Crystallisation Behaviour of Pharmaceutical Compounds Confined within Mesoporous Silicon



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. Drug Crystallisation and Amorphisation
1.2. Drug Confinement into Porous Substrates
1.3. Crystallisation Characterisation Techniques
1.4. Porous Silicon
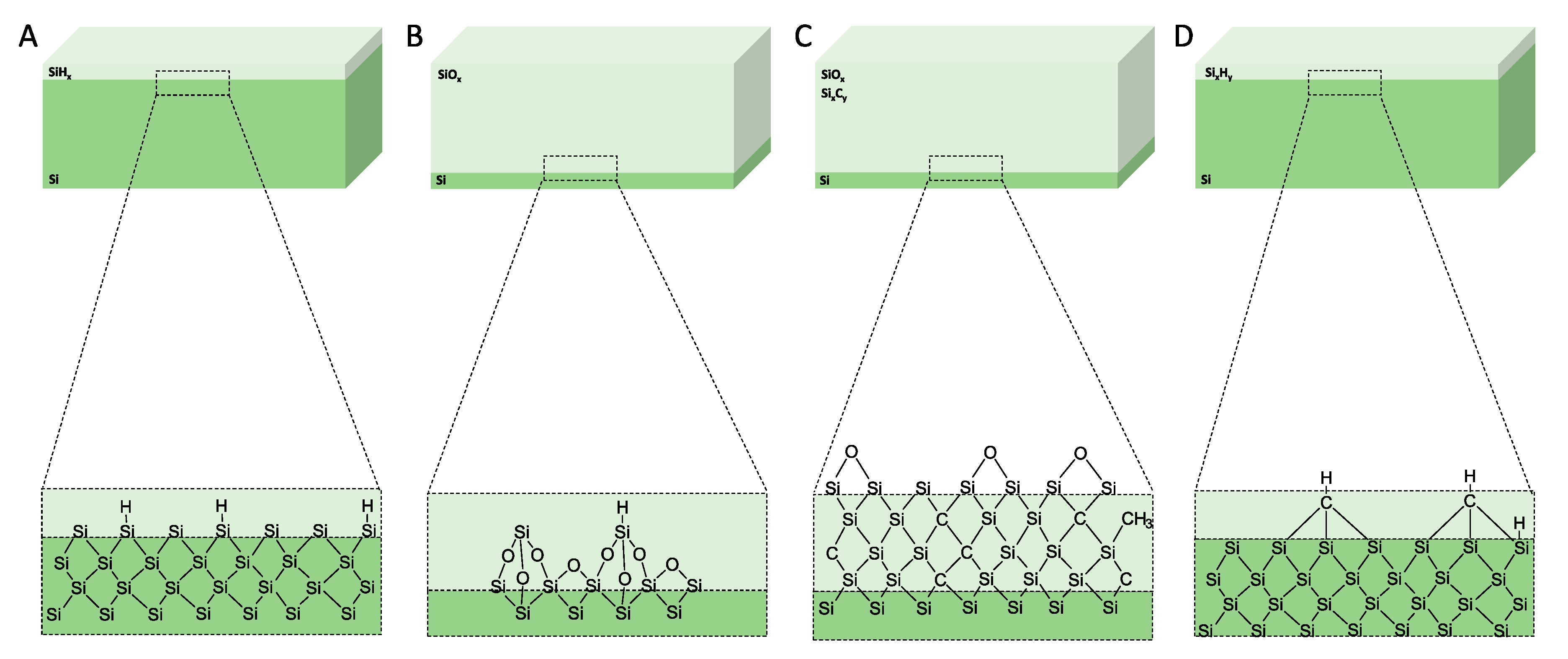
1.5. Porous Silicon Surface Stabilisation
1.5.1. Thermal Oxidation
1.5.2. Hydrosilylation
1.5.3. Thermal Carbonisation
1.5.4. Thermal Hydrocarbonisation
1.6. Metalothermic Reduction
1.7. Drug Loading and Release from pSi
2. Summary
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Singh, A.; Worku, Z.A.; Van Den Mooter, G. Oral formulation strategies to improve solubility of poorly water-soluble drugs. Expert Opin. Drug Deliv. 2011, 8, 1361–1378. [Google Scholar] [CrossRef] [PubMed]
- Seto, J.; Romero, P.A. Shaping it up: Design and engineering of biominerals and crystalline materials from the bottom up. In Biomineralization and Biomaterials: Fundamentals and Applications; Woodhead Publishing: Cambridge, UK, 2015; pp. 3–50. ISBN 9781782423560. [Google Scholar]
- Briuglia, M.L.; Sefcik, J.; Horst, J.H.T. Measuring secondary nucleation through single crystal seeding. Cryst. Growth Des. 2019, 19, 421–429. [Google Scholar] [CrossRef] [Green Version]
- Lynch, M.B.; Lawrence, S.E.; Nolan, M. Predicting nucleation of isonicotinamide from the solvent-solute interactions of isonicotinamide in common organic solvents. J. Phys. Chem. A 2018, 122, 3301–3312. [Google Scholar] [CrossRef] [PubMed]
- Erdemir, D.; Lee, A.Y.; Myerson, A.S. Nucleation of crystals from solution: Classical and two-step models. Acc. Chem. Res. 2009, 42, 621–629. [Google Scholar] [CrossRef]
- Ter Horst, J.H.; Schmidt, C.; Ulrich, J. Fundamentals of industrial crystallization. In Handbook of Crystal Growth: Bulk Crystal Growth, 2nd ed.; Elsevier: Amsterdam, The Netherlands, 2015; Volume 2, pp. 1317–1349. ISBN 9780444633033. [Google Scholar]
- Nicoud, L.; Licordari, F.; Myerson, A.S. Estimation of the solubility of metastable polymorphs: A critical review. Cryst. Growth Des. 2018, 18, 7228–7237. [Google Scholar] [CrossRef]
- Ferrari, E.S.; Davey, R.J.; Cross, W.I.; Gillon, A.L.; Towler, C.S. Crystallization in polymorphic systems: The solution-mediated transformation of β to α glycine. Cryst. Growth Des. 2003, 3, 53–60. [Google Scholar] [CrossRef]
- Chen, J.; Sarma, B.; Evans, J.M.B.B.; Myerson, A.S. Pharmaceutical crystallization. Cryst. Growth Des. 2011, 11, 887–895. [Google Scholar] [CrossRef] [Green Version]
- Grohganz, H.; Löbmann, K.; Priemel, P.; Jensen, K.; Graeser, K.; Strachan, C.; Rades, T. Amorphous drugs and dosage forms. J. Drug Deliv. Sci. Technol. 2013, 23, 403–408. [Google Scholar] [CrossRef]
- Garcia-Bennett, A.E.; Lau, M.; Bedford, N. Probing the amorphous state of pharmaceutical compounds within mesoporous material using pair distribution function analysis. J. Pharm. Sci. 2018, 107, 2216–2224. [Google Scholar] [CrossRef]
- Hancock, B.C.; Zografi, G. Characteristics and significance of the amorphous state in pharmaceutical systems. J. Pharm. Sci. 2002, 86, 1–12. [Google Scholar] [CrossRef]
- Hancock, B.C.; Parks, M. What is the true solubility advantage for amorphous pharmaceuticals? Pharm. Res. 2000, 17, 397–404. [Google Scholar] [CrossRef] [PubMed]
- Vallet-Regí, M.; Balas, F.; Arcos, D. Mesoporous materials for drug delivery. Angew. Chem. Int. Ed. 2007, 46, 7548–7558. [Google Scholar] [CrossRef] [PubMed]
- Brás, A.R.; Merino, E.G.; Neves, P.D.; Fonseca, I.M.; Dionísio, M.; Schönhals, A.; Correia, N.T. Amorphous ibuprofen confined in nanostructured silica materials: A dynamical approach. J. Phys. Chem. C 2011, 115, 4616–4623. [Google Scholar] [CrossRef]
- Uejo, F.; Limwikrant, W.; Moribe, K.; Yamamoto, K. Dissolution improvement of fenofibrate by melting inclusion in mesoporous silica. Asian J. Pharm. Sci. 2013, 8, 329–335. [Google Scholar] [CrossRef] [Green Version]
- Kiwilsza, A.; Milanowski, B.; Drużbicki, K.; Coy, L.E.; Grzeszkowiak, M.; Jarek, M.; Mielcarek, J.; Lulek, J.; Pajzderska, A.; Wkasicki, J. Mesoporous drug carrier systems for enhanced delivery rate of poorly water-soluble drug: Nimodipine. J. Porous Mater. 2015, 22, 817–829. [Google Scholar] [CrossRef] [Green Version]
- Bavnhøj, C.G.; Knopp, M.M.; Madsen, C.M.; Löbmann, K. The role interplay between mesoporous silica pore volume and surface area and their effect on drug loading capacity. Int. J. Pharm. X 2019, 1, 100008. [Google Scholar] [CrossRef]
- Mäkilä, E.; Ferreira, M.P.A.A.; Kivelä, H.; Niemi, S.M.; Correia, A.; Shahbazi, M.A.; Kauppila, J.; Hirvonen, J.; Santos, H.A.; Salonen, J. Confinement effects on drugs in thermally hydrocarbonized porous silicon. Langmuir 2014, 30, 2196–2205. [Google Scholar] [CrossRef]
- Qian, K.K.; Bogner, R.H. Application of mesoporous silicon dioxide and silicate in oral amorphous drug delivery systems. J. Pharm. Sci. 2012, 101, 444–463. [Google Scholar] [CrossRef]
- Kaukonen, A.M.; Laitinen, L.; Salonen, J.; Tuura, J.; Heikkilä, T.; Limnell, T.; Hirvonen, J.; Lehto, V.P. Enhanced in vitro permeation of furosemide loaded into thermally carbonized mesoporous silicon (TCPSi) microparticles. Eur. J. Pharm. Biopharm. 2007, 66, 348–356. [Google Scholar] [CrossRef]
- Brouwers, J.; Brewster, M.E.; Augustijns, P. Supersaturating drug delivery systems: The answer to solubility-limited oral bioavailability? J. Pharm. Sci. 2009, 98, 2549–2572. [Google Scholar] [CrossRef]
- Mellaerts, R.; Aerts, C.A.; Van Humbeeck, J.; Augustijns, P.; Van Den Mooter, G.; Martens, J.A. Enhanced release of itraconazole from ordered mesoporous SBA-15 silica materials. Chem. Commun. 2007, 1375–1377. [Google Scholar] [CrossRef]
- Van Speybroeck, M.; Barillaro, V.; Thi, T.D.; Mellaerts, R.; Martens, J.; Van Humbeeck, J.; Vermant, J.; Annaert, P.; Van Den Mooter, G.; Augustijns, P. Ordered mesoporous silica material SBA-15: A broad-spectrum formulation platform for poorly soluble drugs. J. Pharm. Sci. 2009, 98, 2648–2658. [Google Scholar] [CrossRef] [PubMed]
- Ha, J.-M.; Wolf, J.H.; Hillmyer, M.A.; Ward, M.D. Polymorph selectivity under nanoscopic confinement. J. Am. Chem. Soc. 2004, 126, 3382–3383. [Google Scholar] [CrossRef] [PubMed]
- Choi, K.-M.; Kuroda, K. Polymorph control of calcium carbonate on the surface of mesoporous silica. Cryst. Growth Des. 2012, 12, 887–893. [Google Scholar] [CrossRef]
- Sliwinska-Bartkowiak, M.; Dudziak, G.; Gras, R.; Sikorski, R.; Radhakrishnan, R.; Gubbins, K. Freezing behavior in porous glasses and MCM-41. Colloids Surf. A Physicochem. Eng. Asp. 2001, 187, 523–529. [Google Scholar] [CrossRef]
- Dwyer, L.M.; Michaelis, V.K.; O’Mahony, M.; Griffin, R.G.; Myerson, A.S. Confined crystallization of fenofibrate in nanoporous silica. CrystEngComm 2015, 17, 7922–7929. [Google Scholar] [CrossRef] [Green Version]
- Nartowski, K.P.; Tedder, J.; Braun, D.E.; Fábián, L.; Khimyak, Y.Z. Building solids inside nano-space: From confined amorphous through confined solvate to confined ‘metastable’ polymorph. Phys. Chem. Chem. Phys. 2015, 17, 24761–24773. [Google Scholar] [CrossRef] [Green Version]
- Salonen, J.; Paski, J.; Vähä-Heikkilä, K.; Heikkilä, T.; Björkqvist, M.; Lehto, V.-P. Determination of drug load in porous silicon microparticles by calorimetry. Phys. Status Solidi 2005, 202, 1629–1633. [Google Scholar] [CrossRef]
- Santana, M.; Estevez, J.O.; Agarwal, V.; Herrera-Becerra, R. Room temperature crystallization of hydroxyapatite in porous silicon structures. Nanoscale Res. Lett. 2016, 11, 497. [Google Scholar] [CrossRef] [Green Version]
- Kalluri, J.R.; West, J.; Akkaraju, G.R.; Canham, L.T.; Coffer, J.L. Plant-derived tandem drug/mesoporous silicon microcarrier structures for anti-inflammatory therapy. ACS Omega 2019, 4, 8359–8364. [Google Scholar] [CrossRef]
- Chayen, N.E.; Saridakis, E.; El-Bahar, R.; Nemirovsky, Y. Porous silicon: An effective nucleation-inducing material for protein crystallization. J. Mol. Biol. 2001, 312, 591–595. [Google Scholar] [CrossRef] [PubMed]
- Webber, J.B.W.; Dore, J.C. Neutron Diffraction Cryoporometry—A measurement technique for studying mesoporous materials and the phases of contained liquids and their crystalline forms. Nucl. Instrum. Methods Phys. Res. Sect. A Accel. Spectrom. Detect. Assoc. Equip. 2008, 586, 356–366. [Google Scholar] [CrossRef] [Green Version]
- Siefker, J.; Biehl, R.; Kruteva, M.; Feoktystov, A.; Coppens, M.-O. Confinement facilitated protein stabilization as investigated by small-angle neutron scattering. J. Am. Chem. Soc. 2018, 140, 12720–12723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pantalei, C.; Senesi, R.; Andreani, C.; Sozzani, P.; Comotti, A.; Bracco, S.; Beretta, M.; Sokol, P.E.; Reiter, G. Interaction of single water molecules with silanols in mesoporous silica. Phys. Chem. Chem. Phys. 2011, 13, 6022–6028. [Google Scholar] [CrossRef]
- Rantanen, J.; Majda, D.; Riikonen, J.; Lehto, V.-P. The atomic local ordering of SBA-15 studied with pair distribution function analysis, and its relationship to porous structure and thermal stability. Acta Mater. 2019, 175, 341–347. [Google Scholar] [CrossRef]
- Rimsza, J.M.; Du, J. Structural and mechanical properties of nanoporous silica. J. Am. Ceram. Soc. 2014, 97, 772–781. [Google Scholar] [CrossRef]
- Hsieh, K.-Y.; Bendeif, E.-E.; Gansmuller, A.; Pillet, S.; Woike, T.; Schaniel, D. Structure and dynamics of guest molecules confined in a mesoporous silica matrix: Complementary NMR and PDF characterisation. RSC Adv. 2013, 3, 26132–26141. [Google Scholar] [CrossRef]
- Nartowski, K.P.; Malhotra, D.; Hawarden, L.E.; Sibik, J.; Iuga, D.; Zeitler, J.A.; Fábián, L.; Khimyak, Y.Z. 19F NMR spectroscopy as a highly sensitive method for the direct monitoring of confined crystallization within nanoporous materials. Angew. Chem. Int. Ed. 2016, 55, 8904–8908. [Google Scholar] [CrossRef]
- Halib, N.; Mohd Amin, M.C.I.; Ahmad, I.; Abrami, M.; Fiorentino, S.; Farra, R.; Grassi, G.; Musiani, F.; Lapasin, R.; Grassi, M. Topological characterization of a bacterial cellulose-acrylic acid polymeric matrix. Eur. J. Pharm. Sci. 2014, 62, 326–333. [Google Scholar] [CrossRef]
- Chui, M.M.; Phillips, R.J.; McCarthy, M.J. Measurement of the porous microstructure of hydrogels by nuclear magnetic resonance. J. Colloid Interface Sci. 1995, 174, 336–344. [Google Scholar] [CrossRef]
- Sørland, G.H.; Djurhuus, K.; Widerøe, H.; Lien, J.; Skauge, A. Absolute pore size distributions from NMR. Diffus. Fundam. 2007, 5, 4.1–4.15. [Google Scholar]
- Uhlir, A. Electrolytic shaping of germanium and silicon. Bell Syst. Tech. J. 1956, 35, 333–347. [Google Scholar] [CrossRef]
- Watanabe, Y.; Sakai, T. Application of a thick anode film to semiconductor devices. Rev. Electr. Commun. Lab. 1971, 19, 899–903. [Google Scholar]
- Canham, L.T. Silicon quantum wire array fabrication by electrochemical and chemical dissolution of wafers. Appl. Phys. Lett. 1990, 57, 1046–1048. [Google Scholar] [CrossRef]
- Lehmann, V.; Gösele, U. Porous silicon formation: A quantum wire effect. Appl. Phys. Lett. 1991, 58, 856–858. [Google Scholar] [CrossRef]
- Canham, L.T. Bioactive silicon structure fabrication through nanoetching techniques. Adv. Mater. 1995, 7, 1033–1037. [Google Scholar] [CrossRef]
- Stewart, M.P.; Buriak, J.M. Chemical and biological applications of porous silicon technology. Adv. Mater. 2000, 12, 859–869. [Google Scholar] [CrossRef]
- Canham, L. Porous silicon as a therapeutic biomaterial. In Proceedings of the 1st Annual International IEEE-EMBS Special Topic Conference on Microtechnologies in Medicine & Biology, Lyon, France, 12–14 October 2000; Dittmar, A., Beebe, D., Eds.; IEEE: Lyon, France, 2000; pp. 109–112. [Google Scholar]
- Bayliss, S.C.; Buckberry, L.D.; Fletcher, I.; Tobin, M.J. The culture of neurons on silicon. Sens. Actuators A Phys. 1999, 74, 139–142. [Google Scholar] [CrossRef]
- Salonen, J.; Kaukonen, A.M.; Hirvonen, J.; Lehto, V.-P. Mesoporous silicon in drug delivery applications. J. Pharm. Sci. 2008, 97, 632–653. [Google Scholar] [CrossRef] [PubMed]
- Tahvanainen, M.; Rotko, T.; Mäkilä, E.A.; Santos, H.; Neves, D.; Laaksonen, T.; Kallonen, A.; Hämäläinen, K.; Peura, M.; Serimaa, R.; et al. Tablet preformulations of indomethacin-loaded mesoporous silicon microparticles. Int. J. Pharm. 2012, 422, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Santos, H.A.; Bimbo, L.M.; Herranz, B.; Shahbazi, M.-A.; Hirvonen, J.; Salonen, J. Nanostructured porous silicon in preclinical imaging: Moving from bench to bedside. J. Mater. Res. 2013, 28, 152–164. [Google Scholar] [CrossRef]
- Canham, L. Routes of formation for porous silicon. In Handbook of Porous Silicon, 2nd ed.; Springer: Berlin/Heidelberg, Germany, 2018; Volumes 1–2, pp. 3–11. ISBN 9783319713816. [Google Scholar]
- Salonen, J.; Björkqvist, M.; Laine, E.; Niinistö, L. Effects of fabrication parameters on porous p+-type silicon morphology. Phys. Status Solidi Appl. Res. 2000, 182, 249–254. [Google Scholar] [CrossRef]
- Salonen, J.; Lehto, V.P. Fabrication and chemical surface modification of mesoporous silicon for biomedical applications. Chem. Eng. J. 2008, 137, 162–172. [Google Scholar] [CrossRef]
- Lehmann, V.; Stengl, R.; Luigart, A. On the morphology and the electrochemical formation mechanism of mesoporous silicon. Mater. Sci. Eng. B Solid-State Mater. Adv. Technol. 2000, 69–70, 11–22. [Google Scholar] [CrossRef]
- Burrows, V.A.; Chabal, Y.J.; Higashi, G.S.; Raghavachari, K.; Christman, S.B. Infrared spectroscopy of Si(111) surfaces after HF treatment: Hydrogen termination and surface morphology. Appl. Phys. Lett. 1988, 53, 998–1000. [Google Scholar] [CrossRef]
- Canham, L.T.; Houlton, M.R.; Leong, W.Y.; Pickering, C.; Keen, J.M. Atmospheric impregnation of porous silicon at room temperature. J. Appl. Phys. 1991, 70, 422–431. [Google Scholar] [CrossRef]
- Loni, A.; Simons, A.J.; Calcott, P.D.J.; Newey, J.P.; Cox, T.I.; Canham, L.T. Relationship between storage media and blue photoluminescence for oxidized porous silicon. Appl. Phys. Lett. 1997, 71, 107–109. [Google Scholar] [CrossRef]
- Hossain, S.M.; Chakraborty, S.; Dutta, S.K.; Das, J.; Saha, H. Stability in photoluminescence of porous silicon. J. Lumin. 2000, 91, 195–202. [Google Scholar] [CrossRef]
- Shabir, Q.; Webb, K.; Nadarassan, D.K.; Loni, A.; Canham, L.T.; Terracciano, M.; De Stefano, L.; Rea, I. Quantification and reduction of the residual chemical reactivity of passivated biodegradable porous silicon for drug delivery applications. Silicon 2018, 10, 349–359. [Google Scholar] [CrossRef] [Green Version]
- Salonen, J.; Laitinen, L.; Kaukonen, A.M.; Tuura, J.; Björkqvist, M.; Heikkilä, T.; Vähä-Heikkilä, K.; Hirvonen, J.; Lehto, V.P. Mesoporous silicon microparticles for oral drug delivery: Loading and release of five model drugs. J. Control. Release 2005, 108, 362–374. [Google Scholar] [CrossRef]
- Sarparanta, M.; Mäkilä, E.; Heikkilä, T.; Salonen, J.; Kukk, E.; Lehto, V.-P.; Santos, H.A.; Hirvonen, J.; Airaksinen, A.J. 18F-labeled modified porous silicon particles for investigation of drug delivery carrier distribution in vivo with positron emission tomography. Mol. Pharm. 2011, 8, 1799–1806. [Google Scholar] [CrossRef] [PubMed]
- Ulin, V.P.; Ulin, N.V.; Soldatenkov, F.Y.; Semenov, A.V.; Bobyl, A.V. Surface of porous silicon under hydrophilization and hydrolytic degradation. Semiconductors 2014, 48, 1211–1216. [Google Scholar] [CrossRef]
- Petrova-Koch, V.; Muschik, T.; Kux, A.; Meyer, B.K.; Koch, F.; Lehmann, V. Rapid-thermal-oxidized porous Si-The superior photoluminescent Si. Appl. Phys. Lett. 1992, 61, 943–945. [Google Scholar] [CrossRef]
- Salonen, J.; Lehto, V.P.; Laine, E. Thermal oxidation of free-standing porous silicon films. Appl. Phys. Lett. 1997, 70, 637–639. [Google Scholar] [CrossRef]
- Mawhinney, D.B.; Glass, J.A.; Yates, J.T. FTIR study of the oxidation of porous silicon. J. Phys. Chem. B 1997, 101, 1202–1206. [Google Scholar] [CrossRef]
- Kumar, R.; Kitoh, Y.; Hara, K. Effect of surface treatment on visible luminescence of porous silicon: Correlation with hydrogen and oxygen terminators. Appl. Phys. Lett. 1993, 63, 3032–3034. [Google Scholar] [CrossRef]
- Takazawa, A.; Tamura, T.; Yamada, M. Photoluminescence mechanisms of porous Si oxidized by dry oxygen. J. Appl. Phys. 1994, 75, 2489–2495. [Google Scholar] [CrossRef]
- Dancil, K.P.S.; Greiner, D.P.; Sailor, M.J. A porous silicon optical biosensor: Detection of reversible binding of IgG to a protein A-modified surface. J. Am. Chem. Soc. 1999, 121, 7925–7930. [Google Scholar] [CrossRef]
- Song, J.H.; Sailor, M.J. Dimethyl sulfoxide as a mild oxidizing agent for porous silicon and its effect on photoluminescence. Inorg. Chem. 1998, 37, 3355–3360. [Google Scholar] [CrossRef]
- Rao, B.V.R.M.; Basu, P.K.; Biswas, J.C.; Lahiri, S.K.; Ghosh, S.; Bose, D.N. Large enhancement of photoluminescence from porous silicon films by post-anodization treatment in boiling hydrogen peroxide. Solid State Commun. 1996, 97, 417–418. [Google Scholar] [CrossRef]
- Linford, M.R.; Chidsey, C.E.D. Alkyl monolayers covalently bonded to silicon surfaces. J. Am. Chem. Soc. 1993, 115, 12631–12632. [Google Scholar] [CrossRef]
- Linford, M.R.; Chidsey, C.E.D.; Fenter, P.; Eisenberger, P.M. Alkyl monolayers on silicon prepared from 1-alkenes and hydrogen-terminated silicon. J. Am. Chem. Soc. 1995, 117, 3145–3155. [Google Scholar] [CrossRef]
- Ponpon, J.P.; Bourdon, B. Oxidation of glow discharge a-Si:H. Solid State Electron. 1982, 25, 875–876. [Google Scholar] [CrossRef]
- Lehner, A.; Steinhoff, G.; Brandt, M.S.; Eickhoff, M.; Stutzmann, M. Hydrosilylation of crystalline silicon (111) and hydrogenated amorphous silicon surfaces: A comparative X-ray photoelectron spectroscopy study. J. Appl. Phys. 2003, 94, 2289–2294. [Google Scholar] [CrossRef]
- Stewart, M.P.; Buriak, J.M. Photopatterned hydrosilylation on porous silicon. Angew. Chem. Int. Ed. 1998, 37, 3257–3260. [Google Scholar] [CrossRef]
- Buriak, J.M.; Stewart, M.P.; Geders, T.W.; Allen, M.J.; Choi, H.C.; Smith, J.; Raftery, D.; Canham, L.T. Lewis acid mediated hydrosilylation on porous silicon surfaces. J. Am. Chem. Soc. 1999, 121, 11491–11502. [Google Scholar] [CrossRef]
- Holland, J.M.; Stewart, M.P.; Allen, M.J.; Buriak, J.M. Metal mediated reactions on porous silicon surfaces. J. Solid State Chem. 1999, 147, 251–258. [Google Scholar] [CrossRef]
- Zazzera, L.A.; Evans, J.F.; Deruelle, M.; Tirrell, M.; Kessel, C.R.; Mckeown, P. Bonding organic molecules to hydrogen-terminated silicon wafers. J. Electrochem. Soc. 1997, 144, 2184–2188. [Google Scholar] [CrossRef]
- Gurtner, C.; Wun, A.W.; Sailor, M.J. Surface modification of porous silicon by electrochemical reduction of organo halides. Angew. Chem. Int. Ed. 1999, 38, 1966–1968. [Google Scholar] [CrossRef]
- Lees, I.N.; Lin, H.; Canaria, C.A.; Gurtner, C.; Sailor, M.J.; Miskelly, G.M. Chemical stability of porous silicon surfaces electrochemically modified with functional alkyl species. Langmuir 2003, 19, 9812–9817. [Google Scholar] [CrossRef]
- Salonen, J.; Kaasalainen, M.; Rauhala, O.P.; Lassila, L.; Hakamies, M.; Jalkanen, T.; Hahn, R.; Schmuki, P.; Mäkilä, E. Thermal carbonization of porous silicon: The current status and recent applications. ECS Trans. 2015, 69, 167–176. [Google Scholar] [CrossRef]
- Kostishko, B.M.; Atazhanov, S.R.; Mikov, S.N.; Koltsova, L.V.; Puzov, I.P. Photoluminescence and degradation properties of the carbonized porous silicon. Tech. Phys. Lett. 1998, 24, 633–635. [Google Scholar] [CrossRef]
- Seo, Y.J.; Cheon, H.J.; Choi, D.J. Enhancement of the thermal stability of photoluminescence by the carbonization of porous silicon. J. Mater. Sci. Lett. 1998, 17, 313–315. [Google Scholar] [CrossRef]
- Buriak, J.M. Silicon-carbon bonds on porous silicon surfaces. Adv. Mater. 1999, 11, 265–267. [Google Scholar] [CrossRef]
- Buriak, J.M.; Allen, M.J. Lewis acid mediated functionalization of porous silicon with substituted alkenes and alkynes. J. Am. Chem. Soc. 1998, 120, 1339–1340. [Google Scholar] [CrossRef]
- Kim, N.Y.; Laibinis, P.E. Derivatization of porous silicon by Grignard reagents at room temperature. J. Am. Chem. Soc. 1998, 120, 4516–4517. [Google Scholar] [CrossRef]
- Bansal, A.; Li, X.; Lauermann, I.; Lewis, N.S.; Yi, S.I.; Weinberg, W.H. Alkylation of Si surfaces using a two-step halogenation/Grignard route. J. Am. Chem. Soc. 1996, 118, 7225–7226. [Google Scholar] [CrossRef]
- Björkqvist, M.; Salonen, J.; Paski, J.; Laine, E. Characterization of thermally carbonized porous silicon humidity sensor. Sens. Actuators A Phys. 2004, 112, 244–247. [Google Scholar] [CrossRef]
- Sciacca, B.; Alvarez, S.D.; Geobaldo, F.; Sailor, M.J. Bioconjugate functionalization of thermally carbonized porous silicon using a radical coupling reaction. Dalton Trans. 2010, 39, 10847–10853. [Google Scholar] [CrossRef]
- Salonen, J.; Björkqvist, M.; Laine, E.; Niinistö, L. Stabilization of porous silicon surface by thermal decomposition of acetylene. Appl. Surf. Sci. 2004, 225, 389–394. [Google Scholar] [CrossRef]
- Wang, C.F.; Mäkilä, E.M.; Bonduelle, C.; Rytkönen, J.; Raula, J.; Almeida, S.; Närvänen, A.; Salonen, J.J.; Lecommandoux, S.; Hirvonen, J.T.; et al. Functionalization of alkyne-terminated thermally hydrocarbonized porous silicon nanoparticles with targeting peptides and antifouling polymers: Effect on the human plasma protein adsorption. ACS Appl. Mater. Interfaces 2015, 7, 2006–2015. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Balasubramanian, V.; Bhat, C.; Vahermo, M.; Mäkilä, E.; Kemell, M.; Fontana, F.; Janoniene, A.; Petrikaite, V.; Salonen, J.; et al. Quercetin-based modified porous silicon nanoparticles for enhanced inhibition of doxorubicin-resistant cancer cells. Adv. Healthc. Mater. 2017, 6, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Canham, L. Porous silicon formation by porous silica reduction. In Handbook of Porous Silicon, 2nd ed.; Springer: Berlin/Heidelberg, Germany, 2018; pp. 1–8. ISBN 9783319713816. [Google Scholar]
- Filsinger, D.H.; Bourrie, D.B. Silica to silicon: Key carbothermic reactions and kinetics. J. Am. Ceram. Soc. 1990, 73, 1726–1732. [Google Scholar] [CrossRef]
- Banerjee, H.D.; Sen, S.; Acharya, H.N. Investigations on the production of silicon from rice husks by the magnesium method. Mater. Sci. Eng. 1982, 52, 173–179. [Google Scholar] [CrossRef]
- Bose, D.N.; Govindacharyulu, P.A.; Banerjee, H.D. Large grain polycrystalline silicon from rice husk. Sol. Energy Mater. 1982, 7, 319–321. [Google Scholar] [CrossRef]
- Haouli, S.; Boudebane, S.; Slipper, I.J.; Lemboub, S.; Gębara, P.; Mezrag, S. Combustion synthesis of silicon by magnesiothermic reduction. Phosphorus. Sulfur. Silicon Relat. Elem. 2018, 193, 280–287. [Google Scholar] [CrossRef]
- Kim, K.H.; Lee, D.J.; Cho, K.M.; Kim, S.J.; Park, J.K.; Jung, H.T. Complete magnesiothermic reduction reaction of vertically aligned mesoporous silica channels to form pure silicon nanoparticles. Sci. Rep. 2015, 5, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Barati, M.; Sarder, S.; McLean, A.; Roy, R. Recovery of silicon from silica fume. J. Non. Cryst. Solids 2011, 357, 18–23. [Google Scholar] [CrossRef]
- Xu, K.; Zhang, Z.; Su, W.; Wei, Z.; Zhong, G.; Wang, C.; Huang, X. Alumina coated nano silicon synthesized by aluminothermic reduction as anodes for lithium ion batteries. Funct. Mater. Lett. 2017, 10, S1793604716500739. [Google Scholar] [CrossRef]
- Lai, Y.; Thompson, J.R.; Dasog, M. Metallothermic reduction of silica nanoparticles to porous silicon for drug delivery using new and existing reductants. Chem. A Eur. J. 2018, 24, 7913–7920. [Google Scholar] [CrossRef] [Green Version]
- Riikonen, J. Solvent loading of porous silicon. In Handbook of Porous Silicon, 2nd ed.; Springer: Berlin/Heidelberg, Germany, 2018; Volume 2, pp. 913–925. ISBN 9783319713816. [Google Scholar]
- Bimbo, L.M.; Mäkilä, E.; Raula, J.; Laaksonen, T.; Laaksonen, P.; Strommer, K.; Kauppinen, E.I.; Salonen, J.; Linder, M.B.; Hirvonen, J.; et al. Functional hydrophobin-coating of thermally hydrocarbonized porous silicon microparticles. Biomaterials 2011, 32, 9089–9099. [Google Scholar] [CrossRef]
- Bimbo, L.M.; Denisova, O.V.; Mäkilä, E.; Kaasalainen, M.; De Brabander, J.K.; Hirvonen, J.; Salonen, J.; Kakkola, L.; Kainov, D.; Santos, H.A. Inhibition of influenza A virus infection in vitro by saliphenylhalamide- loaded porous silicon nanoparticles. ACS Nano 2013, 7, 6884–6893. [Google Scholar] [CrossRef] [PubMed]
- Loni, A. Melt intrusion in porous silicon. In Handbook of Porous Silicon, 2nd ed.; Springer: Berlin/Heidelberg, Germany, 2018; Volume 2, pp. 945–950. ISBN 9783319713816. [Google Scholar]
- Nadarassan, D.K.; Loni, A.; Kelly, C.; O’Brien, H.; Caffull, E.; Webb, K.; Canham, L.T.; Maniruzamman, M.; Trivedi, V.; Douroumis, D. Ultrahigh drug loading and release from biodegradable porous silicon aerocrystals. In Proceedings of the 2015 Controlled Release Society Meeting, San Diego, CA, USA, 26–29 July 2015; pp. 294–306. [Google Scholar]
- Lehto, V.P.; Vähä-Heikkilä, K.; Paski, J.; Salonen, J. Use of thermoanalytical methods in quantification of drug load in mesoporous silicon microparticles. J. Therm. Anal. Calorim. 2005, 80, 393–397. [Google Scholar] [CrossRef]
- Prestidge, C.A.; Barnes, T.J.; MierczynsKa-Vasilev, A.; Kempson, I.; Peddie, F.; Barnett, C. Peptide and protein loading into porous silicon wafers. Phys. Status Solidi Appl. Mater. Sci. 2008, 205, 311–315. [Google Scholar] [CrossRef]
- Wang, M.; Hartman, P.S.; Loni, A.; Canham, L.T.; Bodiford, N.; Coffer, J.L. Influence of surface chemistry on the release of an antibacterial drug from nanostructured porous silicon. Langmuir 2015, 31, 6179–6185. [Google Scholar] [CrossRef]
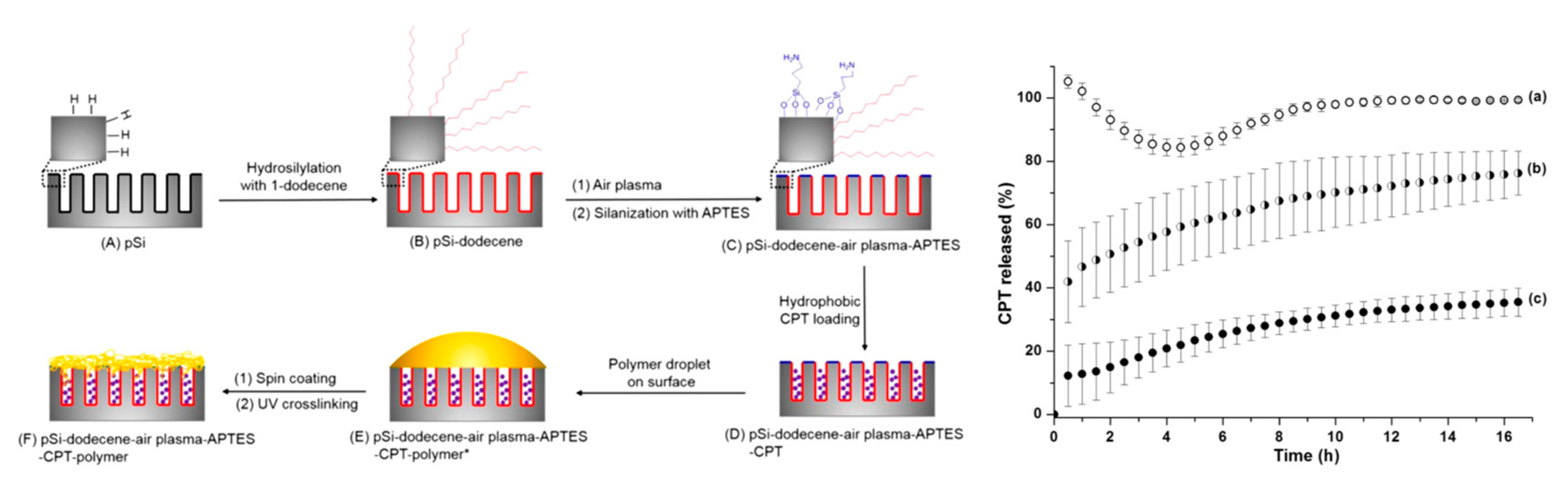
- Zhang, D.-X.; Yoshikawa, C.; Welch, N.G.; Pasic, P.; Thissen, H.; Voelcker, N.H. Spatially controlled surface modification of porous silicon for sustained drug delivery applications. Sci. Rep. 2019, 9, 1367. [Google Scholar] [CrossRef] [Green Version]
- Krepker, M.A.; Segal, E. Porous silicon-polymer composites B. In Handbook of Porous Silicon; Canham, L., Ed.; Springer International Publishing: Cham, Switzerland, 2017; pp. 1–12. ISBN 978-3-319-04508-5. [Google Scholar]
- Beavers, K.R.; Werfel, T.A.; Shen, T.; Kavanaugh, T.E.; Kilchrist, K.V.; Mares, J.W.; Fain, J.S.; Wiese, C.B.; Vickers, K.C.; Weiss, S.M.; et al. Porous silicon and polymer nanocomposites for delivery of peptide nucleic acids as anti-microrna therapies. Adv. Mater. 2016, 28, 7984–7992. [Google Scholar] [CrossRef] [Green Version]
- Nelson, C.E.; Kintzing, J.R.; Hanna, A.; Shannon, J.M.; Gupta, M.K.; Duvall, C.L. Balancing cationic and hydrophobic content of PEGylated siRNA polyplexes enhances endosome escape, stability, blood circulation time, and bioactivity in vivo. ACS Nano 2013, 7, 8870–8880. [Google Scholar] [CrossRef] [Green Version]
- Adolph, E.J.; Nelson, C.E.; Werfel, T.A.; Guo, R.; Davidson, J.M.; Guelcher, S.A.; Duvall, C.L. Enhanced performance of plasmid dna polyplexes stabilized by a combination of core hydrophobicity and surface PEGylation. J. Mater. Chem. B 2014, 2, 8154–8164. [Google Scholar] [CrossRef]
- Xu, W.; Riikonen, J.; Lehto, V.P. Mesoporous systems for poorly soluble drugs. Int. J. Pharm. 2013, 453, 181–197. [Google Scholar] [CrossRef]
- Bimbo, L.M.; Mäkilä, E.; Laaksonen, T.; Lehto, V.-P.; Salonen, J.; Hirvonen, J.; Santos, H.A. Drug permeation across intestinal epithelial cells using porous silicon nanoparticles. Biomaterials 2011, 32, 2625–2633. [Google Scholar] [CrossRef] [PubMed]
- Bimbo, L.M.; Sarparanta, M.; Mäkilä, E.; Laaksonen, T.; Laaksonen, P.; Salonen, J.; Linder, M.B.; Hirvonen, J.; Airaksinen, A.J.; Santos, H.A. Cellular interactions of surface modified nanoporous silicon particles. Nanoscale 2012, 4, 3184–3192. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Bimbo, L.M.; Mäkilä, E.; Villanova, F.; Kaasalainen, M.; Herranz-Blanco, B.; Caramella, C.M.; Lehto, V.-P.; Salonen, J.; Herzig, K.-H.; et al. Co-delivery of a hydrophobic small molecule and a hydrophilic peptide by porous silicon nanoparticles. J. Control. Release 2013, 170, 268–278. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.; Lewik, G.; Ratcliffe, J.C.; Choi, C.H.J.; Mäkilä, E.; Tong, W.Y.; Voelcker, N.H. Systematic evaluation of transferrin-modified porous silicon nanoparticles for targeted delivery of doxorubicin to glioblastoma. ACS Appl. Mater. Interfaces 2019, 11, 33637–33649. [Google Scholar] [CrossRef]
- Nanev, C.N.; Saridakis, E.; Chayen, N.E. Protein crystal nucleation in pores. Sci. Rep. 2017, 7, 35821. [Google Scholar] [CrossRef] [PubMed]
- Gundogdu, N.; Cetin, M. Chitosan-poly (lactide-co-glycolide) (CS-PLGA) nanoparticles containing metformin HCl: Preparation and in vitro evaluation. Pak. J. Pharm. Sci. 2014, 27, 1923–1929. [Google Scholar] [PubMed]
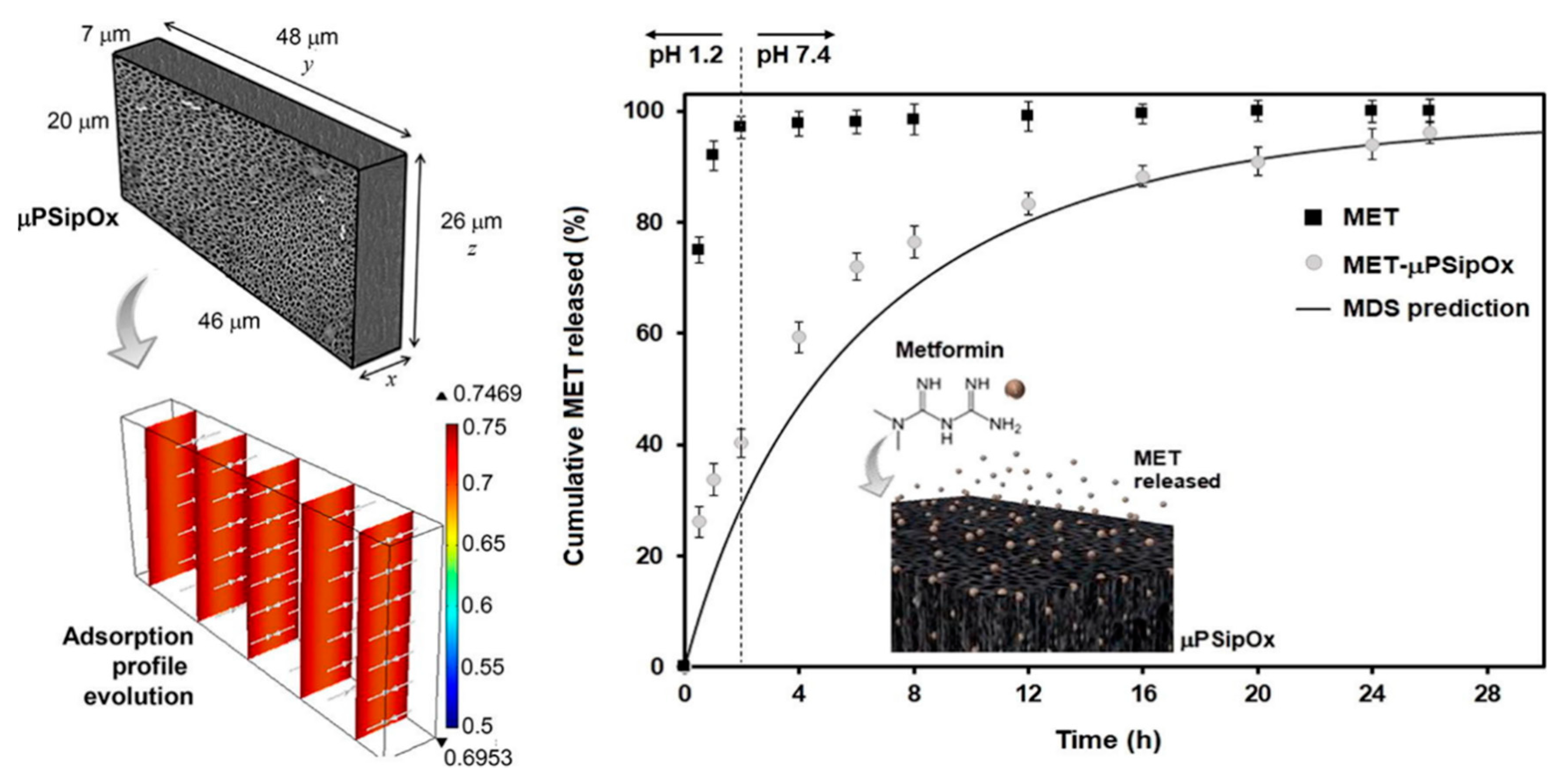
- García-Briones, G.S.; Ocampo, R.; Gómez-Durán, C.F.A.; Neri-Gómez, T.; Palestino, G. Porous silicon microcarriers for extended release of metformin: Design, biological evaluation and 3D kinetics modeling. Chem. Eng. J. 2019, 365, 415–428. [Google Scholar] [CrossRef]
- Kamakura, R.; Kovalainen, M.; Riikonen, J.; Nissinen, T.; Shere Raza, G.; Walkowiak, J.; Lehto, V.-P.; Herzig, K.-H. Inorganic mesoporous particles for controlled α-linolenic acid delivery to stimulate GLP-1 secretion in vitro. Eur. J. Pharm. Biopharm. 2019, 144, 132–138. [Google Scholar] [CrossRef]
- Karhunen, L.J.; Juvonen, K.R.; Huotari, A.; Purhonen, A.K.; Herzig, K.H. Effect of protein, fat, carbohydrate and fibre on gastrointestinal peptide release in humans. Regul. Pept. 2008, 149, 70–78. [Google Scholar] [CrossRef]
- Nettleton, J.A. Omega-3 fatty acids: Comparison of plant and seafood sources in human nutrition. J. Am. Diet. Assoc. 1991, 91, 331–337. [Google Scholar]
- Huotari, A.; Xu, W.; Mönkäre, J.; Kovalainen, M.; Herzig, K.H.; Lehto, V.P.; Järvinen, K. Effect of surface chemistry of porous silicon microparticles on glucagon-like peptide-1 (GLP-1) loading, release and biological activity. Int. J. Pharm. 2013, 454, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Kaasalainen, M.; Rytkönen, J.; Mäkilä, E.; Närvänen, A.; Salonen, J. Electrostatic interaction on loading of therapeutic peptide GLP-1 into porous silicon nanoparticles. Langmuir 2015, 31, 1722–1729. [Google Scholar] [CrossRef] [PubMed]
- Polkovnikova, Y.A.; Len’shin, A.S.; Slivkin, A.I. Quantum-chemical modeling of vinpocetine desorption from silicon and silicon-dioxide particle surfaces. Pharm. Chem. J. 2019, 53, 170–174. [Google Scholar] [CrossRef]
- Bimbo, L.M.; Peltonen, L.; Hirvonen, J.; Santos, H.A. Toxicological profile of therapeutic nanodelivery systems. Curr. Drug Metab. 2012, 13, 1068–1086. [Google Scholar] [CrossRef] [PubMed]
- Park, J.-H.; Gu, L.; von Maltzahn, G.; Ruoslahti, E.; Bhatia, S.N.; Sailor, M.J. Biodegradable luminescent porous silicon nanoparticles for in vivo applications. Nat. Mater. 2009, 8, 331–336. [Google Scholar] [CrossRef] [PubMed]
- Low, S.P.; Voelcker, N.H.; Canham, L.T.; Williams, K.A. The biocompatibility of porous silicon in tissues of the eye. Biomaterials 2009, 30, 2873–2880. [Google Scholar] [CrossRef]
- Low, S.P.; Voelcker, N.H. Biocompatibility of porous silicon. In Handbook of Porous Silicon, 2nd ed.; Springer: Berlin/Heidelberg, Germany, 2018; pp. 533–545. ISBN 9783319045085. [Google Scholar]
- Shahbazi, M.A.; Hamidi, M.; Mäkilä, E.M.; Zhang, H.; Almeida, P.V.; Kaasalainen, M.; Salonen, J.J.; Hirvonen, J.T.; Santos, H.A. The mechanisms of surface chemistry effects of mesoporous silicon nanoparticles on immunotoxicity and biocompatibility. Biomaterials 2013, 34, 7776–7789. [Google Scholar] [CrossRef]
- Liu, D.; Shahbazi, M.-A.; Bimbo, L.M.; Hirvonen, J.; Santos, H.A. Biocompatibility of porous silicon for biomedical applications. In Porous Silicon for Biomedical Applications; Woodhead publishing: Cambridge, UK, 2014; pp. 129–181. ISBN 9780857097118. [Google Scholar]
- Tölli, M.A.; Ferreira, M.P.A.; Kinnunen, S.M.; Rysä, J.; Mäkilä, E.M.; Szabó, Z.; Serpi, R.E.; Ohukainen, P.J.; Välimäki, M.J.; Correia, A.M.R.; et al. In vivo biocompatibility of porous silicon biomaterials for drug delivery to the heart. Biomaterials 2014, 35, 8394–8405. [Google Scholar] [CrossRef]
- Bimbo, L.M.; Sarparanta, M.; Santos, H.A.; Airaksinen, A.J.; Mäkilä, E.; Laaksonen, T.; Peltonen, L.; Lehto, V.-P.; Hirvonen, J.; Salonen, J. Biocompatibility of thermally hydrocarbonized porous silicon nanoparticles and their biodistribution in rats. ACS Nano 2010, 4, 3023–3032. [Google Scholar] [CrossRef]
- Shabir, Q. Biodegradability of porous silicon. In Handbook of Porous Silicon, 2nd ed.; Springer: Berlin/Heidelberg, Germany, 2018; Volumes 1–2, pp. 547–554. [Google Scholar]
- Decuzzi, P.; Godin, B.; Tanaka, T.; Lee, S.Y.; Chiappini, C.; Liu, X.; Ferrari, M. Size and shape effects in the biodistribution of intravascularly injected particles. J. Control. Release 2010, 141, 320–327. [Google Scholar] [CrossRef]
- Anglin, E.J.; Schwartz, M.P.; Ng, V.P.; Perelman, L.A.; Sailor, M.J. Engineering the chemistry and nanostructure of porous silicon Fabry-Pérot films for loading and release of a steroid. Langmuir 2004, 20, 11264–11269. [Google Scholar] [CrossRef] [PubMed]
- Tasciotti, E.; Liu, X.; Bhavane, R.; Plant, K.; Leonard, A.D.; Price, B.K.; Cheng, M.M.C.; Decuzzi, P.; Tour, J.M.; Robertson, F.; et al. Mesoporous silicon particles as a multistage delivery system for imaging and therapeutic applications. Nat. Nanotechnol. 2008, 3, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Popplewell, J.F.; King, S.J.; Day, J.P.; Ackrill, P.; Fifield, L.K.; Cresswell, R.G.; Di Tada, M.L.; Liu, K. Kinetics of uptake and elimination of silicic acid by a human subject: A novel application of 32Si and accelerator mass spectrometry. J. Inorg. Biochem. 1998, 69, 177–180. [Google Scholar] [CrossRef]
- Jugdaohsingh, R.; Anderson, S.H.C.; Tucker, K.L.; Elliott, H.; Kiel, D.P.; Thompson, R.P.H.; Powell, J.J. Dietary silicon intake and absorption. Am. J. Clin. Nutr. 2002, 75, 887–893. [Google Scholar] [CrossRef]
- Jugdaohsingh, R.; Tucker, K.L.; Qiao, N.; Cupples, L.A.; Kiel, D.P.; Powell, J.J. Dietary silicon intake is positively associated with bone mineral density in men and premenopausal women of the Framingham offspring cohort. J. Bone Miner. Res. 2004, 19, 297–307. [Google Scholar] [CrossRef] [Green Version]
- Reffitt, D.M.; Jugdaohsingh, R.; Thompson, R.P.H.; Powell, J.J. Silicic acid: Its gastrointestinal uptake and urinary excretion in man and effects on aluminium excretion. J. Inorg. Biochem. 1999, 76, 141–147. [Google Scholar] [CrossRef]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jones, E.C.L.; Bimbo, L.M. Crystallisation Behaviour of Pharmaceutical Compounds Confined within Mesoporous Silicon. Pharmaceutics 2020, 12, 214. https://doi.org/10.3390/pharmaceutics12030214
Jones ECL, Bimbo LM. Crystallisation Behaviour of Pharmaceutical Compounds Confined within Mesoporous Silicon. Pharmaceutics. 2020; 12(3):214. https://doi.org/10.3390/pharmaceutics12030214
Chicago/Turabian StyleJones, Eleanor C. L., and Luis M. Bimbo. 2020. "Crystallisation Behaviour of Pharmaceutical Compounds Confined within Mesoporous Silicon" Pharmaceutics 12, no. 3: 214. https://doi.org/10.3390/pharmaceutics12030214