Improved Stability of Rifampicin in the Presence of Gastric-Resistant Isoniazid Microspheres in Acidic Media


Abstract
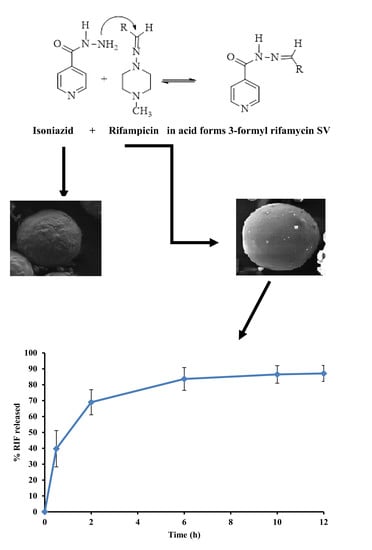
:
1. Introduction
2. Materials and Methods
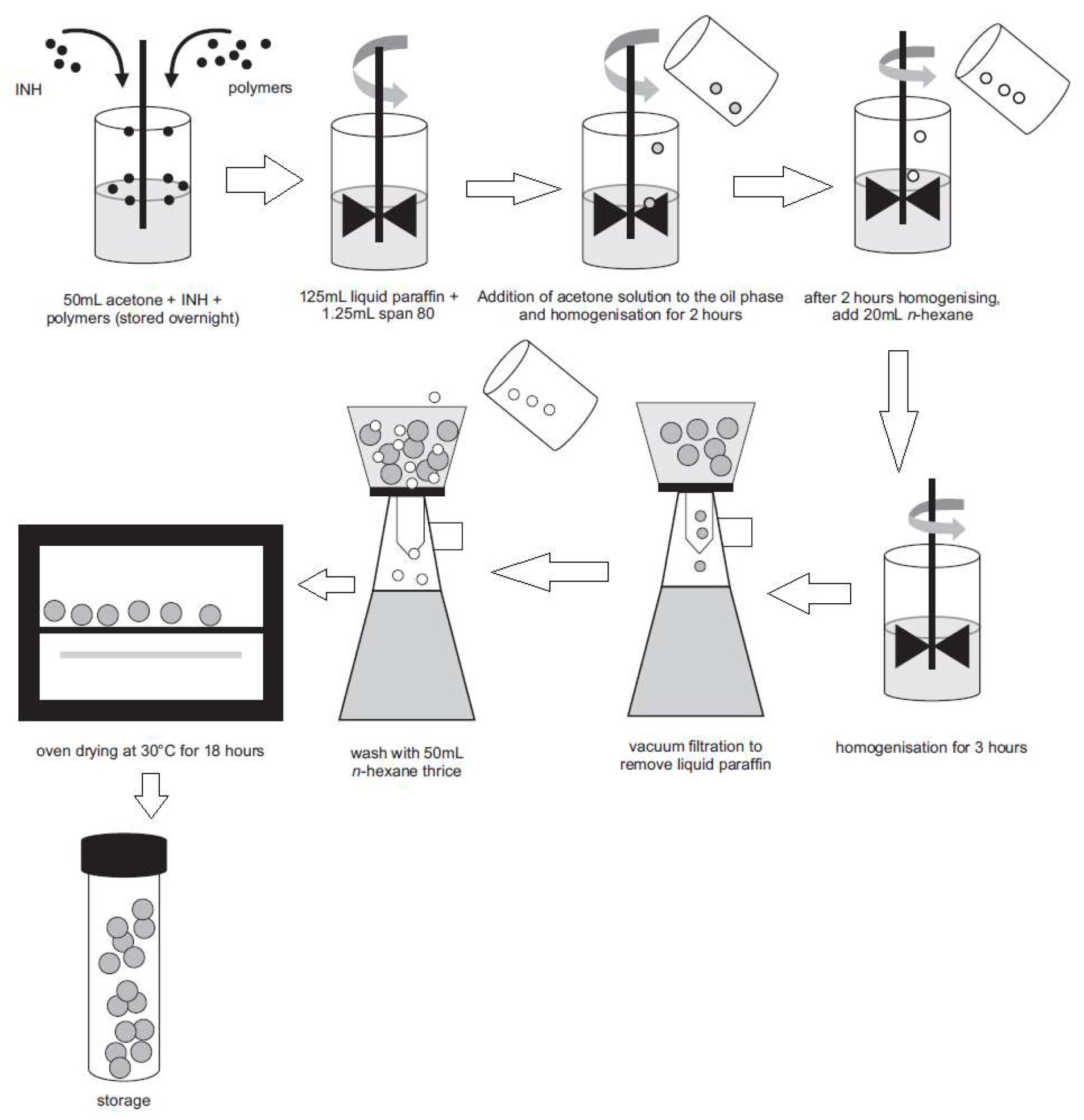
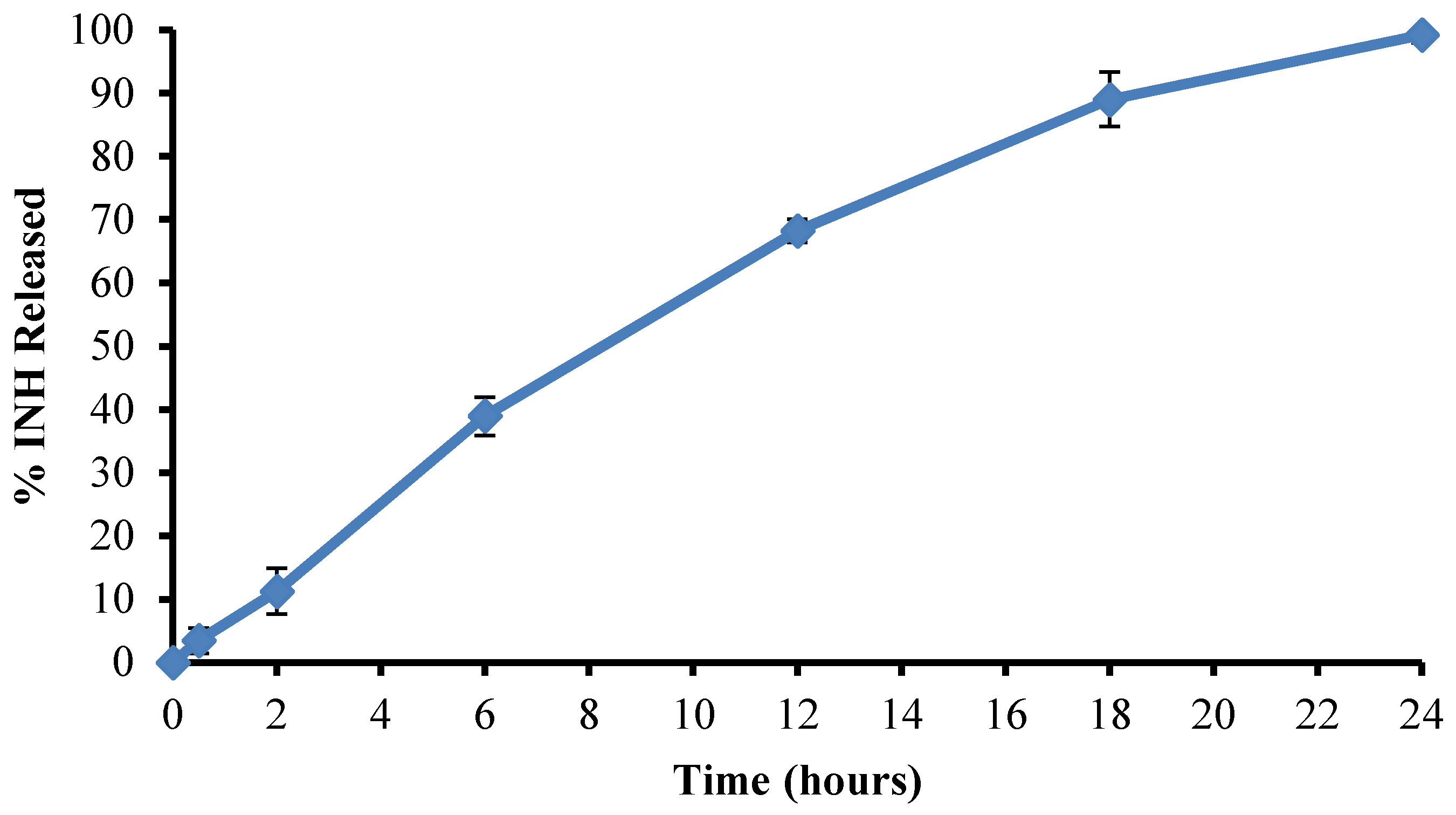
2.1. Manufacture and Dissolution Testing of Gastric Resistant Sustained Release Isoniazid (INH) Microcapsules
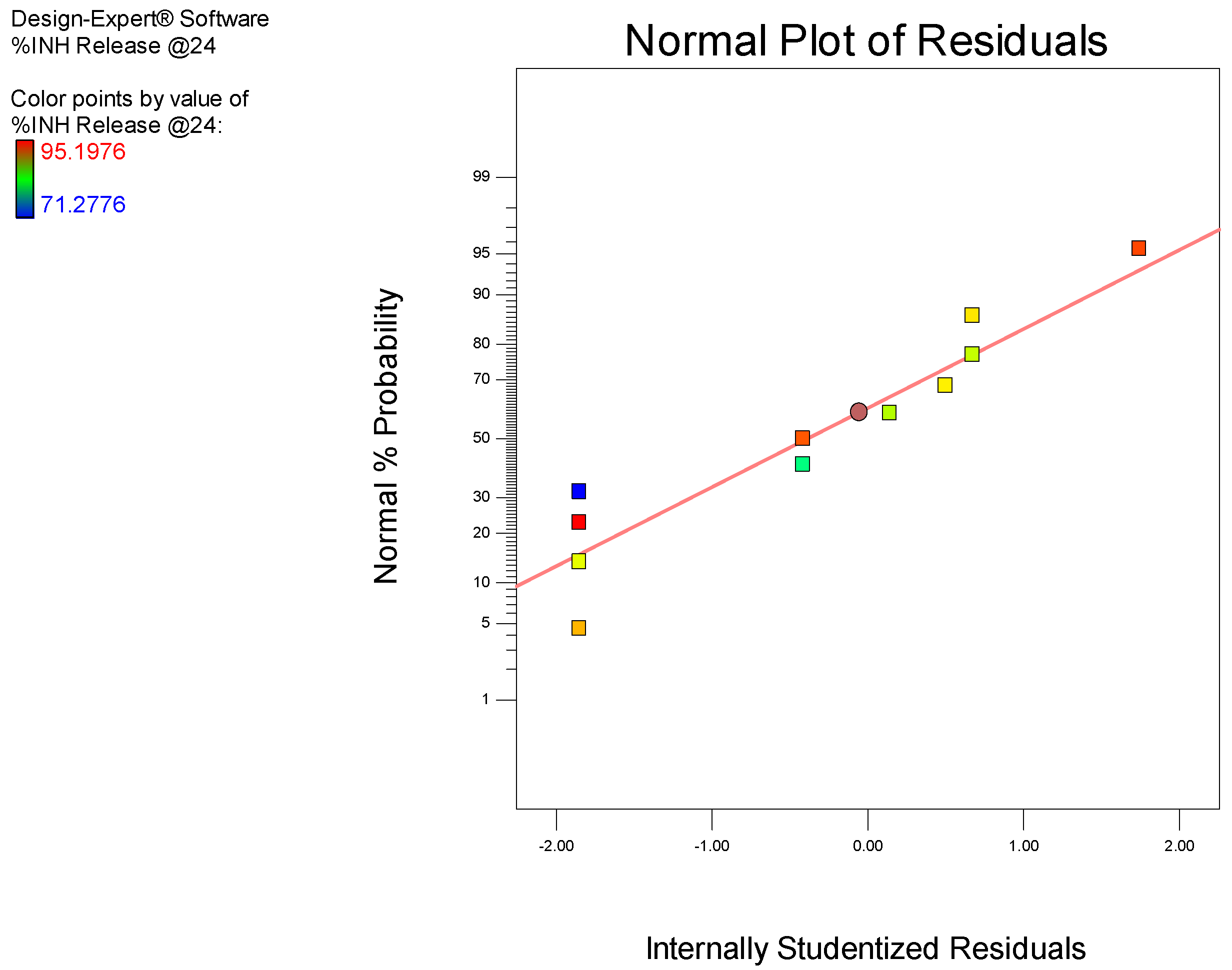
2.2. Optimization of INH Microspheres and Evaluation of Model Adequacy
2.3. Manufacture and Dissolution Testing of Microporous Floating Sustained Release RIF Microspheres
2.4. Optimization of RIF Microspheres and Evaluation of Model Adequacy
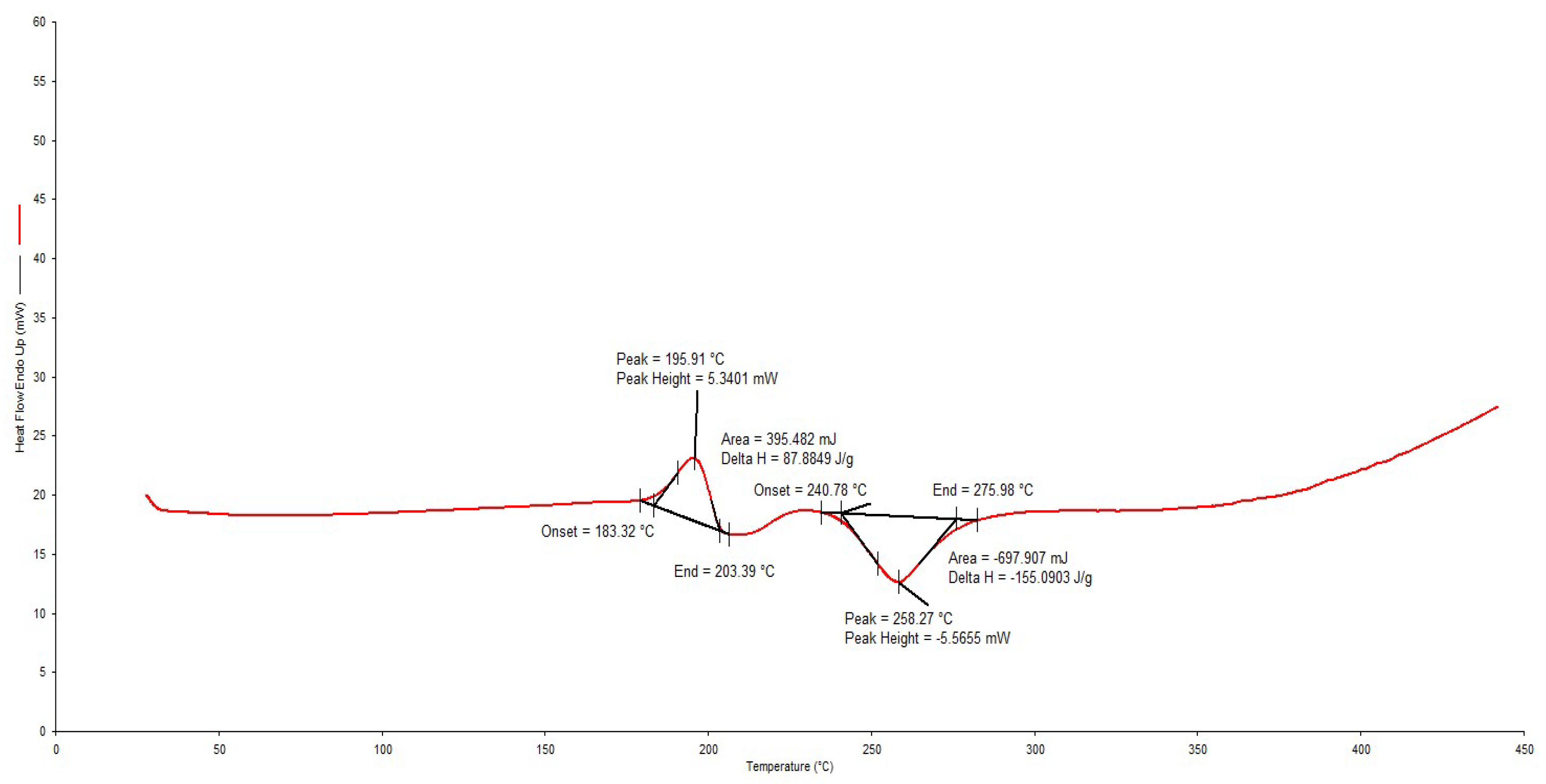
2.5. Differential Scanning Calorimetry
2.6. Fourier Transform Raman Spectroscopy (FTRS)
2.7. Determination of the Particle Size Distribution
2.8. Bulk and Tapped Density
2.9. Analytical Method and Chromatographic Conditions
2.10. Encapsulation Efficiency
2.11. Scanning Electron Microscopy
2.12. Assessment of Dissolution and Stability of Rifampicin in the Presence of Isoniazid
2.13. In Vitro Buoyancy Studies
3. Results and Discussion
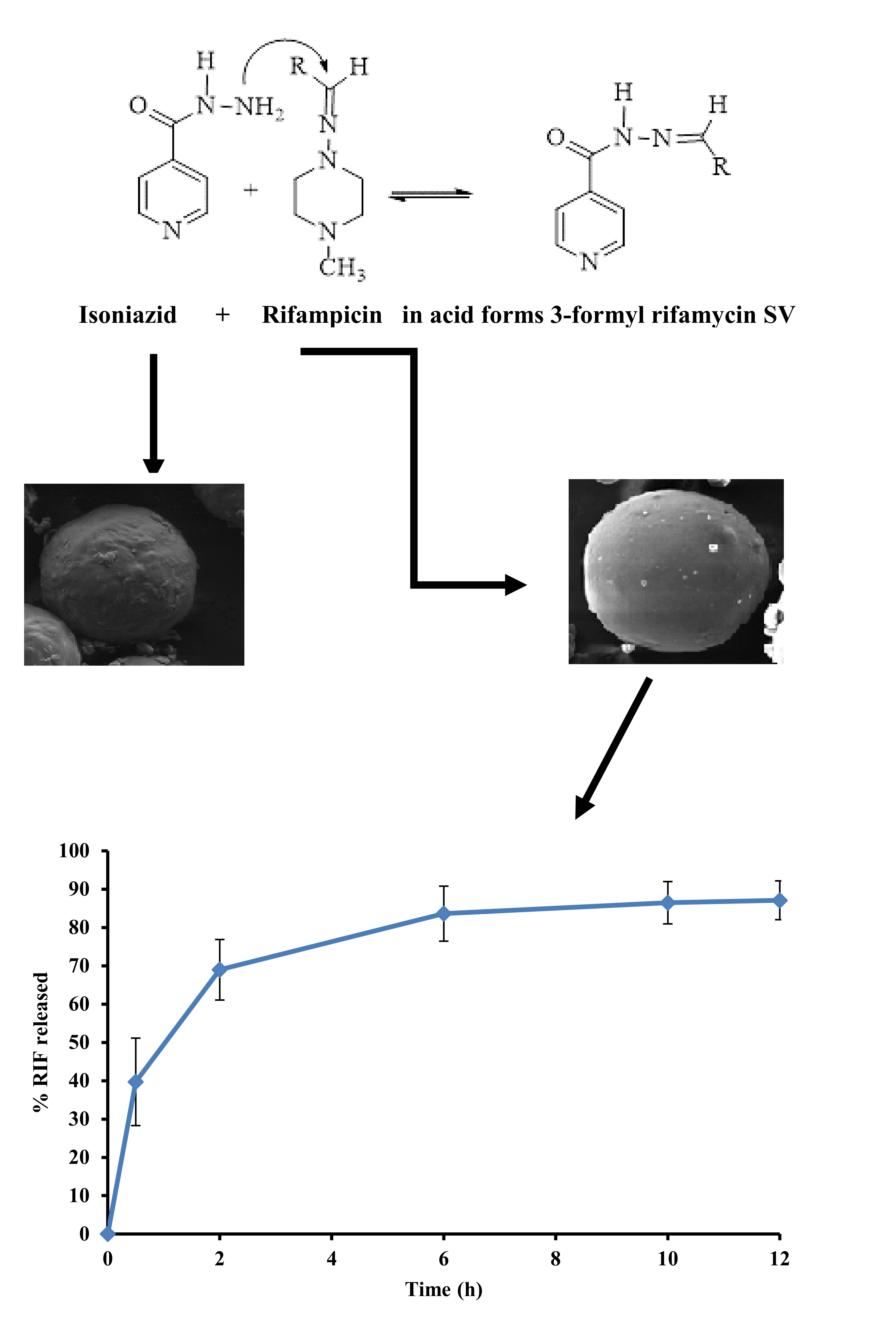
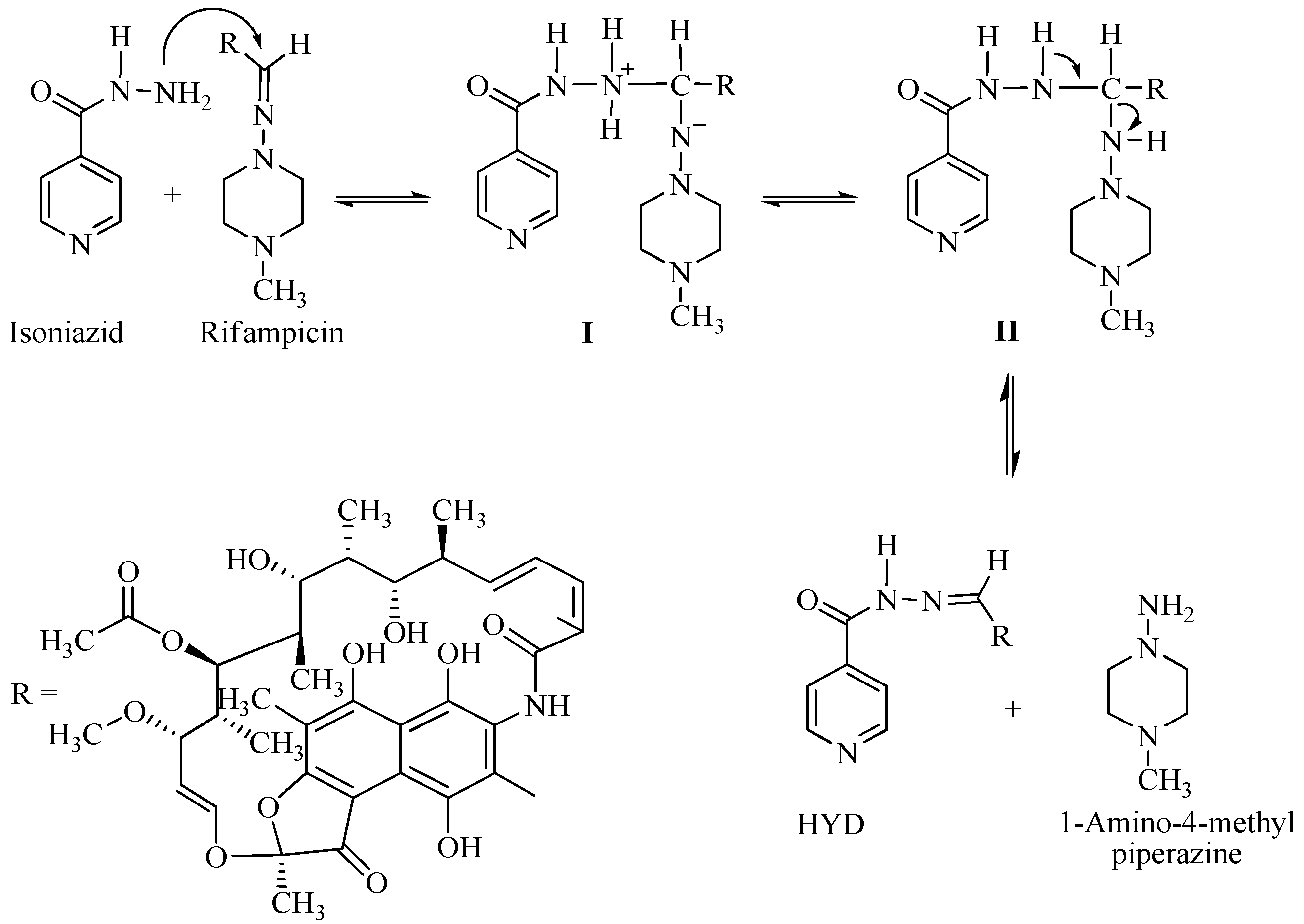
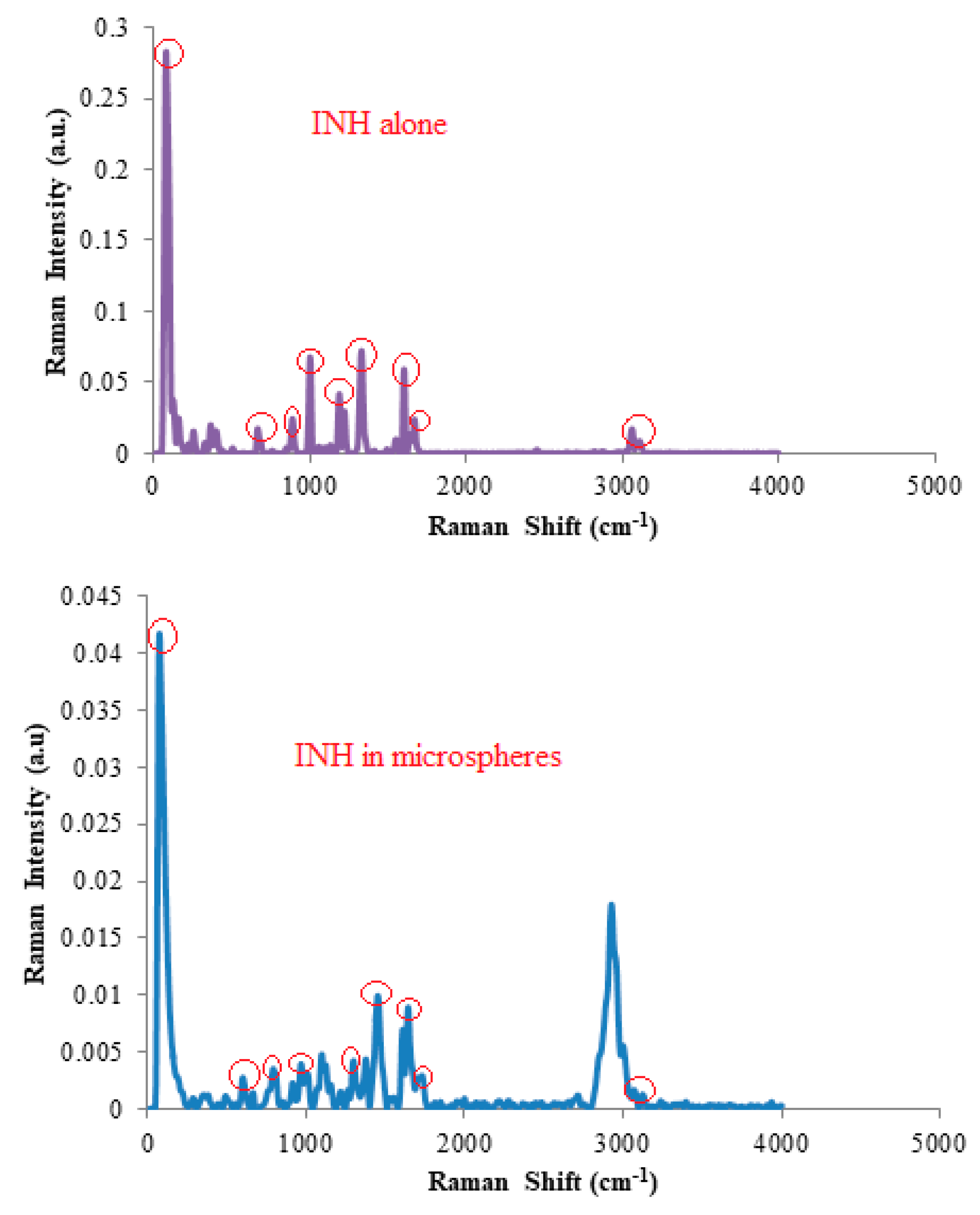
3.1. Drug-Excipient Compatibility Studies
3.2. Manufacture of Isoniazid Microspheres, Evaluation of Model Adequacy, and Formulation Optimization
3.3. Manufacture of RIF Microspheres, Evaluation of Model Adequacy, and Formulation Optimization
3.4. Particle Size Distribution
3.5. Flowability—Carr’s Index (CI) and Hausner Ratio (HR)
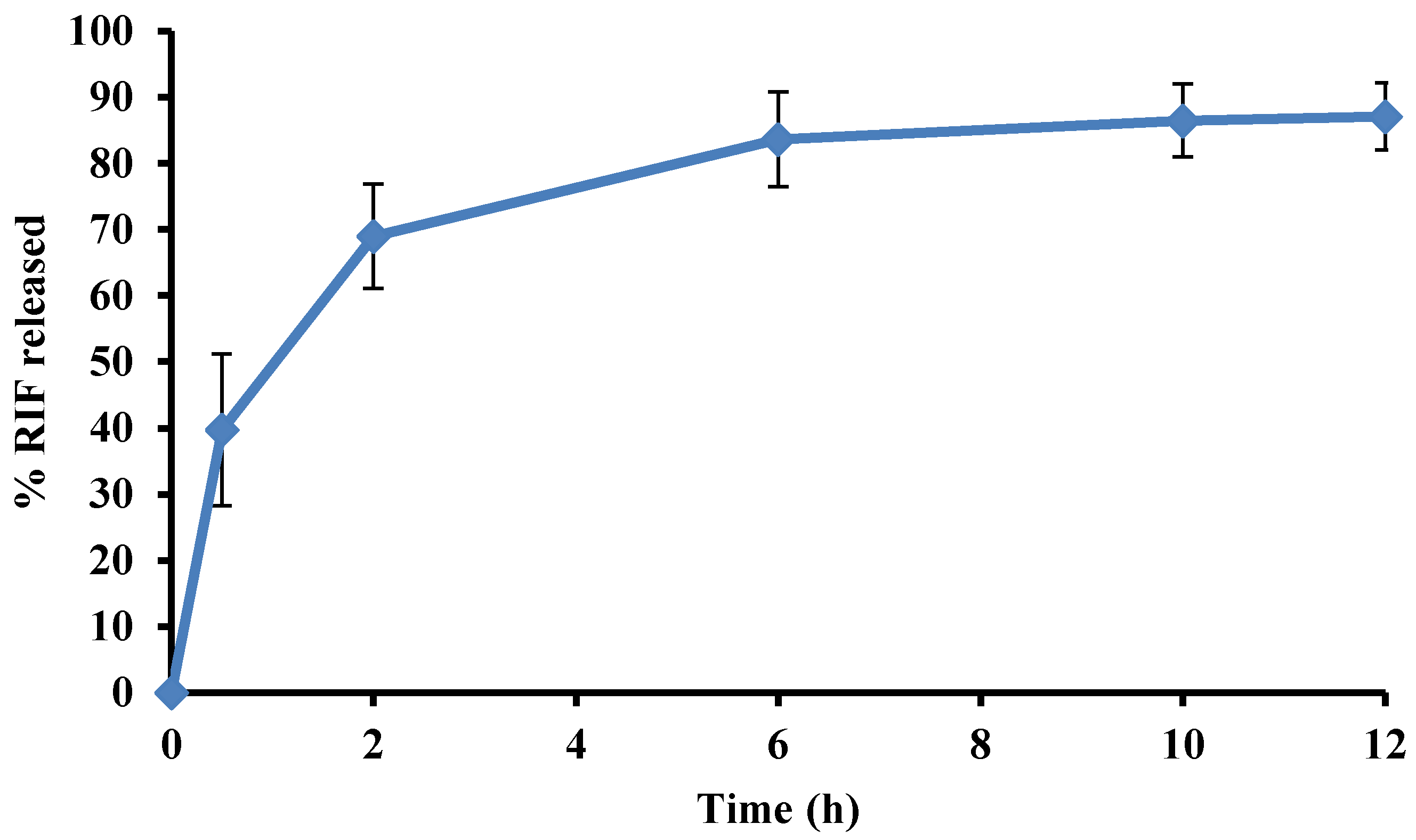
3.6. Stability of RIF in Presence of INH
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Agrawal, S.; Singh, I.; Kaur, K.J.; Bhade, S.R.; Kaul, C.L.; Panchagnula, R. Comparative bioavailability of rifampicin, isoniazid and pyrazinamide from a four drug fixed dose combination with separate formulations at the same dose levels. Int. J. Pharm. 2004, 276, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Pillai, G.; Fourie, P.B.; Padayatchi, N.; Onyebujoh, P.C.; Mcllleron, H.; Smith, P.J.; Gabriels, G.R. Recent bioequivalence studies on fixed dose combination antituberculosis drug formulations available on the global market. Int. J. Tuberc. Lung Dis. 1999, 3, S309–S316. [Google Scholar] [PubMed]
- Coupe, A.J.; Davis, S.S.; Wilding, I.R. Variation in gastrointestinal transit of pharmaceutical dosage forms in healthy subjects. Pharm. Res. 1991, 8, 360–364. [Google Scholar] [CrossRef] [PubMed]
- Shishoo, C.J.; Shah, S.A.; Rathod, I.S.; Savale, S.S.; Vora, M.J. Impaired bioavailability of rifampicin in presence of isoniazid from fixed dose combination (FDC) formulation. Int. J. Pharm. 2001, 228, 53–67. [Google Scholar] [CrossRef]
- Sankar, R.; Sharda, N.; Singh, S. Behaviour of decomposition of rifampicin in the presence of isoniazid on the pH range 1-3. Drug Dev. Ind. Pharm. 2003, 29, 733–738. [Google Scholar] [CrossRef]
- Shishoo, C.J.; Shah, S.A.; Rathod, I.S.; Savale, S.S.; Kotecha, J.S.; Shah, P.B. Stability of rifampicin in dissolution medium in presence of isoniazid. Int. J. Pharm. 1999, 190, 109–123. [Google Scholar] [CrossRef]
- Singh, S.; Mariappan, T.T.; Sharda, N.; Chakraborti, A.K. The reason for an increase in decomposition of rifampicin in the presence of isoniazid under acid conditions. Pharm. Pharm. Comm. 2000, 6, 405–410. [Google Scholar] [CrossRef]
- Maggi, N.; Pasqualucci, C.R.; Ballota, R.; Sensi, P. Rifampicin, a new orally active rifamycin. Chemotherapia 1966, 11, 285–292. [Google Scholar] [CrossRef]
- Sensi, P.; Maggi, N.; Furesz, S.; Maffi, G. Chemical modifications and biological properties of rifamycins. Antimicrob. Agents Chemother. 1966, 6, 699–714. [Google Scholar]
- Singh, S.; Mariappan, T.T.; Shankar, R.; Sarda, N.; Singh, B. A critical review of the probable reasons for the poor variable bioavailability of rifampicin from anti-tubercular fixed-dose combination (FDC) products, and the likely solutions to the problem. Int. J. Pharm. 2001, 228, 5–17. [Google Scholar] [CrossRef]
- Mariappan, T.T.; Singh, S. Regional gastrointestinal permeability of rifampicin and isoniazid (alone and their combination) in the rat. Int. J. Tuberc. Lung Dis. 2003, 7, 797–803. [Google Scholar] [PubMed]
- Bhise, S.B.; More, A.B.; Malayandi, R. Formulation and In Vitro Evaluation of Rifampicin Loaded Porous Microspheres. Sci. Pharm. 2010, 78, 291–302. [Google Scholar] [CrossRef] [Green Version]
- Freire, D.F.; Aragao, S.C.F. Thermal Studies of Isoniazid and Mixtures with Rifampicin. J. Therm. Anal. Calorim. 2009, 97, 333–336. [Google Scholar] [CrossRef]
- Gohel, C.M.; Krishnakant, G.S. A Novel Solid Dosage Form of Rifampicin and Isoniazid with Improved Functionality. AAPS Pharmscitech. 2007, 8, 8. [Google Scholar] [CrossRef] [PubMed]
- Hadassah, M.; Swarna, K.C.H.; Prasanthi, D.; Sai, K.I.; Vijaya, R.J. Formulation and Evaluation of Floating Bilayered Tablets of Rifampicin, Isoniazid and Pyrazinamide. Int. J. Pharm. Technol. 2012, 4, 4931–4951. [Google Scholar]
- Krishna, T.V.; Reddy, M.S. Formulation and Evaluation of Enteric Coated Pellets of Rifampicin and Isoniazid with Improved Rifampicin Stability. Asian J. Pharm. Res. 2014, 7, 154–156. [Google Scholar]
- Kumar, T.U.; Vasudevan, M. Novel Sustained Release Swellable Bioadhesive Floating Gastroretentive Drug Delivery System of Bilayer Tablets Containing Rifampicin and Isoniazid. Int. J. Pharm. Sci. Lett. 2012, 1, 21–24. [Google Scholar]
- Pund, S.; Joshi, A.; Vasu, K.; Nivsarkar, M.; Shishoo, C. Gastroretentive delivery of rifampicin: In vitro mucoadhesion and in vivo gamma scintigraphy. Int. J. Pharm. 2011, 411, 106–112. [Google Scholar] [CrossRef]
- Singh, H.; Bhadari, R.; Kaur, I.P. Encapsulation of Rifampicin in Solid Lipid Nanoparticulate System to Limit its Degradation and Interaction with Isoniazid at Acidic pH. Int. J. Pharm. 2013, 446, 106–111. [Google Scholar] [CrossRef]
- Sullad, A.G.; Manjeshwar, L.S.; Aminabhavi, T.M. Novel pH-Sensitive Hydrogels Prepared from the Blends of (Poly(vinyl alcohol) with Acrylic Acid-graft-Guar Gum Matrixes for Isoniazid Delivery. Ind. Eng. Chem. Res. 2010, 49, 7323–7329. [Google Scholar] [CrossRef]
- Khamanga, S.M.; Parfitt, N.; Nyamuhiwa, T.; Haidula, H.; Walker, R.B. The Evaluation of Eudragit microcapsules manufactured by solvent evaporation using USP Apparatus 1. Dissolut. Technol. 2009, 16, 15–22. [Google Scholar] [CrossRef]
- Hwisa, N.T.; Katakam, P.; Chandu, B.R.; Adiki, S.K. Solvent Evaporat ion Techniques as Promising Advancement in Microencapsulation. Vedic Res. Int. Biol. Med. Chem. 2013, 1, 8–22. [Google Scholar] [CrossRef]
- Prakash, K.; Raju, P.N.; Shanta, K.K.; Lakshmi, M.N. Preparation and characterization of lamivudine microcapsules using various cellulose polymers. Trop. J. Pharm Res. 2007, 6, 841–847. [Google Scholar] [CrossRef] [Green Version]
- Mateovic, T.; Kriznar, B.; Bogataj, M.; Mrhar, A. The influence of stirring rate on biopharmaceutical properties of Eudragit RS microspheres. J. Microencapsul. 2002, 19, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Noordin, M.Y.; Venkatesh, V.C.; Sharif, S.; Elting, S.; Abdullah, A. Application of response surface methodology in describing the performance of coated carbide tools when turning AISI 1045 steel. J. Mater. Process. Technol. 2004, 145, 46–58. [Google Scholar] [CrossRef] [Green Version]
- Hill, W.J.; Hunter, W.G. A Review of response surface methodology, a literature survey. Technometrics 1966, 8, 571–590. [Google Scholar] [CrossRef]
- Kirkwood, T.B.L. Geometric means and measures of dispersion. Biometrics 1979, 35, 908–909. [Google Scholar]
- Hendricks., W.A.; Robey, K.W. The Sampling Distribution of the Coefficient of Variation. Ann. Math. Stat. 1936, 7, 129–132. [Google Scholar] [CrossRef]
- Barmpalexis, P.; Kanaze, F.I.; Georgarakis, E. Developing and optimising a validated iscocratic reversed-phase high-performance liquid chromatography separation of nimodipine and impurities in tablets using experimental design methodology. J. Pharm. Biomed. Anal. 2009, 49, 1192–1202. [Google Scholar] [CrossRef]
- Box, G.E.P.; Wilson, K.B. On the Experimental Attainment of Optimum Multifactorial Conditions. R. Stat. Soc. 1951, 13, 1–12. [Google Scholar]
- Carr, R.L. Classifying flow properties of solids. Chem. Eng. 1965, 72, 69–72. [Google Scholar]
- Hausner, H.H. Friction conditions in a mass of metal powders. Int. J. Powder Metall. 1967, 3, 7–13. [Google Scholar]
- Zhang, Z.; Feng, S. The drug encapsulation efficiency, in vitro release, cellular uptake and cytotoxicity of paclitaxel-loaded poly(lactide)-tocopheryl polyethylene glycol succinate nanoparticles. Biomaterials 2006, 27, 4025–4033. [Google Scholar] [CrossRef] [PubMed]
- Mwila, C.; Khamanga, S.M.M.; Walker, R.B. Development and Assessment of a USP Apparatus 3 Dissolution Test Method for Sustained-Release Nevirapine Matrix Tablets. Dissol Technol. 2016, 23, 22–30. [Google Scholar] [CrossRef]
- Ozdemir, N.; Ordu, S.; Ozkan, Y. Studies of floating dosage forms of furosemide, in vitro and in vivo evaluations of bilayer tablet formulations. Drug Dev. Ind. Pharm. 2000, 28, 857–866. [Google Scholar] [CrossRef] [PubMed]
- Harikumar, S.L.; Sharma, A. Development and evaluation of bromhexine hydrochloride floating microparticulates. Asian J. Pharm. 2012, 6, 38–43. [Google Scholar] [CrossRef]
- Gunasekaran, S.; Sailatha, E.; Seshadri, S.; Kumaresan, S. FTIR, FT Raman Spectra and molecular structural confirmation of isoniazid. Indian. J. Pure Appl. Phys. 2009, 47, 12–18. [Google Scholar]
- Agrawal, S.; Ashokraj, Y.; Bharatam, P.V.; Pillai, O.; Panchangnula, R. Solid-state characterisation of rifampicin samples and its biopharmaceutic relevance. Eur. J. Pharm. Sci. 2004, 22, 127–144. [Google Scholar] [CrossRef]
- Kenechukwu, F.C.; Momoh, M.A. Formulation, characterisation and evaluation of the effect of polymer concentration on the release behavior of insulin-loaded Eudragit®-entrapped mucoadhesive microspheres. Int. J. Pharm. Investig. 2016, 6, 69–77. [Google Scholar] [CrossRef] [Green Version]
- Xiao, Z.; Liu, W.; Zhu, G.; Zhou, R.; Niu, Y. Production and characterization of multinuclear microcapsules encapsulating lavender oil by complex coacervation. Flavour Fragr. J. 2014, 29, 166–172. [Google Scholar] [CrossRef]
- Rowe, R.C.; Sheskey, P.J.; Owen, S.C. Polymethacrylates; Pharmaceutical Press and American Pharmacists Association: London, UK, 2006; pp. 525–533. [Google Scholar]
- Friesen, D.T.; Shanker, R.; Crew, M.; Smithey, D.T.; Curatolo, W.J.; Nightingale, J.A.S. Hydroxypropyl Methylcellulose Acetate Succinate-Based Spray-Dried Dispersions: An Overview. Mol. Pharm. 2008, 5, 1003–1019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, X.; Ping, Q.; Gao, Y. Effects of formulation factors on encapsulation efficiency and release behaviour in vitro of huperzine A-PLGA microspheres. J. Microencapsul. 2005, 22, 705–714. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Deng, X.; Yuan, M.; Xiong, C.; Huang, Z.; Zhang, Y.; Jia, W. Investigation on process parameters involved in preparation of poly-DL-lactide-poly(ethylene glycol) microspheres containing Leptospira Interrogans antigens. Int. J. Pharm. 1999, 178, 245–255. [Google Scholar] [CrossRef]
- Mehta, R.C.; Thanoo, B.C.; Deluca, P.P. Peptide containing microspheres from low molecular weight and hydrophilic poly(d,l-lactide-co-glycolide). J. Control. Release 1996, 41, 249–257. [Google Scholar] [CrossRef]
- Raval, J.P.; Naik, D.R.; Amin, K.A.; Patel, P.S. Controlled-release and antibacterial studies of doxycycline-loaded poly(å-caprolactone) microspheres. J. Saudi Chem. Soc. 2014, 18, 566–573. [Google Scholar] [CrossRef] [Green Version]
- Rafati, H.; Combes, A.G.A.; Adler, J.; Davis, S.S. Protein-loaded poly(DL-lactide-co-glycolide) microparticles for oral administration, Formulation, structural and release characteristics. J. Control. Release 1997, 43, 89–102. [Google Scholar] [CrossRef]
- Bodmier, R.; McGinity, J.W. Solvent selection in the preparation of PLA microspheres prepared by the solvent evaporation method. Int. J. Pharm. 1988, 43, 179–186. [Google Scholar] [CrossRef]
- Wilson, B.; Babubhai, P.P.; Sajeev, M.S.; Jenita, J.L.; Priyadarshini, S.R. Sustained release enteric coated tablets of pantoprazole: Formulation, in vitro and in vivo evaluation. Acta Pharma 2013, 63, 131–140. [Google Scholar] [CrossRef]
- Kim, B.K.; Hwang, S.J.; Park, J.B.; Park, H.J. Preparation and characterization of drug-loaded polymethacrylate microspheres by an emulsion solvent evaporation method. J. Microencapsul. 2002, 19, 811–822. [Google Scholar] [CrossRef]
- Viswanathan, N.B.; Thomas, P.A.; Pandit, J.K.; Kulkarni, M.G.; Mashelkar, R.A. Preparation of non-porous microspheres with high entrapment efficiency of proteins by a (water-in-oil)-in-oil emulsion technique. J. Control. Release 1999, 58, 9–20. [Google Scholar] [CrossRef]
- Jelvehgari, M.; Dastmalch, S.; Nazila, D. Theophylline-ethylcellulose microparticles, screening of the process and formulation variables for preparation of sustained release particles. Iran. J. Med. Sci. 2012, 15, 608–624. [Google Scholar]
- Jelvehgari, M.; Montazam, S.H. Comparison of microencapsulation by emulsion-solvent extraction/evaporation technique using derivatives cellulose and acrylate-methacrylate copolymer as carriers. J. Nat. Pharm. Prod. 2012, 7, 144–152. [Google Scholar]
- Jain, S.; Srinath, M.S.; Narendra, C.; Reddy, S.N.; Sindhu, A. Development of a floating dosage form of ranitidine hydrochloride by statistical optimisation technique. J. Young Pharm. 2010, 2, 342–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kilacarslan, M.; Baykara, T. The effect of drug polymer ratio on the properties of the verapamil HCl loaded microspheres. Int. J. Pharm. 2003, 252, 99–109. [Google Scholar] [CrossRef]
- Chandran, S.; Asghar, L.F.A.; Mantha, N. Design and evaluation of ethyl cellulose based matrix tablets of ibuprofen with pH modulated release kinetics. Ind J. Pharm. Sci. 2008, 70, 596–602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ozturk, A.G.; Ozturk, S.S.; Palsson, B.O.; Wheatley, T.A.; Dressman, J.B. Mechanism of release from pellets coated with an ethylcellulose-based film. J. Control. Release. 1990, 14, 202–213. [Google Scholar] [CrossRef] [Green Version]
- Bodmeir, R.; Paeratakul, O. The effect of curing on drug release and morphological properties of ethylcellulose pseudolatex-coated beads. Drug Dev. Ind. Pharm. 1994, 20, 1517–1533. [Google Scholar] [CrossRef]
- Khan, G.M.; Meidan, V.M. Drug release kinetics from tablet matrices based upon ethylcellulose ether-derivatives, a comparison between different formulations. Drug Dev. Ind. Pharm. 2007, 33, 627–639. [Google Scholar] [CrossRef]
- Sengel-Turk, C.T.; Hascicek, C.; Gonul, N. Ethylcellulose-based matrix-type microspheres, influence of plasticizer ratio as pore-forming agent. AAPS Pharmscitech. 2011, 12, 1127–1135. [Google Scholar] [CrossRef] [Green Version]
- Schnieders, J.; Gbureck, U.; Vorndran, E.; Schossik, M.; Kissel, T. The effect of porosity on drug release kinetics from vancomycin microsphere/calcium phosphate cement composites. J. Mater. Res. 2011, 99, 391–398. [Google Scholar] [CrossRef]
- Emeje, M.O.; Kunle, O.O.; Ofoefule, S.I. Compaction characteristics of ethylcellulose in the presence of some chanelling agents, technical note. AAPS Pharmscitech. 2006, 7, 58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhi-wei, Y.; Patrick, R.; Jean, P.R.; Chris, V. Correlation between permeability of metoprolol tartrate through plasticized isolated ethylcellulose/hydroxypropyl methylcellulose films and drug release from reservoir pellets. Eur. J. Pharm. Biopharm. 2007, 67, 485–490. [Google Scholar]
- Rowe, R.C. The effect of the molecular weight of ethyl cellulose on the drug release properties of mixed films of ethyl cellulose and hydroxypropylmethylcellulose. Int. J. Pharm. 1986, 29, 37–41. [Google Scholar] [CrossRef]
- Murtasa, G. Ethylcellulose microparticles, a review. Acta Pol. Pharm.-Drug Res. 2012, 69, 11–22. [Google Scholar]
- Katayoun, D.; Marzieh, S. Formulation and in vitro evaluation of nifedipine controlled release tablet: Influence of combination of hydrophylic and hydrophobic matrix forms. Asian J. Pharm. 2010, 4, 185–193. [Google Scholar]
- Kang, F.; Singh, J. Effect of additives on the release of a model protein from PLGA microspheres. AAPS PharmSciTech 2001, 2, 30. [Google Scholar] [CrossRef]
- Patra, C.N.; Kumar, A.B.; Pandit, H.K.; Singh, S.P.; Devi, M.V. Design and evaluation of sustained release bilayer tablets of propranolol hydrochloride. Acta Pharm. 2007, 57, 479–489. [Google Scholar] [CrossRef] [Green Version]
- Khandai, M.; Chakraborty, S.; Sharma, A.; Panda, D.; Khanam, N.; Panda, S.K. Development of propranolol hydrochloride matrix tablets, an investigation on effects of combination of hydrophilic and hydrophobic matrix formers using multiple comparison analysis. Int. J. Pharm. Sci. Rev. Res. 2010, 1, 1–7. [Google Scholar]
- Jagtap, Y.M.; Bhujbal, R.K.; Ranade, A.N.; Ranpise, N.S. Effect of various polymers concentrations on physicochemical properties of floating microspheres. Ind. J. Pharm. Sci. 2012, 74, 512–520. [Google Scholar] [CrossRef] [Green Version]
- Pande, A.V.; Vaidya, P.D.; Arora, A.; Dhoka, M.V. In vitro and in vivo evaluation of ethyl cellulose based floating microspheres of cefpodoxime proxetil. Int. J. Biomed. Res. 2010, 1, 122–128. [Google Scholar]
- de la Cruz, G.V.; Torres, J.A.; Martin-Polo, M.O. Temperature effect on the moisture sorption isotherms for methylcellulose and ethylcellulose films. J. Food Eng. 2001, 48, 91–94. [Google Scholar] [CrossRef]
- Saravanan, M.; Anupama, B. Development and evaluation of ethylcellulose floating microspheres loaded with ranitidine hydrochloride by novel solvent evaporation-matrix erosion method. Carbohyd. Polym. 2011, 85, 592–598. [Google Scholar] [CrossRef]
- Pandya, N.; Pandya, M.; Bhaskar, V.H. Preparation and in vitro characterisation of porous carrier–based glipizide floating microspheres for gastric delivery. J. Young Pharm. 2011, 3, 97–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amin, L.; Ahmed, T.; Mannan, A. Development of floating-mucoadhesive microsphere for site specific release of metronidazole. Adv. Pharm. Bull. 2016, 6, 195–200. [Google Scholar] [CrossRef] [Green Version]
- Akbuga, J. Preparation and evaluation of controlled release furosemide microspheres by spherical crystallisation. Int. J. Pharm. 1989, 53, 99–105. [Google Scholar] [CrossRef]
- Chemtob, C.; Chaumeil, J.C.; N’Dongo, M. Microencapsulation by ethylcellulose phase separation: Microcapsule characteristics. Int. J. Pharm. 1986, 29, 1–7. [Google Scholar] [CrossRef]
- Huang, X.; Cheng, X.; Luo, Y.; Guo, Z.; Zhong, C.; Zhang, Y. Effect of homogenisation speed on morphology and release of protein loaded PLGA microspheres made by W/O/W method. Acta Sci. Nat. Univ. Sunyatseni 2015, 54, 119–124. [Google Scholar]
- Kim, K.K.; Pack, D.W. Microspheres for Drug delivery. In BioMEMS and Biomedical Nanotechnology; Ferrari, M., Lee, A.P., Lee, L.J., Eds.; Springer International Publishing AG: Boston, MA, USA, 2006; Volume 1, pp. 19–50. [Google Scholar]
- Stevens, R.E.; Dorantes, A.; Gray, V.; Pham, L. Scientific and Regulatory Standards for Assessing Product Performance Using the Similarity Factor, f2. AAPS J. 2015, 17, 301–306. [Google Scholar] [CrossRef] [Green Version]
- Prasanthi, B.; D.Prasanthi, D.; Bhavani, B.; Ratna, J.V. A novel drug delivery system designed for modulating the release kinetics of antitubercular drugs. Int. J. Pharm. 2014, 4, 189–196. [Google Scholar]
- Acocella, G. Clinical pharmacokinetics of rifampicin. Clin. Pharm. 1978, 3, 108–127. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Factor | Code | Coded Level | ||||
|---|---|---|---|---|---|---|
| Independent/Input | ||||||
| Level | −1.4142 | −1 | 0 | +1 | +1.4142 | |
| HMPC-AS (g) | A | 0.585 | 1.000 | 2.000 | 3.000 | 3.414 |
| Eudragit®L100 (g) | B | 0.585 | 1.000 | 2.000 | 3.000 | 3.414 |
| Homogenization Speed (rpm × 100) | C | 0.5 | 1.0 | 2.0 | 3.0 | 3.4 |
| Dependent/Output | Constraint | |||||
| Y1 = Percent yield | Maximize | |||||
| Y2 = % INH released in 0.1 M HCl at 4 h | Minimize | |||||
| Y3 = % INH released in buffer pH 6.8 at 12 h | Maximize | |||||
| Y4 = Encapsulation efficiency | Maximize | |||||
| Y5 = % INH released in buffer pH 6.8 at 24 h | Maximize | |||||
| Factor | Code | Coded Level | ||
|---|---|---|---|---|
| Independent | ||||
| Level | [−1] | [0] | [+1] | |
| Eudragit® RLPO content (g) | A | 1 | 2 | 3 |
| Ethylcellulose content (g) | B | 1 | 1.5 | 2 |
| Anhydrous d-glucose content (g) | C | 0.25 | 0.5 | 0.75 |
| Dependent | Constraints | |||
| % RIF released at 0.5 h = R1 | R1 ≤ 15% | |||
| % RIF released at 2 h = R2 | R2 = maximize | |||
| % RIF released at 4 h = R3 | R3 = maximize | |||
| % RIF released at 6 h = R4 | R4 = maximize | |||
| % RIF released at 8 h = R5 | R5 = maximize | |||
| Encapsulation efficiency (%) = R6 | R6 = maximize | |||
| Buoyancy = R7 | R7 = maximize | |||
| Lag time (seconds) = R8 | R8 = minimize | |||
| % RIF released at 12h = R9 | R9 = maximize | |||
| % Yield = R10 | R10 = maximize | |||
| Parameter | Setting | ||
| Chromatographic parameters: | - | ||
| Flow rate (mL/min) | 1.0 | ||
| Buffer pH | 3.0 | ||
| Starting % MeOH | 3.0 | ||
| Ending % MeOH | 75 | ||
| Curve profile | 6 | ||
| Gradient duration (min) | 4 | ||
| Injection volume (µL) | 10 | ||
| Column temperature (°C) | 22 | ||
| Wavelength (nm) | 254 (RIF), 265 (INH), 268 (PZA) | ||
| Mobile phase composition | Methanol (3% v/v) and 10 mM phosphate buffer (97% v/v) | ||
| System Suitability Parameters | INH | PZA | RIF |
| Theoretical number of plates (N) | 8262.616 | 10,993.81 | 83,450.80 |
| Capacity factor | 1.06560 | 4.316160 | 7.77262 |
| Resolution factor | - | 21.54936 | 8.09081 |
| Tailing factor | 1.08756 | 1.099920 | 1.65720 |
| Retention time | 3.261 | 7.630 | 12.793 |
| Correlation coefficient (R2) | 0.9991 | 0.9995 | 0.9990 |
| Limit of Detection (LOD) n = 5 | 0.3 ± 0.17 | 0.3 ± 1.87 | 5.0 ± 0.75 |
| Limit of quantitation (LOQ) | 0.1 | 0.1 | 0.5 |
| Functional Group | RIF | 1:1 Mixture of RIF and | ||||
|---|---|---|---|---|---|---|
| Ethylcellulose | Eudragit® RLPO | d-Glucose | Sodium Bicarbonate | Citric Acid | ||
| –CH3 stretching | 2941.77 | 2936.70 | 2936.70 | 2941.77 | 2941.77 | 2941.78 |
| –CH3O asymmetric stretching | 2875.94 | 2875.94 | 2875.94 | 2881.01 | 2875.94 | 2875.94 |
| –C=O acetyl stretching | 1735.99 | 1735.99 | 1735.99 | 1735.99 | 1735.35 | 1735.99 |
| –C=N asymmetric bending | 1673.87 | 1676.40 | 1676.40 | 1676.40 | 1676.40 | 1673.87 |
| –C=C stretching | 1562.64 | 1560.11 | 1560.11 | 1560.11 | 1562.40 | 1562.64 |
| –C–N– stretching | 1427.81 | 1426.10 | 1426.12 | 1426.12 | 1426.12 | 1427.44 |
| Run | HPMC-AS (g) | Eudragit®L100 (g) | Homogenisation Speed (rpm) | Yield (%) | INH Released at 4 h (%) | INH Released at 12 h (%) | Encapsulation Efficiency (%) | INH Released at 24 h (%) |
|---|---|---|---|---|---|---|---|---|
| 1 | 3.41 | 2.00 | 1500 | 51.32 | 17.45 | 45.35 | 89.67 | 80.26 |
| 2 | 2.00 | 2.00 | 2000 | 43.30 | 29.74 | 64.44 | 72.57 | 89.56 |
| 3 | 2.00 | 2.00 | 500 | 56.52 | 23.30 | 55.35 | 77.42 | 87.45 |
| 4 | 3.00 | 3.00 | 2500 | 48.32 | 19.05 | 36.25 | 65.16 | 71.28 |
| 5 | 0.485 | 2.00 | 1500 | 21.91 | 38.42 | 65.41 | 50.51 | 93.16 |
| 6 | 3.00 | 1.00 | 2500 | 45.83 | 39.33 | 53.99 | 70.99 | 88.67 |
| 7 | 1.00 | 3.00 | 2500 | 40.00 | 13.02 | 59.45 | 68.65 | 95.20 |
| 8 | 2.00 | 0.485 | 1500 | 21.14 | 71.04 | 55.78 | 55.48 | 89.81 |
| 9 | 2.00 | 2.00 | 3500 | 31.83 | 36.66 | 68.89 | 60.84 | 93.58 |
| 10 | 1.00 | 1.00 | 2500 | 22.27 | 45.86 | 67.51 | 66.72 | 90.96 |
| 11 | 2.00 | 3.41 | 1500 | 47.17 | 8.82 | 47.75 | 89.41 | 87.87 |
| Source | Sum of Squares | Df | Mean Square | F-Value | p-Value | Prob > F |
|---|---|---|---|---|---|---|
| Panel A | ||||||
| Model | 420.28 | 6 | 70.05 | 6.37 | 0.0427 | Significant |
| A-HPMC-AS | 246.99 | 1 | 246.99 | 22.48 | 0.009 | Significant |
| B-Eudragit® L100 | 31.57 | 1 | 31.57 | 2.87 | 0.1654 | - |
| C-Homogenisation speed | 3.29 | 1 | 3.29 | 0.30 | 0.6134 | - |
| AB | 116.98 | 1 | 116.98 | 10.65 | 0.0310 | Significant |
| AC | 7.93 | 1 | 7.93 | 0.72 | 0.4434 | - |
| BC | 13.53 | 1 | 13.53 | 1.23 | 0.3294 | - |
| Residual | 43.96 | 4 | 10.99 | - | - | - |
| Cor total | 464.24 | 10 | - | - | - | - |
| Panel B | ||||||
| Std. deviation | 3.31 | - | - | - | - | - |
| Mean | 87.98 | - | - | - | - | - |
| % C.V | 3.77 | - | - | - | - | - |
| Press | 1655.17 | - | - | - | - | - |
| R2 | 0.9053 | - | - | - | - | - |
| Adjusted R2 | 0.7633 | - | - | - | - | - |
| Adeq precision | 9.045 | - | - | - | - | - |
| Response | Fitting Model | Significant Factors | Equation |
|---|---|---|---|
| %Yield-Y1 | Linear | A-HPMC-AS B-Eudragit® L100 | Y1 = 39.06 + 9.18A + 7.13B − 3.05C |
| % INH released at 4 h in pH 1.2 0.1M HCl-Y2 | Linear | B-Eudragit® L100 | Y2 = 31.15 − 3.77A − 17.64B + 0.73C |
| %INH released at 24 h in pH 6.8 0.1 M phosphate buffer-Y3 | 2FI | A-HPMC-AS B-Eudragit® L100 | Y3 = 87.98 − 5.56A − 1.99B + 0.64C − 5.41AB − 1.41AC − 1.84BC |
| Encapsulation efficiency (%EE)-Y4 | 2FI | A-HPMC-AS B-Eudragit® L100 C-Homogenisation speed | Y4 = 72.49 + 5.77A + 6.76B − 1.48C − 9.44AB − 11.42AC − 7.40BC |
| Batch | Response | Predicted Value | Observed Value | Residual | % P.E. |
|---|---|---|---|---|---|
| INH-012 | % Yield | 51.35 | 50.41 | 0.940 | 1.830 |
| % INH released at 4 h in pH1.2 | 5.745 | 8.510 | 5.989 | 48.13 | |
| Encapsulation efficiency (%) | 90.81 | 87.37 | 3.440 | 3.788 | |
| % INH released at 24 h in pH 6.8 | 86.62 | 90.96 | 4.340 | 5.010 | |
| INH-013 | % Yield | 51.35 | 52.89 | 1.540 | 2.999 |
| % INH released at 4 h in pH 1.2 | 5.745 | 8.810 | 3.219 | 53.35 | |
| Encapsulation efficiency (%) | 90.81 | 80.79 | 10.02 | 11.03 | |
| % INH released at 24 h in pH 6.8 | 86.62 | 99.20 | 12.58 | 14.52 | |
| INH-014 | % Yield | 51.35 | 53.92 | 2.570 | 5.004 |
| % INH released at 4 h in pH 1.2 | 5.745 | 8.720 | 2.975 | 51.78 | |
| Encapsulation efficiency (%) | 90.81 | 80.77 | 10.04 | 11.06 | |
| % INH released at 24 h in pH 6.8 | 86.62 | 87.87 | 1.250 | 1.443 |
| Source | Sum of Squares | Df | Mean Square | F-Value | p-Value | Prob > F |
|---|---|---|---|---|---|---|
| Panel A | ||||||
| Model | 444.24 | 9 | 49.36 | 75.19 | <0.0001 | Significant |
| A-Eudragit® RLPO | 38.36 | 1 | 38.36 | 58.43 | 0.0001 | Significant |
| B-Ethylcellulose | 334.76 | 1 | 334.76 | 509.90 | <0.0001 | Significant |
| C-d-glucose | 8.88 | 1 | 8.88 | 13.52 | 0.0079 | Significant |
| AB | 2.34 | 1 | 2.34 | 3.56 | 0.1011 | - |
| AC | 0.078 | 1 | 0.078 | 0.12 | 0.7399 | - |
| BC | 0.80 | 1 | 0.80 | 1.22 | 0.3053 | - |
| A2 | 5.24 | 1 | 5.24 | 7.98 | 0.0256 | Significant |
| B2 | 53.64 | 1 | 53.64 | 81.70 | <0.0001 | Significant |
| C2 | 2.57 | 1 | 2.57 | 3.92 | 0.0881 | - |
| Residual | 4.60 | 7 | 0.66 | - | - | - |
| Lack of fit | 1.79 | 1 | 1.79 | 0.89 | 0.7200 | - |
| Pure error | 0.46 | 4 | 0.12 | - | - | - |
| Cor total | 448.84 | 16 | - | - | - | - |
| Panel B | ||||||
| Std. deviation | 0.81 | - | - | - | - | - |
| Mean | 8.24 | - | - | - | - | - |
| % C.V | 9.83 | - | - | - | - | - |
| Press | 66.87 | - | - | - | - | - |
| R2 | 0.9898 | - | - | - | - | - |
| Adjusted R2 | 0.9766 | - | - | - | - | - |
| Predicted R2 | 0.8510 | - | - | - | - | - |
| Adeq precision | 27.866 | - | - | - | - | - |
| Response | Fitting Model | Significant Factors | Equation |
|---|---|---|---|
| % RIF released at 0.5 h = R1 | Quadratic | A-Eudragit® RLPO B-Ethylcellulose C-d-glucose | R1 = 7.46 − 2.19 − 6.47B + 1.05C + 0.76AB − 0.14AC + 0.45BC − 1.12A2 + 3.57B2 − 0.78C2 |
| % RIF released at 2 h = R2 | Quadratic | A-Eudragit® RLPO B-Ethylcellulose | R2 =34.44 − 6.59A − 6.42B + 2.42 C + 4.93AB − 1.14 AC + 1.55BC − 6.00A2 − 7.86B2 − 4.37C2 |
| % RIF released at 4 h = R3 | Quadratic | B-Ethylcellulose C-d-glucose | R3 = 47.3 − 0.54A − 8.47B + 2.03 C + 0.41AB − 0.53AC + 2.06BC − 3.94A2 − 2.29B2 − 4.65C2 |
| % RIF released at 6 h = R4 | Linear | B-Ethylcellulose C-d-glucose | R4 = 67.00 − 2.15A − 7.44B + 3.32C |
| % RIF released at 8 h = R5 | Quadratic | A-Eudragit® RLPO B-Ethylcellulose C-d-glucose | R5 = 83.95 − 3.85A - 8.78B + 2.28C − 0.76AB + 1.12AC + 0.038BC − 3.01A2 − 4.23B2 − 1.92C2 |
| Encapsulation efficiency (%) = R6 | Quadratic | A-Eudragit® RLPO B-Ethylcellulose C-d-glucose | R6 = 77.46 + 9.44A + 4.29B − 1.26C − 2.51AB + 1.37AC + 0.32BC − 0.59A2 + 2.14B2 + 4.77C2 |
| Buoyancy = R7 | Quadratic | A-Eudragit® RLPO B-Ethylcellulose | R7 = 60.17 − 2.60A + 5.10B + 2.07C |
| Lag time (s) = R8 | Quadratic | A-Eudragit® RLPO B-Ethylcellulose C-d-glucose | R8 =151.00 + 52.88A − 19.75B + 13.13C − 39.25AB − 2.50AC + 13.75BC − 19.00A2 − 30.25B2 − 14.50C2 |
| % RIF released at 12 h = R9 | Quadratic | A-Eudragit® RLPO B-Ethylcellulose C-d-glucose | R9 = 90.37 − 2.12A − 7.80B + 3.04 C + 0.17AB + 1.54 AC + 1.61BC − 4.14A2 + 0.74B2 − 1.29C2 |
| % Yield = R10 | Quadratic | A-Eudragit® RLPO B-Ethylcellulose | R10 = 71.16 + 8.60A + 5.34B − 0.90C − 0.80AB − 0.65AC + 1.04BC + 2.23A2 − 2.07B2 + 1.93C2 |
| Batch | Response | Prediction Accuracy | |||
|---|---|---|---|---|---|
| Predicted | Actual | Residual | % P.E. | ||
| A | % RIF released at 0.5 h (R1) | 15.39 | 15.65 | 0.26 | 1.66 |
| % RIF released at 2 h (R2) | 36.99 | 38.04 | 1.05 | 2.76 | |
| % RIF released at 4 h (R3) | 48.58 | 49.18 | 0.6 | 1.22 | |
| % RIF released at 6 h (R4) | 68.71 | 65.70 | 3.01 | 4.58 | |
| % RIF released at 8 h (R5) | 76.55 | 78.55 | 2 | 2.55 | |
| Encapsulation efficiency (%) (R6) | 94.71 | 94.80 | 0.09 | 0.095 | |
| % Buoyancy (R7) | 62.23 | 58.62 | 3.61 | 6.16 | |
| Floating lag time (seconds) (R8) | 149.63 | 144 | 5.63 | 3.91 | |
| % RIF released at 12 h (R9) | 92.12 | 87.89 | 4.23 | 4.81 | |
| % Yield (R10) | 82.19 | 81.95 | 0.24 | 0.29 | |
| B | % RIF released at 0.5 h (R1) | 15.39 | 15.81 | 0.42 | 2.66 |
| % RIF released at 2 h (R2) | 36.99 | 38.58 | 1.59 | 4.12 | |
| % RIF released at 4 h (R3) | 48.58 | 49.11 | 0.53 | 1.08 | |
| % RIF released at 6 h (R4) | 68.71 | 64.57 | 4.14 | 6.41 | |
| % RIF released at 8 h (R5) | 76.55 | 77.08 | 0.53 | 0.688 | |
| Encapsulation efficiency (%) (R6) | 94.71 | 99.19 | 4.48 | 4.52 | |
| % Buoyancy (R7) | 62.23 | 56.22 | 6.01 | 10.69 | |
| Floating lag time (seconds) (R8) | 149.63 | 140 | 9.63 | 6.88 | |
| % RIF released at 12 h (R9) | 92.12 | 86.44 | 5.68 | 6.57 | |
| % Yield (R10) | 82.19 | 87.97 | 5.78 | 6.57 | |
| C | % RIF released at 0.5 h (R1) | 15.39 | 16.03 | 0.64 | 3.99 |
| % RIF released at 2 h (R2) | 36.99 | 39.32 | 2.33 | 5.93 | |
| % RIF released at 4 h (R3) | 48.58 | 47.22 | 1.36 | 2.88 | |
| % RIF released at 6 h (R4) | 68.71 | 66.73 | 1.98 | 2.97 | |
| % RIF released at 8 h (R5) | 76.55 | 78.44 | 1.89 | 2.41 | |
| Encapsulation efficiency (%) (R6) | 94.71 | 96.99 | 2.28 | 2.35 | |
| % Buoyancy (R7) | 62.23 | 55.81 | 6.42 | 11.50 | |
| Floating lag time (seconds) (R8) | 149.63 | 142 | 7.63 | 5.37 | |
| % RIF released at 12 h (R9) | 92.12 | 94.71 | 2.59 | 2.74 | |
| % Yield (R10) | 82.19 | 85.88 | 3.69 | 4.20 | |
| Run | Mean Particle Size | Standard Deviation | % RSD | % Mass Retained on Each Sieve | |||
|---|---|---|---|---|---|---|---|
| 315 µm | 800 µm | 1250 µm | 2000 µm | ||||
| 1 | 745.99 | 104.26 | 13.98 | - | 44.7 | 55.3 | - |
| 2 | 680.60 | 367.07 | 53.93 | - | 52.4 | 47.6 | - |
| 3 | 891.28 | 161.02 | 18.07 | - | 0 | 56.1 | 43.9 |
| 4 | 615.85 | 84.20 | 13.67 | - | 58.9 | 41.1 | - |
| 5 | 494.15 | 113.84 | 23.04 | - | 79.6 | 19.4 | - |
| 6 | 496.82 | 105.24 | 21.18 | - | 59.5 | 40.5 | - |
| 7 | 515.07 | 103.39 | 20.07 | - | 58.8 | 41.2 | - |
| 8 | 903.35 | 197.10 | 21.82 | - | 78.7 | 21.3 | |
| 9 | 659.30 | 206.35 | 31.30 | 44.3 | 47.2 | 8.5 | - |
| 10 | 415.76 | 76.93 | 18.50 | - | 100 | - | - |
| 11 | 693.43 | 154.20 | 22.24 | - | 45.3 | 54.7 | - |
| 12 | 695.22 | 124.90 | 17.97 | ||||
| Batch | Mean Particle Size | Standard Deviation | % RSD | % Mass of Microspheres Retained per Sieve Size (µm) | |
|---|---|---|---|---|---|
| 800 | 1250 | ||||
| RIF001 | 515.08 | 209.79 | 40.72 | 95.50 | 4.50 |
| RIF002 | 489.77 | 115.63 | 23.61 | 97.27 | 2.73 |
| RIF003 | 476.59 | 188.78 | 39.61 | 92.54 | 7.46 |
| RIF004 | 459.02 | 145.89 | 31.78 | 94.91 | 5.09 |
| RIF005 | 540.00 | 125.39 | 23.22 | 97.33 | 2.67 |
| RIF006 | 481.42 | 92.81 | 19.28 | 98.42 | 1.58 |
| RIF007 | 541.87 | 108.41 | 20.01 | 96.71 | 3.29 |
| RIF008 | 423.19 | 121.86 | 28.80 | 97.88 | 2.12 |
| RIF009 | 545.96 | 185.57 | 33.99 | 96.33 | 3.67 |
| RIF010 | 452.69 | 88.34 | 19.51 | 100 | 0.00 |
| RIF011 | 499.55 | 177.19 | 35.47 | 98.11 | 1.89 |
| RIF012 | 601.71 | 167.94 | 27.91 | 81.83 | 18.17 |
| RIF013 | 620.07 | 102.67 | 16.56 | 72.78 | 27.22 |
| RIF014 | 537.71 | 128.39 | 23.88 | 92.64 | 7.36 |
| RIF015 | 576.73 | 95.39 | 16.54 | 93.12 | 6.88 |
| RIF016 | 485.38 | 170.33 | 35.09 | 54.18 | 45.82 |
| RIF017 | 477.58 | 102.58 | 21.48 | 96.67 | 3.33 |
| RIF018 | 547.22 | 108.63 | 19.85 | 94.45 | 5.55 |
| RIF021 | 526.49 | 91.88 | 17.45 | 96.77 | 3.23 |
| Product | Initial µg/mL | Final µg/mL | % Recovery ± SD (n = 6) | % Degradation |
|---|---|---|---|---|
| Rimactane® 150 | 461.5 | 444.5 | 96.3 ± 0.2 | 3.70 |
| Rimactane® 150 in presence of BE-TABS Isoniazid 100 | 461.5 | 333.6 | 74.4 ± 0.2 | 21.9 |
| Rimactane® 150 in presence of INH API | 461.5 | 335.8 | 72.8 ± 0.2 | 24.4 |
| RIF API | 461.5 | 444.9 | 96.4 ± 0.2 | 3.59 |
| RIF API in presence of INH API | 461.5 | 367.3 | 79.6 ± 0.1 | 19.1 |
| Rimactane® 150 in presence INH microspheres | 461.5 | 426.6 | 92.4 ± 0.3 | 4.05 |
| RIF API in presence of INH microspheres | 461.5 | 436.7 | 94.6 ± 0.2 | 3.86 |
| RIF microspheres in presence of INH microspheres | 416.9 | 398.4 | 95.6.0 ± 0.1 | 4.44 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mwila, C.; Walker, R.B. Improved Stability of Rifampicin in the Presence of Gastric-Resistant Isoniazid Microspheres in Acidic Media. Pharmaceutics 2020, 12, 234. https://doi.org/10.3390/pharmaceutics12030234
Mwila C, Walker RB. Improved Stability of Rifampicin in the Presence of Gastric-Resistant Isoniazid Microspheres in Acidic Media. Pharmaceutics. 2020; 12(3):234. https://doi.org/10.3390/pharmaceutics12030234
Chicago/Turabian StyleMwila, Chiluba, and Roderick B. Walker. 2020. "Improved Stability of Rifampicin in the Presence of Gastric-Resistant Isoniazid Microspheres in Acidic Media" Pharmaceutics 12, no. 3: 234. https://doi.org/10.3390/pharmaceutics12030234