbell, T.B.; Ito, S.; Graham-Evans, B.; Cooper, S.; Broxmeyer, H.E.; Steinman, R.; Steinman, R.; et al. TGF-beta combined with M-CSF and IL-4 induces generation of immune inhibitory cord blood dendritic cells capable of enhancing cytokine-induced ex vivo expansion of myeloid progenitors. Blood 2007, 110, 2872–2879. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibody Clone | Specificity | Technique | Source | Reference |
|---|---|---|---|---|
| W6/32 | Recognizes antigenic determinant common to HLA-A, B and C antigens when in their three-dimensional configuration | FC/WB and IP | ImmunoTools, Germany/Pierce, Thermo Fisher Scientific (Waltham, MA, USA) | [19] |
| HC10 | Recognizes free HLA class I heavy chains. HC10 reacts mostly with HLA-B and HLA-C heavy chains and some HLA-A (HLA-A10, HLA-A28, HLA-A29, HLA-A30, HLA-A31, HLA-A32, HLA-A33) | FC and WB | Hybridoma supernatants | [20] |
| BBM.1 | Recognizes both free and class I bound β2m | FC and WB | Hybridoma supernatants | [21] |
| BB7.2 | Recognizes the α subunit of HLA-A2 which is a subset of MHC-class I molecules encoded by A*02 alleles | FC | BD Bioscience (CA, USA) | [22] |
| 246-B8.E7 | Recognizes monomorphic determinant of human MHC- I antigens (HLA-A, B, and C) | CM | Thermo Fisher Scientific (Waltham, MA, USA) | Not found |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Silva, Z.; Ferro, T.; Almeida, D.; Soares, H.; Ferreira, J.A.; Deschepper, F.M.; Hensbergen, P.J.; Pirro, M.; van Vliet, S.J.; Springer, S.; et al. MHC Class I Stability is Modulated by Cell Surface Sialylation in Human Dendritic Cells. Pharmaceutics 2020, 12, 249. https://doi.org/10.3390/pharmaceutics12030249
Silva Z, Ferro T, Almeida D, Soares H, Ferreira JA, Deschepper FM, Hensbergen PJ, Pirro M, van Vliet SJ, Springer S, et al. MHC Class I Stability is Modulated by Cell Surface Sialylation in Human Dendritic Cells. Pharmaceutics. 2020; 12(3):249. https://doi.org/10.3390/pharmaceutics12030249
Chicago/Turabian StyleSilva, Zélia, Tiago Ferro, Danielle Almeida, Helena Soares, José Alexandre Ferreira, Fanny M. Deschepper, Paul J. Hensbergen, Martina Pirro, Sandra J. van Vliet, Sebastian Springer, and et al. 2020. "MHC Class I Stability is Modulated by Cell Surface Sialylation in Human Dendritic Cells" Pharmaceutics 12, no. 3: 249. https://doi.org/10.3390/pharmaceutics12030249