Current Status and Challenges Associated with CNS-Targeted Gene Delivery across the BBB


Abstract
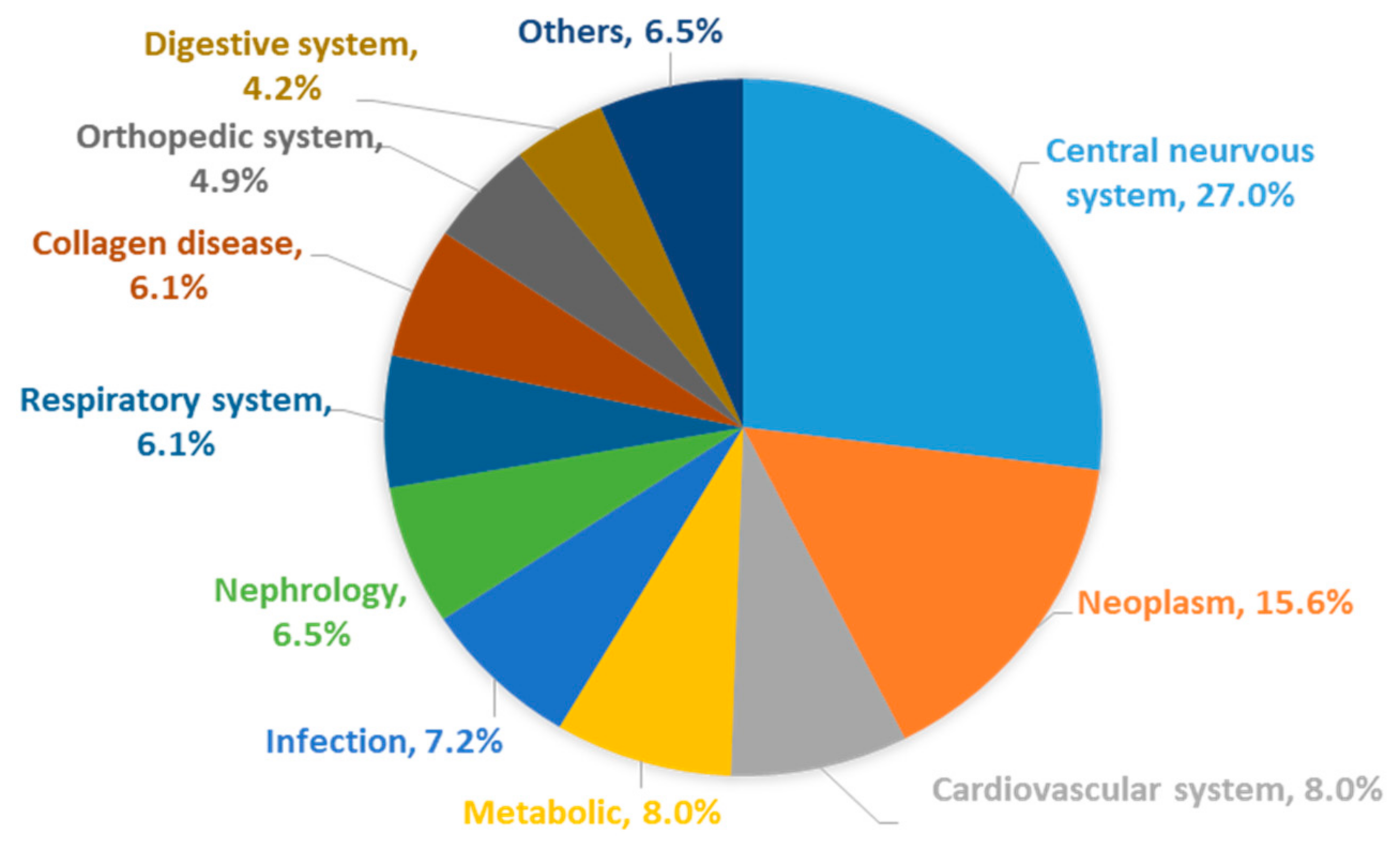
:1. Introduction
2. BBB Function
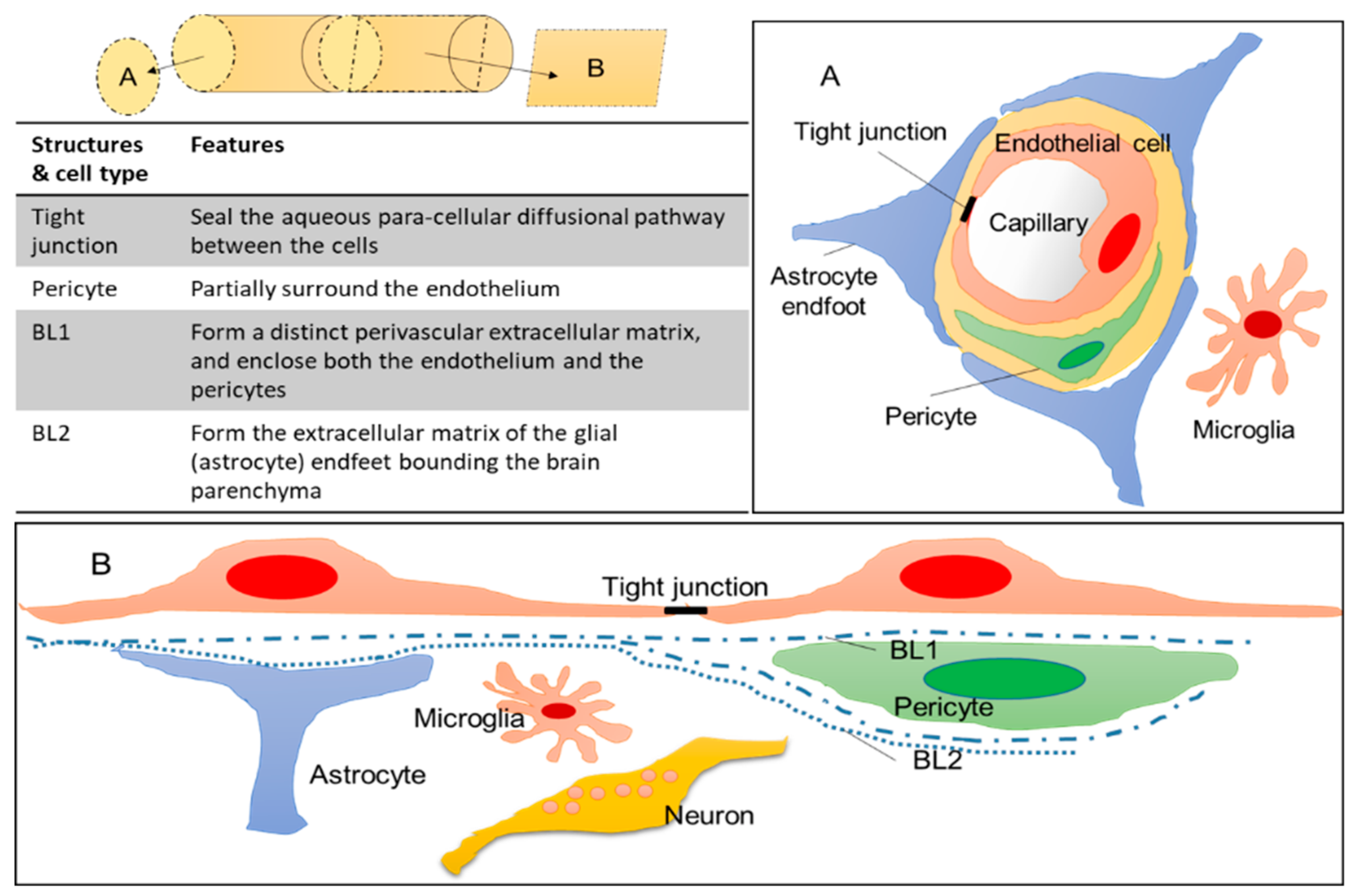
2.1. General Structure of the BBB
2.2. Transporters Expressed on the BBB
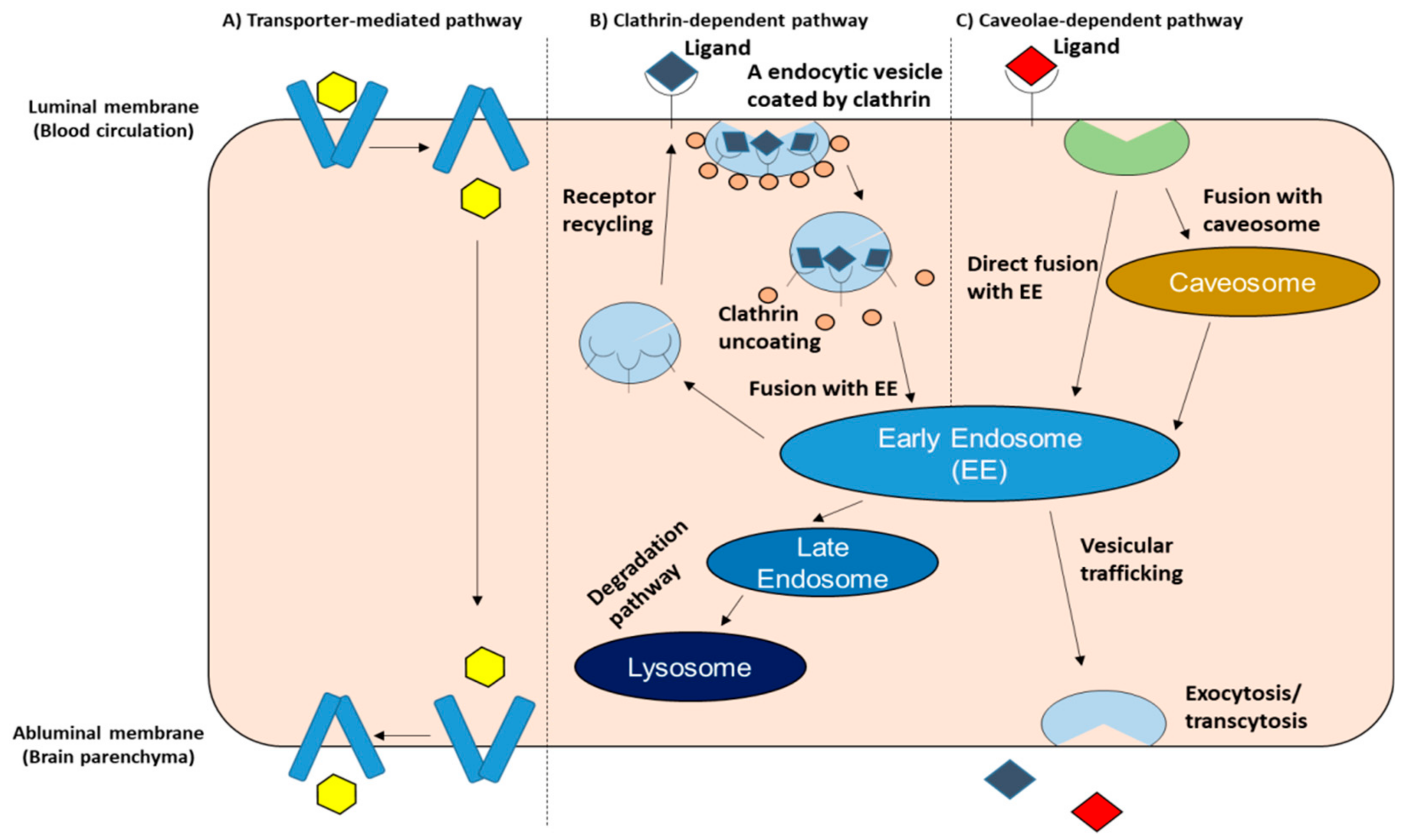
2.3. Mechanism of BBB Crossing (Immune Cells, AAV Vector)
2.4. BBB Breakdown under Pathological Conditions
3. AAV Vector; Currently the Most Advanced Gene Delivery Vector
3.1. Brain-Targeted AAV Vectors Developed So Far
3.2. Zolgensma; AAV9-Based Gene Therapy to Treat SMA
3.3. Controversy Concerning the Use of AAV Vectors
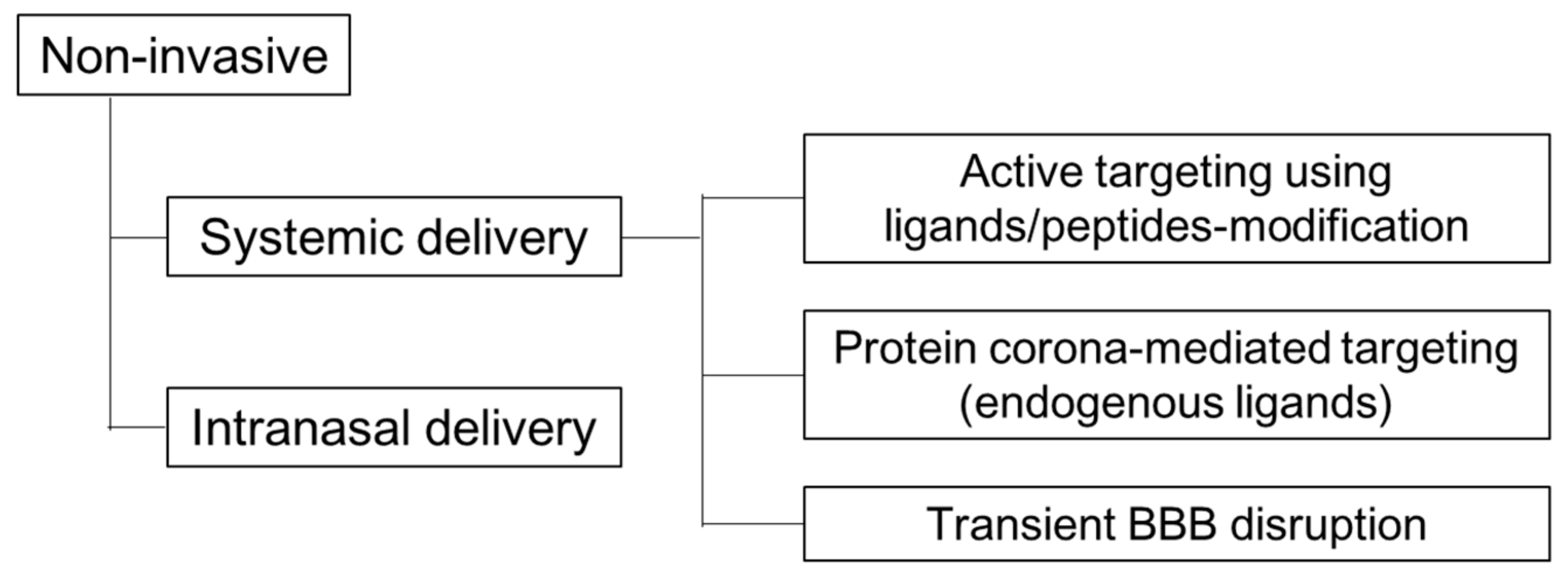
4. Non-Viral Brain Targeting by Non-Invasive Methods
4.1. Active Targeting Using Ligands/Peptides-Modification
4.2. Protein Corona (Endogenous Ligands)
4.3. Transient BBB Disruption
5. Summary and Future Directions
Funding
Acknowledgments
Conflicts of Interest
References
- Feigin, V.L.; Vos, T.; Nichols, E.; Owolabi, M.O.; Carroll, W.M.; Dichgans, M.; Deuschl, G.; Parmar, P.; Brainin, M.; Murray, C. The global burden of neurological disorders: Translating evidence into policy. Lancet Neurol. 2020, 19, 255–265. [Google Scholar] [CrossRef]
- Japan Health Sciences Foundation. 2019年度国内基盤技術調査報告書「60疾患に関する医療ニーズ調査」. Available online: http://www.jhsf.or.jp/paper/report.html#top_2019 (accessed on 16 July 2020).
- Pardridge, W.M. The blood-brain barrier: Bottleneck in brain drug development. NeuroRx 2005, 2, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Saunus, J.M.; McCart Reed, A.E.; Lim, Z.L.; Lakhani, S.R. Breast Cancer Brain Metastases: Clonal Evolution in Clinical Context. Int. J. Mol. Sci. 2017, 18, 152. [Google Scholar] [CrossRef] [PubMed]
- Chiriboga, C.A. Nusinersen for the treatment of spinal muscular atrophy. Expert Rev. Neurother. 2017, 17, 955–962. [Google Scholar] [CrossRef] [PubMed]
- スピンラザ髄注12mg. Available online: https://www.info.pmda.go.jp/go/pack/1190403A1022_1_03/ (accessed on 22 July 2020).
- Khorkova, O.; Wahlestedt, C. Oligonucleotide therapies for disorders of the nervous system. Nat. Biotechnol. 2017, 35, 249–263. [Google Scholar] [CrossRef] [PubMed]
- Ginn, S.L.; Amaya, A.K.; Alexander, I.E.; Edelstein, M.; Abedi, M.R. Gene Therapy Clinical Trials Worldwide to 2017: An Update. J. Gene Med. 2018, 20. [Google Scholar] [CrossRef] [PubMed]
- Piguet, F.; Alves, S.; Cartier, N. Clinical Gene Therapy for Neurodegenerative Diseases: Past, Present, and Future. Hum. Gene Ther. 2017, 28, 988–1003. [Google Scholar] [CrossRef] [PubMed]
- Dunbar, C.E.; High, K.A.; Joung, J.K.; Kohn, D.B.; Ozawa, K.; Sadelain, M. Gene therapy comes of age. Science 2018, 359, eaan4672. [Google Scholar] [CrossRef] [Green Version]
- Choong, C.J.; Baba, K.; Mochizuki, H. Gene therapy for neurological disorders. Expert Opin. Biol. Ther. 2016, 16, 143–159. [Google Scholar] [CrossRef]
- Deverman, B.E.; Ravina, B.M.; Bankiewicz, K.S.; Paul, S.M.; Sah, D.W.Y. Gene therapy for neurological disorders: Progress and prospects. Nat. Rev. Drug Discov. 2018, 17, 641–659. [Google Scholar] [CrossRef]
- Gessler, D.J.; Gao, G. Gene Therapy for the Treatment of Neurological Disorders: Metabolic Disorders. Methods Mol. Biol. 2016, 1382, 429–465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ingusci, S.; Verlengia, G.; Soukupova, M.; Zucchini, S.; Simonato, M. Gene Therapy Tools for Brain Diseases. Front. Pharmacol. 2019, 10, 724. [Google Scholar] [CrossRef] [PubMed]
- Sudhakar, V.; Richardson, R.M. Gene Therapy for Neurodegenerative Diseases. Neurotherapeutics 2019, 16, 166–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eberling, J.L.; Jagust, W.J.; Christine, C.W.; Starr, P.; Larson, P.; Bankiewicz, K.S.; Aminoff, M.J. Results from a phase I safety trial of hAADC gene therapy for Parkinson disease. Neurology 2008, 70, 1980–1983. [Google Scholar] [CrossRef] [PubMed]
- LeWitt, P.A.; Rezai, A.R.; Leehey, M.A.; Ojemann, S.G.; Flaherty, A.W.; Eskandar, E.N.; Kostyk, S.K.; Thomas, K.; Sarkar, A.; Siddiqui, M.S.; et al. AAV2-GAD gene therapy for advanced Parkinson’s disease: A double-blind, sham-surgery controlled, randomised trial. Lancet Neurol. 2011, 10, 309–319. [Google Scholar] [CrossRef]
- Hardy, J.; Allsop, D. Amyloid deposition as the central event in the aetiology of Alzheimer’s disease. Trends Pharmacol. Sci. 1991, 12, 383–388. [Google Scholar] [CrossRef]
- Holtzman, D.M.; Herz, J.; Bu, G. Apolipoprotein E and apolipoprotein E receptors: Normal biology and roles in Alzheimer disease. Cold Spring Harb Perspect Med. 2012, 2, a006312. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; Yamada, K.; Liddelow, S.A.; Smith, S.T.; Zhao, L.; Luo, W.; Tsai, R.M.; Spina, S.; Grinberg, L.T.; Rojas, J.C.; et al. ApoE4 markedly exacerbates tau-mediated neurodegeneration in a mouse model of tauopathy. Nature 2017, 549, 523–527. [Google Scholar] [CrossRef]
- Huang, Y.A.; Zhou, B.; Wernig, M.; Südhof, T.C. ApoE2, ApoE3, and ApoE4 Differentially Stimulate APP Transcription and Aβ Secretion. Cell 2017, 168, 427–441.e21. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.; Carlson-Stevermer, J.; Das, U.; Shen, M.; Delenclos, M.; Snead, A.M.; Koo, S.Y.; Wang, L.; Qiao, D.; Loi, J.; et al. CRISPR/Cas9 editing of APP C-terminus attenuates β-cleavage and promotes α-cleavage. Nat. Commun. 2019, 10, 53. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, H.; Kondo, K.; Chen, X.; Homma, H.; Tagawa, K.; Kerever, A.; Aoki, S.; Saito, T.; Saido, T.; Muramatsu, S.I.; et al. The intellectual disability gene PQBP1 rescues Alzheimer’s disease pathology. Mol. Psychiatry 2018, 23, 2090–2110. [Google Scholar] [CrossRef] [Green Version]
- Mendell, J.R.; Al-Zaidy, S.; Shell, R.; Arnold, W.D.; Rodino-Klapac, L.R.; Prior, T.W.; Lowes, L.; Alfano, L.; Berry, K.; Church, K.; et al. Single-Dose Gene-Replacement Therapy for Spinal Muscular Atrophy. N. Engl. J. Med. 2017, 377, 1713–1722. [Google Scholar] [CrossRef] [PubMed]
- Hoy, S.M. Onasemnogene Abeparvovec: First Global Approval. Drugs 2019, 79, 1255–1262. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Fang, H. Clinical trials with oncolytic adenovirus in China. Curr. Cancer Drug Targets 2007, 7, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Bryant, L.M.; Christopher, D.M.; Giles, A.R.; Hinderer, C.; Rodriguez, J.L.; Smith, J.B.; Traxler, E.A.; Tycko, J.; Wojno, A.P.; Wilson, J.M. Lessons learned from the clinical development and market authorization of Glybera. Hum. Gene Ther. Clin. Dev. 2013, 24, 55–64. [Google Scholar] [CrossRef]
- Pol, J.; Kroemer, G.; Galluzzi, L. First oncolytic virus approved for melanoma immunotherapy. Oncoimmunology 2016, 5, e1115641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schimmer, J.; Breazzano, S. Investor Outlook: Rising from the Ashes; GSK’s European Approval of Strimvelis for ADA-SCID. Hum. Gene Ther. Clin. Dev. 2016, 27, 57–61. [Google Scholar] [CrossRef] [PubMed]
- Sheridan, C. First approval in sight for Novartis’ CAR-T therapy after panel vote. Nat. Biotechnol. 2017, 35, 691–693. [Google Scholar] [CrossRef]
- FDA Approves Second CAR T-cell Therapy. Cancer Discov. 2018, 8, 5–6. [CrossRef] [Green Version]
- Schuessler-Lenz, M.; Enzmann, H.; Vamvakas, S. Regulators’ Advice Can Make a Difference: European Medicines Agency Approval of Zynteglo for Beta Thalassemia. Clin. Pharmacol. Ther. 2020, 107, 492–494. [Google Scholar] [CrossRef] [Green Version]
- Morabito, G.; Giannelli, S.G.; Ordazzo, G.; Bido, S.; Castoldi, V.; Indrigo, M.; Cabassi, T.; Cattaneo, S.; Luoni, M.; Cancellieri, C.; et al. AAV-PHP.B-Mediated Global-Scale Expression in the Mouse Nervous System Enables GBA1 Gene Therapy for Wide Protection from Synucleinopathy. Mol. Ther. 2017, 25, 2727–2742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zharikov, A.D.; Cannon, J.R.; Tapias, V.; Bai, Q.; Horowitz, M.P.; Shah, V.; El Ayadi, A.; Hastings, T.G.; Greenamyre, J.T.; Burton, E.A. shRNA targeting α-synuclein prevents neurodegeneration in a Parkinson’s disease model. J. Clin. Investig. 2015, 125, 2721–2735. [Google Scholar] [CrossRef] [Green Version]
- Dodart, J.C.; Marr, R.A.; Koistinaho, M.; Gregersen, B.M.; Malkani, S.; Verma, I.M.; Paul, S.M. Gene delivery of human apolipoprotein E alters brain Abeta burden in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2005, 102, 1211–1216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, W.; Zhao, L.; Blackman, B.; Parmar, M.; Wong, M.Y.; Woo, T.; Yu, F.; Chiuchiolo, M.J.; Sondhi, D.; Kaminsky, S.M.; et al. Vectored Intracerebral Immunization with the Anti-Tau Monoclonal Antibody PHF1 Markedly Reduces Tau Pathology in Mutant Tau Transgenic Mice. J. Neurosci. 2016, 36, 12425–12435. [Google Scholar] [CrossRef] [Green Version]
- Gaj, T.; Ojala, D.S.; Ekman, F.K.; Byrne, L.C.; Limsirichai, P.; Schaffer, D.V. In Vivo genome editing improves motor function and extends survival in a mouse model of ALS. Sci. Adv. 2017, 3, eaar3952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mis, M.S.C.; Brajkovic, S.; Tafuri, F.; Bresolin, N.; Comi, G.P.; Corti, S. Development of Therapeutics for C9ORF72 ALS/FTD-Related Disorders. Mol. Neurobiol. 2017, 54, 4466–4476. [Google Scholar] [CrossRef]
- Becker, L.A.; Huang, B.; Bieri, G.; Ma, R.; Knowles, D.A.; Jafar-Nejad, P.; Messing, J.; Kim, H.J.; Soriano, A.; Auburger, G.; et al. Therapeutic reduction of ataxin-2 extends lifespan and reduces pathology in TDP-43 mice. Nature 2017, 544, 367–371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pardridge, W.M. Blood-Brain Barrier and Delivery of Protein and Gene Therapeutics to Brain. Front. Aging Neurosci. 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Rabiei, M.; Kashanian, S.; Samavati, S.S.; Jamasb, S.; McInnes, S.J.P. Active Targeting Towards and Inside the Brain based on Nanoparticles: A Review. Curr. Pharm. Biotechnol. 2020, 21, 374–383. [Google Scholar] [CrossRef]
- Moura, R.P.; Martins, C.; Pinto, S.; Sousa, F.; Sarmento, B. Blood-brain barrier receptors and transporters: An insight on their function and how to exploit them through nanotechnology. Expert Opin. Drug Deliv. 2019, 16, 271–285. [Google Scholar] [CrossRef]
- Wiley, D.T.; Webster, P.; Gale, A.; Davis, M.E. Transcytosis and brain uptake of transferrin-containing nanoparticles by tuning avidity to transferrin receptor. Proc. Natl. Acad. Sci. USA 2013, 110, 8662–8667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, F.; Wang, Y.; He, S.; Ku, S.; Gu, W.; Ye, L. Transferrin-conjugated, fluorescein-loaded magnetic nanoparticles for targeted delivery across the blood-brain barrier. J. Mater. Sci. Mater. Med. 2013, 24, 2371–2379. [Google Scholar] [CrossRef] [PubMed]
- Clark, A.J.; Davis, M.E. Increased brain uptake of targeted nanoparticles by adding an acid-cleavable linkage between transferrin and the nanoparticle core. Proc. Natl. Acad. Sci. USA 2015, 112, 12486–12491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dos Santos Rodrigues, B.; Arora, S.; Kanekiyo, T.; Singh, J. Efficient neuronal targeting and transfection using RVG and transferrin-conjugated liposomes. Brain Res. 2020, 1734, 146738. [Google Scholar] [CrossRef] [PubMed]
- Gupta, Y.; Jain, A.; Jain, S.K. Transferrin-conjugated solid lipid nanoparticles for enhanced delivery of quinine dihydrochloride to the brain. J. Pharm. Pharmacol. 2007, 59, 935–940. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Wu, H.; McBride, J.L.; Jung, K.E.; Kim, M.H.; Davidson, B.L.; Lee, S.K.; Shankar, P.; Manjunath, N. Transvascular delivery of small interfering RNA to the central nervous system. Nature 2007, 448, 39–43. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Erviti, L.; Seow, Y.; Yin, H.; Betts, C.; Lakhal, S.; Wood, M.J. Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat. Biotechnol. 2011, 29, 341–345. [Google Scholar] [CrossRef]
- Hwang, D.W.; Son, S.; Jang, J.; Youn, H.; Lee, S.; Lee, D.; Lee, Y.S.; Jeong, J.M.; Kim, W.J.; Lee, D.S. A brain-targeted rabies virus glycoprotein-disulfide linked PEI nanocarrier for delivery of neurogenic microRNA. Biomaterials 2011, 32, 4968–4975. [Google Scholar] [CrossRef]
- Dal Magro, R.; Albertini, B.; Beretta, S.; Rigolio, R.; Donzelli, E.; Chiorazzi, A.; Ricci, M.; Blasi, P.; Sancini, G. Artificial apolipoprotein corona enables nanoparticle brain targeting. Nanomedicine 2018, 14, 429–438. [Google Scholar] [CrossRef]
- Shubar, H.M.; Dunay, I.R.; Lachenmaier, S.; Dathe, M.; Bushrab, F.N.; Mauludin, R.; Müller, R.H.; Fitzner, R.; Borner, K.; Liesenfeld, O. The role of apolipoprotein E in uptake of atovaquone into the brain in murine acute and reactivated toxoplasmosis. J. Drug Target 2009, 17, 257–267. [Google Scholar] [CrossRef]
- Molino, Y.; David, M.; Varini, K.; Jabès, F.; Gaudin, N.; Fortoul, A.; Bakloul, K.; Masse, M.; Bernard, A.; Drobecq, L.; et al. Use of LDL receptor-targeting peptide vectors for in vitro and in vivo cargo transport across the blood-brain barrier. Faseb J. 2017, 31, 1807–1827. [Google Scholar] [CrossRef]
- Zhang, Z.; Guan, J.; Jiang, Z.; Yang, Y.; Liu, J.; Hua, W.; Mao, Y.; Li, C.; Lu, W.; Qian, J.; et al. Brain-targeted drug delivery by manipulating protein corona functions. Nat. Commun. 2019, 10, 3561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arcella, A.; Palchetti, S.; Digiacomo, L.; Pozzi, D.; Capriotti, A.L.; Frati, L.; Oliva, M.A.; Tsaouli, G.; Rota, R.; Screpanti, I.; et al. Brain Targeting by Liposome-Biomolecular Corona Boosts Anticancer Efficacy of Temozolomide in Glioblastoma Cells. ACS Chem. Neurosci. 2018. [Google Scholar] [CrossRef] [PubMed]
- Anraku, Y.; Kuwahara, H.; Fukusato, Y.; Mizoguchi, A.; Ishii, T.; Nitta, K.; Matsumoto, Y.; Toh, K.; Miyata, K.; Uchida, S.; et al. Glycaemic control boosts glucosylated nanocarrier crossing the BBB into the brain. Nat. Commun. 2017, 8, 1001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Min, H.S.; Kim, H.J.; Naito, M.; Ogura, S.; Toh, K.; Hayashi, K.; Kim, B.S.; Fukushima, S.; Anraku, Y.; Miyata, K.; et al. Systemic Brain Delivery of Antisense Oligonucleotides across the Blood-Brain Barrier with a Glucose-Coated Polymeric Nanocarrier. Angew. Chem. Int. Ed. Engl. 2020, 59, 8173–8180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sweeney, M.D.; Zhao, Z.; Montagne, A.; Nelson, A.R.; Zlokovic, B.V. Blood-Brain Barrier: From Physiology to Disease and Back. Physiol. Rev. 2019, 99, 21–78. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, M.D.; Sagare, A.P.; Zlokovic, B.V. Blood-brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat. Rev. Neurol. 2018, 14, 133–150. [Google Scholar] [CrossRef] [PubMed]
- Zlokovic, B.V. The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron 2008, 57, 178–201. [Google Scholar] [CrossRef] [Green Version]
- Hama, S.; Akita, H.; Ito, R.; Mizuguchi, H.; Hayakawa, T.; Harashima, H. Quantitative comparison of intracellular trafficking and nuclear transcription between adenoviral and lipoplex systems. Mol. Ther. 2006, 13, 786–794. [Google Scholar] [CrossRef]
- Hama, S.; Akita, H.; Iida, S.; Mizuguchi, H.; Harashima, H. Quantitative and mechanism-based investigation of post-nuclear delivery events between adenovirus and lipoplex. Nucleic Acids Res. 2007, 35, 1533–1543. [Google Scholar] [CrossRef]
- Varga, C.M.; Tedford, N.C.; Thomas, M.; Klibanov, A.M.; Griffith, L.G.; A Lauffenburger, D. Quantitative comparison of polyethylenimine formulations and adenoviral vectors in terms of intracellular gene delivery processes. Gene Ther. 2005, 12, 1023–1032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osten, P.; Grinevich, V.; Cetin, A. Viral vectors: A wide range of choices and high levels of service. Handb. Exp. Pharmacol. 2007, 178, 177–202. [Google Scholar] [CrossRef]
- Hocquemiller, M.; Giersch, L.; Audrain, M.; Parker, S.; Cartier, N. Adeno-Associated Virus-Based Gene Therapy for CNS Diseases. Hum. Gene Ther. 2016, 27, 478–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.; Samulski, R.J. Engineering adeno-associated virus vectors for gene therapy. Nat. Rev. Genet. 2020, 21, 255–272. [Google Scholar] [CrossRef]
- Naso, M.F.; Tomkowicz, B.; Perry, W.L., 3rd; Strohl, W.R. Adeno-Associated Virus (AAV) as a Vector for Gene Therapy. BioDrugs 2017, 31, 317–334. [Google Scholar] [CrossRef] [Green Version]
- Ramamoorth, M.; Narvekar, A. Non viral vectors in gene therapy—An overview. J. Clin. Diagn Res. 2015, 9, Ge01-06. [Google Scholar] [CrossRef]
- Helal, N.A.; Osami, A.; Helmy, A.; McDonald, T.; Shaaban, L.A.; Nounou, M.I. Non-viral gene delivery systems: Hurdles for bench-to-bedside transformation. Pharmazie 2017, 72, 627–693. [Google Scholar] [CrossRef]
- Yin, H.; Kanasty, R.L.; Eltoukhy, A.A.; Vegas, A.J.; Dorkin, J.R.; Anderson, D.G. Non-viral vectors for gene-based therapy. Nat. Rev. Genet. 2014, 15, 541–555. [Google Scholar] [CrossRef]
- Meng, F.; Wang, J.; Ping, Q.; Yeo, Y. Quantitative Assessment of Nanoparticle Biodistribution by Fluorescence Imaging, Revisited. ACS Nano 2018, 12, 6458–6468. [Google Scholar] [CrossRef]
- Varga, C.M.; Hong, K.; Lauffenburger, D.A. Quantitative analysis of synthetic gene delivery vector design properties. Mol. Ther. 2001, 4, 438–446. [Google Scholar] [CrossRef]
- Uchida, S.; Kataoka, K. Design concepts of polyplex micelles for in vivo therapeutic delivery of plasmid DNA and messenger RNA. J. Biomed. Mater. Res. A 2019, 107, 978–990. [Google Scholar] [CrossRef] [PubMed]
- Buck, J.; Grossen, P.; Cullis, P.R.; Huwyler, J.; Witzigmann, D. Lipid-Based DNA Therapeutics—Hallmarks of Non-Viral Gene Delivery. ACS Nano 2019. [Google Scholar] [CrossRef] [PubMed]
- Glebova, K.V.; Marakhonov, A.V.; Baranova, A.V.; Skublov, M.I. [Types of Non-Viral Delivery Systems of Small Interfering RNA]. Mol. Biol. 2012, 46, 387–401. [Google Scholar] [CrossRef]
- Chen, J.; Guo, Z.; Tian, H.; Chen, X. Production and Clinical Development of Nanoparticles for Gene Delivery. Mol. Ther. Methods Clin. Dev. 2016, 3. [Google Scholar] [CrossRef]
- Hoy, S.M. Patisiran: First Global Approval. Drugs 2018, 78, 1625–1631. [Google Scholar] [CrossRef]
- Daneman, R.; Prat, A. The blood-brain barrier. Cold Spring Harb Perspect Biol. 2015, 7, a020412. [Google Scholar] [CrossRef] [Green Version]
- Abbott, N.J.; Rönnbäck, L.; Hansson, E. Astrocyte-endothelial interactions at the blood-brain barrier. Nat. Rev. Neurosci. 2006, 7, 41–53. [Google Scholar] [CrossRef]
- Stamatovic, S.M.; Keep, R.F.; Andjelkovic, A.V. Brain endothelial cell-cell junctions: How to “open” the blood brain barrier. Curr. Neuropharmacol. 2008, 6, 179–192. [Google Scholar] [CrossRef] [Green Version]
- Serlin, Y.; Shelef, I.; Knyazer, B.; Friedman, A. Anatomy and physiology of the blood-brain barrier. Semin. Cell Dev. Biol. 2015, 38, 2–6. [Google Scholar] [CrossRef] [Green Version]
- Preston, J.E.; Joan Abbott, N.; Begley, D.J. Transcytosis of macromolecules at the blood-brain barrier. Adv. Pharmacol. 2014, 71, 147–163. [Google Scholar] [CrossRef]
- Azevedo, F.A.; Carvalho, L.R.; Grinberg, L.T.; Farfel, J.M.; Ferretti, R.E.; Leite, R.E.; Jacob Filho, W.; Lent, R.; Herculano-Houzel, S. Equal numbers of neuronal and nonneuronal cells make the human brain an isometrically scaled-up primate brain. J. Comp. Neurol. 2009, 513, 532–541. [Google Scholar] [CrossRef] [PubMed]
- Von Bartheld, C.S.; Bahney, J.; Herculano-Houzel, S. The search for true numbers of neurons and glial cells in the human brain: A review of 150 years of cell counting. J. Comp. Neurol. 2016, 524, 3865–3895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, W.S.; Allen, N.J.; Eroglu, C. Astrocytes Control Synapse Formation, Function, and Elimination. Cold Spring Harb Perspect Biol. 2015, 7, a020370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathiisen, T.M.; Lehre, K.P.; Danbolt, N.C.; Ottersen, O.P. The perivascular astroglial sheath provides a complete covering of the brain microvessels: An electron microscopic 3D reconstruction. Glia 2010, 58, 1094–1103. [Google Scholar] [CrossRef]
- Haddad-Tóvolli, R.; Dragano, N.R.V.; Ramalho, A.F.S.; Velloso, L.A. Development and Function of the Blood-Brain Barrier in the Context of Metabolic Control. Front. Neurosci. 2017, 11, 224. [Google Scholar] [CrossRef]
- Simard, M.; Nedergaard, M. The neurobiology of glia in the context of water and ion homeostasis. Neuroscience 2004, 129, 877–896. [Google Scholar] [CrossRef]
- Tao-Cheng, J.H.; Brightman, M.W. Development of membrane interactions between brain endothelial cells and astrocytes in vitro. Int. J. Dev. Neurosci. 1988, 6, 25–37. [Google Scholar] [CrossRef]
- Broux, B.; Gowing, E.; Prat, A. Glial regulation of the blood-brain barrier in health and disease. Semin. Immunopathol. 2015, 37, 577–590. [Google Scholar] [CrossRef]
- Sims, D.E. The pericyte—A review. Tissue Cell 1986, 18, 153–174. [Google Scholar] [CrossRef]
- Armulik, A.; Genové, G.; Betsholtz, C. Pericytes: Developmental, physiological, and pathological perspectives, problems, and promises. Dev. Cell 2011, 21, 193–215. [Google Scholar] [CrossRef] [Green Version]
- Díaz-Flores, L.; Gutiérrez, R.; Madrid, J.F.; Varela, H.; Valladares, F.; Acosta, E.; Martín-Vasallo, P.; Díaz-Flores, L., Jr. Pericytes. Morphofunction, interactions and pathology in a quiescent and activated mesenchymal cell niche. Histol. Histopathol. 2009, 24, 909–969. [Google Scholar] [CrossRef] [PubMed]
- Sims, D.; Horne, M.M.; Creighan, M.; Donald, A. Heterogeneity of pericyte populations in equine skeletal muscle and dermal microvessels: A quantitative study. Anat. Histol. Embryol. 1994, 23, 232–238. [Google Scholar] [CrossRef] [PubMed]
- Armulik, A.; Genové, G.; Mäe, M.; Nisancioglu, M.H.; Wallgard, E.; Niaudet, C.; He, L.; Norlin, J.; Lindblom, P.; Strittmatter, K.; et al. Pericytes regulate the blood-brain barrier. Nature 2010, 468, 557–561. [Google Scholar] [CrossRef] [Green Version]
- Nakagawa, S.; Deli, M.A.; Nakao, S.; Honda, M.; Hayashi, K.; Nakaoke, R.; Kataoka, Y.; Niwa, M. Pericytes from brain microvessels strengthen the barrier integrity in primary cultures of rat brain endothelial cells. Cell Mol. Neurobiol. 2007, 27, 687–694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ginhoux, F.; Greter, M.; Leboeuf, M.; Nandi, S.; See, P.; Gokhan, S.; Mehler, M.F.; Conway, S.J.; Ng, L.G.; Stanley, E.R.; et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 2010, 330, 841–845. [Google Scholar] [CrossRef] [Green Version]
- Dudvarski Stankovic, N.; Teodorczyk, M.; Ploen, R.; Zipp, F.; Schmidt, M.H.H. Microglia-blood vessel interactions: A double-edged sword in brain pathologies. Acta Neuropathol. 2016, 131, 347–363. [Google Scholar] [CrossRef]
- Thurgur, H.; Pinteaux, E. Microglia in the Neurovascular Unit: Blood-Brain Barrier-microglia Interactions After Central Nervous System Disorders. Neuroscience 2019, 405, 55–67. [Google Scholar] [CrossRef]
- Haruwaka, K.; Ikegami, A.; Tachibana, Y.; Ohno, N.; Konishi, H.; Hashimoto, A.; Matsumoto, M.; Kato, D.; Ono, R.; Kiyama, H.; et al. Dual microglia effects on blood brain barrier permeability induced by systemic inflammation. Nat. Commun. 2019, 10, 5816. [Google Scholar] [CrossRef] [Green Version]
- Abbott, N.J.; Patabendige, A.A.; Dolman, D.E.; Yusof, S.R.; Begley, D.J. Structure and function of the blood-brain barrier. Neurobiol Dis 2010, 37, 13–25. [Google Scholar] [CrossRef]
- Daneman, R.; Zhou, L.; Agalliu, D.; Cahoy, J.D.; Kaushal, A.; Barres, B.A. The mouse blood-brain barrier transcriptome: A new resource for understanding the development and function of brain endothelial cells. PLoS ONE 2010, 5, e13741. [Google Scholar] [CrossRef] [Green Version]
- Munji, R.N.; Soung, A.L.; Weiner, G.A.; Sohet, F.; Semple, B.D.; Trivedi, A.; Gimlin, K.; Kotoda, M.; Korai, M.; Aydin, S.; et al. Profiling the mouse brain endothelial transcriptome in health and disease models reveals a core blood-brain barrier dysfunction module. Nat. Neurosci. 2019, 22, 1892–1902. [Google Scholar] [CrossRef] [PubMed]
- Zeisel, A.; Muñoz-Manchado, A.B.; Codeluppi, S.; Lönnerberg, P.; La Manno, G.; Juréus, A.; Marques, S.; Munguba, H.; He, L.; Betsholtz, C.; et al. Brain structure. Cell types in the mouse cortex and hippocampus revealed by single-cell RNA-seq. Science 2015, 347, 1138–1142. [Google Scholar] [CrossRef] [PubMed]
- Li, J.Y.; Boado, R.J.; Pardridge, W.M. Blood-brain barrier genomics. J. Cereb. Blood Flow Metab. 2001, 21, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Pardridge, W.M. Blood-brain barrier genomics and the use of endogenous transporters to cause drug penetration into the brain. Curr. Opin. Drug Discov. Devel. 2003, 6, 683–691. [Google Scholar] [PubMed]
- Shusta, E.V. Blood-brain barrier genomics, proteomics, and new transporter discovery. NeuroRx 2005, 2, 151–161. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chen, K.; Sloan, S.A.; Bennett, M.L.; Scholze, A.R.; O’Keeffe, S.; Phatnani, H.P.; Guarnieri, P.; Caneda, C.; Ruderisch, N.; et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J. Neurosci. 2014, 34, 11929–11947. [Google Scholar] [CrossRef]
- Karamanos, Y.; Gosselet, F.; Dehouck, M.P.; Cecchelli, R. Blood-brain barrier proteomics: Towards the understanding of neurodegenerative diseases. Arch. Med. Res. 2014, 45, 730–737. [Google Scholar] [CrossRef]
- Karamanos, Y.; Pottiez, G. Proteomics and the blood-brain barrier: How recent findings help drug development. Expert Rev. Proteomics 2016, 13, 251–258. [Google Scholar] [CrossRef]
- Mäger, I.; Meyer, A.H.; Li, J.; Lenter, M.; Hildebrandt, T.; Leparc, G.; Wood, M.J.A. Targeting blood-brain-barrier transcytosis—Perspectives for drug delivery. Neuropharmacology 2017, 120, 4–7. [Google Scholar] [CrossRef] [Green Version]
- Ohtsuki, S.; Hirayama, M.; Ito, S.; Uchida, Y.; Tachikawa, M.; Terasaki, T. Quantitative targeted proteomics for understanding the blood-brain barrier: Towards pharmacoproteomics. Expert Rev. Proteom. 2014, 11, 303–313. [Google Scholar] [CrossRef]
- Patching, S.G. Glucose Transporters at the Blood-Brain Barrier: Function, Regulation and Gateways for Drug Delivery. Mol. Neurobiol. 2017, 54, 1046–1077. [Google Scholar] [CrossRef] [PubMed]
- Simpson, I.A.; Appel, N.M.; Hokari, M.; Oki, J.; Holman, G.D.; Maher, F.; Koehler-Stec, E.M.; Vannucci, S.J.; Smith, Q.R. Blood-brain barrier glucose transporter: Effects of hypo- and hyperglycemia revisited. J. Neurochem. 1999, 72, 238–247. [Google Scholar] [CrossRef] [PubMed]
- Eyster, C.A.; Higginson, J.D.; Huebner, R.; Porat-Shliom, N.; Weigert, R.; Wu, W.W.; Shen, R.F.; Donaldson, J.G. Discovery of new cargo proteins that enter cells through clathrin-independent endocytosis. Traffic 2009, 10, 590–599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riskin, A.; Nannegari, V.H.; Mond, Y. Acute effectors of GLUT1 glucose transporter subcellular targeting in CIT3 mouse mammary epithelial cells. Pediatr. Res. 2008, 63, 56–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnsen, K.B.; Burkhart, A.; Thomsen, L.B.; Andresen, T.L.; Moos, T. Targeting the transferrin receptor for brain drug delivery. Prog. Neurobiol. 2019, 181, 101665. [Google Scholar] [CrossRef]
- Johnsen, K.B.; Bak, M.; Kempen, P.J.; Melander, F.; Burkhart, A.; Thomsen, M.S.; Nielsen, M.S.; Moos, T.; Andresen, T.L. Antibody affinity and valency impact brain uptake of transferrin receptor-targeted gold nanoparticles. Theranostics 2018, 8, 3416–3436. [Google Scholar] [CrossRef]
- Qian, Z.M.; Li, H.; Sun, H.; Ho, K. Targeted drug delivery via the transferrin receptor-mediated endocytosis pathway. Pharmacol. Rev. 2002, 54, 561–587. [Google Scholar] [CrossRef]
- Kawabata, H.; Yang, R.; Hirama, T.; Vuong, P.T.; Kawano, S.; Gombart, A.F.; Koeffler, H.P. Molecular cloning of transferrin receptor 2. A new member of the transferrin receptor-like family. J. Biol. Chem. 1999, 274, 20826–20832. [Google Scholar] [CrossRef] [Green Version]
- Kawabata, H.; Nakamaki, T.; Ikonomi, P.; Smith, R.D.; Germain, R.S.; Koeffler, H.P. Expression of transferrin receptor 2 in normal and neoplastic hematopoietic cells. Blood 2001, 98, 2714–2719. [Google Scholar] [CrossRef]
- Herbison, C.E.; Thorstensen, K.; Chua, A.C.; Graham, R.M.; Leedman, P.; Olynyk, J.K.; Trinder, D. The role of transferrin receptor 1 and 2 in transferrin-bound iron uptake in human hepatoma cells. Am. J. Physiol. Cell Physiol. 2009, 297, C1567–C1575. [Google Scholar] [CrossRef] [Green Version]
- Kawabata, H. Transferrin and transferrin receptors update. Free Radic. Biol. Med. 2019, 133, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Mills, E.; Dong, X.P.; Wang, F.; Xu, H. Mechanisms of brain iron transport: Insight into neurodegeneration and CNS disorders. Future Med. Chem. 2010, 2, 51–64. [Google Scholar] [CrossRef] [Green Version]
- Dautry-Varsat, A.; Ciechanover, A.; Lodish, H.F. pH and the recycling of transferrin during receptor-mediated endocytosis. Proc. Natl. Acad. Sci. USA 1983, 80, 2258–2262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bickel, U.; Yoshikawa, T.; Pardridge, W.M. Delivery of peptides and proteins through the blood-brain barrier. Adv. Drug Deliv. Rev. 2001, 46, 247–279. [Google Scholar] [CrossRef]
- Skjørringe, T.; Burkhart, A.; Johnsen, K.B.; Moos, T. Divalent metal transporter 1 (DMT1) in the brain: Implications for a role in iron transport at the blood-brain barrier, and neuronal and glial pathology. Front. Mol. Neurosci. 2015, 8, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnsen, K.B.; Burkhart, A.; Melander, F.; Kempen, P.J.; Vejlebo, J.B.; Siupka, P.; Nielsen, M.S.; Andresen, T.L.; Moos, T. Targeting transferrin receptors at the blood-brain barrier improves the uptake of immunoliposomes and subsequent cargo transport into the brain parenchyma. Sci. Rep. 2017, 7, 10396. [Google Scholar] [CrossRef]
- Lee, H.J.; Engelhardt, B.; Lesley, J.; Bickel, U.; Pardridge, W.M. Targeting rat anti-mouse transferrin receptor monoclonal antibodies through blood-brain barrier in mouse. J. Pharmacol. Exp. Ther. 2000, 292, 1048–1052. [Google Scholar]
- Friden, P.M.; Olson, T.S.; Obar, R.; Walus, L.R.; Putney, S.D. Characterization, receptor mapping and blood-brain barrier transcytosis of antibodies to the human transferrin receptor. J. Pharmacol. Exp. Ther. 1996, 278, 1491–1498. [Google Scholar]
- Jefferies, W.A.; Brandon, M.R.; Hunt, S.V.; Williams, A.F.; Gatter, K.C.; Mason, D.Y. Transferrin receptor on endothelium of brain capillaries. Nature 1984, 312, 162–163. [Google Scholar] [CrossRef]
- Pardridge, W.M.; Buciak, J.L.; Friden, P.M. Selective transport of an anti-transferrin receptor antibody through the blood-brain barrier in vivo. J. Pharmacol. Exp. Ther. 1991, 259, 66–70. [Google Scholar]
- Defesche, J.C. Low-density lipoprotein receptor--its structure, function, and mutations. Semin. Vasc. Med. 2004, 4, 5–11. [Google Scholar] [CrossRef] [PubMed]
- Bathori, G.; Cervenak, L.; Karadi, I. Caveolae--an alternative endocytotic pathway for targeted drug delivery. Crit. Rev. Ther. Drug Carrier Syst. 2004, 21, 67–95. [Google Scholar] [CrossRef] [PubMed]
- Kiss, A.L.; Botos, E. Endocytosis via caveolae: Alternative pathway with distinct cellular compartments to avoid lysosomal degradation? J. Cell Mol. Med. 2009, 13, 1228–1237. [Google Scholar] [CrossRef] [Green Version]
- Bertrand, N.; Grenier, P.; Mahmoudi, M.; Lima, E.M.; Appel, E.A.; Dormont, F.; Lim, J.M.; Karnik, R.; Langer, R.; Farokhzad, O.C. Mechanistic understanding of in vivo protein corona formation on polymeric nanoparticles and impact on pharmacokinetics. Nat. Commun. 2017, 8, 777. [Google Scholar] [CrossRef] [PubMed]
- Capriotti, A.L.; Cavaliere, C.; Piovesana, S. Liposome protein corona characterization as a new approach in nanomedicine. Anal. Bioanal. Chem. 2019. [Google Scholar] [CrossRef] [PubMed]
- Caracciolo, G.; Farokhzad, O.C.; Mahmoudi, M. Biological Identity of Nanoparticles In Vivo: Clinical Implications of the Protein Corona. Trends Biotechnol. 2017, 35, 257–264. [Google Scholar] [CrossRef]
- Madathiparambil Visalakshan, R.; González García, L.E.; Benzigar, M.R.; Ghazaryan, A.; Simon, J.; Mierczynska-Vasilev, A.; Michl, T.D.; Vinu, A.; Mailänder, V.; Morsbach, S.; et al. The Influence of Nanoparticle Shape on Protein Corona Formation. Small 2020, 16, e2000285. [Google Scholar] [CrossRef]
- Quagliarini, E.; Di Santo, R.; Palchetti, S.; Ferri, G.; Cardarelli, F.; Pozzi, D.; Caracciolo, G. Effect of Protein Corona on The Transfection Efficiency of Lipid-Coated Graphene Oxide-Based Cell Transfection Reagents. Pharmaceutics 2020, 12, 113. [Google Scholar] [CrossRef] [Green Version]
- Richtering, W.; Alberg, I.; Zentel, R. Nanoparticles in the Biological Context: Surface Morphology and Protein Corona Formation. Small 2020, e2002162. [Google Scholar] [CrossRef]
- Shadmani, P.; Mehrafrooz, B.; Montazeri, A.; Naghdabadi, R. Protein corona impact on nanoparticle-cell interactions: Toward an energy-based model of endocytosis. J. Phys. Condens. Matter. 2020, 32, 115101. [Google Scholar] [CrossRef]
- Xiao, W.; Gao, H. The impact of protein corona on the behavior and targeting capability of nanoparticle-based delivery system. Int. J. Pharm. 2018, 552, 328–339. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.; Zhao, L.; Guo, C.; Yan, B.; Su, G. Regulating Protein Corona Formation and Dynamic Protein Exchange by Controlling Nanoparticle Hydrophobicity. Front. Bioeng. Biotechnol. 2020, 8, 210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ban, Z.; Yuan, P.; Yu, F.; Peng, T.; Zhou, Q.; Hu, X. Machine learning predicts the functional composition of the protein corona and the cellular recognition of nanoparticles. Proc. Natl. Acad. Sci. USA 2020, 117, 10492–10499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Francia, V.; Schiffelers, R.M.; Cullis, P.R.; Witzigmann, D. The Biomolecular Corona of Lipid Nanoparticles for Gene Therapy. Bioconjug. Chem. 2020. [Google Scholar] [CrossRef]
- Salvati, A.; Pitek, A.S.; Monopoli, M.P.; Prapainop, K.; Bombelli, F.B.; Hristov, D.R.; Kelly, P.M.; Aberg, C.; Mahon, E.; Dawson, K.A. Transferrin-functionalized nanoparticles lose their targeting capabilities when a biomolecule corona adsorbs on the surface. Nat. Nanotechnol. 2013, 8, 137–143. [Google Scholar] [CrossRef] [Green Version]
- Aoyama, M.; Hata, K.; Higashisaka, K.; Nagano, K.; Yoshioka, Y.; Tsutsumi, Y. Clusterin in the protein corona plays a key role in the stealth effect of nanoparticles against phagocytes. Biochem. Biophys. Res. Commun 2016, 480, 690–695. [Google Scholar] [CrossRef]
- Caracciolo, G.; Pozzi, D.; Capriotti, A.L.; Cavaliere, C.; Piovesana, S.; Amenitsch, H.; Lagana, A. Lipid composition: A “key factor” for the rational manipulation of the liposome-protein corona by liposome design. RSC Advances 2015, 5, 5967–5975. [Google Scholar] [CrossRef]
- Caracciolo, G.; Palchetti, S.; Colapicchioni, V.; Digiacomo, L.; Pozzi, D.; Capriotti, A.L.; La Barbera, G.; Lagana, A. Stealth effect of biomolecular corona on nanoparticle uptake by immune cells. Langmuir 2015, 31, 10764–10773. [Google Scholar] [CrossRef]
- Chen, D.; Parayath, N.; Ganesh, S.; Wang, W.; Amiji, M. The role of apolipoprotein- and vitronectin-enriched protein corona on lipid nanoparticles for in vivo targeted delivery and transfection of oligonucleotides in murine tumor models. Nanoscale 2019, 11, 18806–18824. [Google Scholar] [CrossRef]
- Lara, S.; Perez-Potti, A.; Herda, L.M.; Adumeau, L.; Dawson, K.A.; Yan, Y. Differential Recognition of Nanoparticle Protein Corona and Modified Low-Density Lipoprotein by Macrophage Receptor with Collagenous Structure. ACS Nano 2018, 12, 4930–4937. [Google Scholar] [CrossRef]
- Schottler, S.; Becker, G.; Winzen, S.; Steinbach, T.; Mohr, K.; Landfester, K.; Mailander, V.; Wurm, F.R. Protein adsorption is required for stealth effect of poly(ethylene glycol)- and poly(phosphoester)-coated nanocarriers. Nat. Nanotechnol. 2016, 11, 372–377. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Tenzer, S.; Storck, W.; Hobernik, D.; Raker, V.K.; Fischer, K.; Decker, S.; Dzionek, A.; Krauthauser, S.; Diken, M.; et al. Protein corona-mediated targeting of nanocarriers to B cells allows redirection of allergic immune responses. J. Allergy Clin. Immunol. 2018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, X.H.; Lin, X.N.; Wei, F.; Feng, W.; Huang, Z.C.; Wang, P.; Ren, L.; Diao, Y. Enhanced brain targeting of temozolomide in polysorbate-80 coated polybutylcyanoacrylate nanoparticles. Int. J. Nanomed. 2011, 6, 445–452. [Google Scholar] [CrossRef] [Green Version]
- Wilson, B.; Samanta, M.K.; Santhi, K.; Kumar, K.P.; Paramakrishnan, N.; Suresh, B. Poly(n-butylcyanoacrylate) nanoparticles coated with polysorbate 80 for the targeted delivery of rivastigmine into the brain to treat Alzheimer’s disease. Brain Res. 2008, 1200, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Xu, N.; Gu, J.; Zhu, Y.; Wen, H.; Ren, Q.; Chen, J. Efficacy of intravenous amphotericin B-polybutylcyanoacrylate nanoparticles against cryptococcal meningitis in mice. Int. J. Nanomed. 2011, 6, 905–913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demeule, M.; Régina, A.; Ché, C.; Poirier, J.; Nguyen, T.; Gabathuler, R.; Castaigne, J.P.; Béliveau, R. Identification and design of peptides as a new drug delivery system for the brain. J. Pharmacol. Exp. Ther. 2008, 324, 1064–1072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demeule, M.; Currie, J.C.; Bertrand, Y.; Ché, C.; Nguyen, T.; Régina, A.; Gabathuler, R.; Castaigne, J.P.; Béliveau, R. Involvement of the low-density lipoprotein receptor-related protein in the transcytosis of the brain delivery vector angiopep-2. J. Neurochem. 2008, 106, 1534–1544. [Google Scholar] [CrossRef]
- Gao, X.; Qian, J.; Zheng, S.; Xiong, Y.; Man, J.; Cao, B.; Wang, L.; Ju, S.; Li, C. Up-regulating blood brain barrier permeability of nanoparticles via multivalent effect. Pharm. Res. 2013, 30, 2538–2548. [Google Scholar] [CrossRef]
- Gao, S.; Tian, H.; Xing, Z.; Zhang, D.; Guo, Y.; Guo, Z.; Zhu, X.; Chen, X. A non-viral suicide gene delivery system traversing the blood brain barrier for non-invasive glioma targeting treatment. J. Control Release 2016, 243, 357–369. [Google Scholar] [CrossRef]
- Huang, S.; Li, J.; Han, L.; Liu, S.; Ma, H.; Huang, R.; Jiang, C. Dual targeting effect of Angiopep-2-modified, DNA-loaded nanoparticles for glioma. Biomaterials 2011, 32, 6832–6838. [Google Scholar] [CrossRef]
- Huang, R.; Ma, H.; Guo, Y.; Liu, S.; Kuang, Y.; Shao, K.; Li, J.; Liu, Y.; Han, L.; Huang, S.; et al. Angiopep-conjugated nanoparticles for targeted long-term gene therapy of Parkinson’s disease. Pharm. Res. 2013, 30, 2549–2559. [Google Scholar] [CrossRef] [PubMed]
- Xin, H.; Sha, X.; Jiang, X.; Zhang, W.; Chen, L.; Fang, X. Anti-glioblastoma efficacy and safety of paclitaxel-loading Angiopep-conjugated dual targeting PEG-PCL nanoparticles. Biomaterials 2012, 33, 8167–8176. [Google Scholar] [CrossRef] [PubMed]
- Kumthekar, P.; Tang, S.C.; Brenner, A.J.; Kesari, S.; Piccioni, D.E.; Anders, C.; Carrillo, J.; Chalasani, P.; Kabos, P.; Puhalla, S.; et al. ANG1005, a Brain-Penetrating Peptide-Drug Conjugate, Shows Activity in Patients with Breast Cancer with Leptomeningeal Carcinomatosis and Recurrent Brain Metastases. Clin. Cancer Res. 2020, 26, 2789–2799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurzrock, R.; Gabrail, N.; Chandhasin, C.; Moulder, S.; Smith, C.; Brenner, A.; Sankhala, K.; Mita, A.; Elian, K.; Bouchard, D.; et al. Safety, pharmacokinetics, and activity of GRN1005, a novel conjugate of angiopep-2, a peptide facilitating brain penetration, and paclitaxel, in patients with advanced solid tumors. Mol. Cancer Ther. 2012, 11, 308–316. [Google Scholar] [CrossRef] [Green Version]
- Drappatz, J.; Brenner, A.; Wong, E.T.; Eichler, A.; Schiff, D.; Groves, M.D.; Mikkelsen, T.; Rosenfeld, S.; Sarantopoulos, J.; Meyers, C.A.; et al. Phase I study of GRN1005 in recurrent malignant glioma. Clin. Cancer Res. 2013, 19, 1567–1576. [Google Scholar] [CrossRef] [Green Version]
- Butcher, E.C. Leukocyte-endothelial cell recognition: Three (or more) steps to specificity and diversity. Cell 1991, 67, 1033–1036. [Google Scholar] [CrossRef]
- Kunkel, E.J.; Butcher, E.C. Chemokines and the tissue-specific migration of lymphocytes. Immunity 2002, 16, 1–4. [Google Scholar] [CrossRef] [Green Version]
- Medawar, P.B. Immunity to homologous grafted skin; the fate of skin homografts transplanted to the brain, to subcutaneous tissue, and to the anterior chamber of the eye. Br. J. Exp. Pathol. 1948, 29, 58–69. [Google Scholar]
- Kleine, T.O.; Benes, L. Immune surveillance of the human central nervous system (CNS): Different migration pathways of immune cells through the blood-brain barrier and blood-cerebrospinal fluid barrier in healthy persons. Cytometry A 2006, 69, 147–151. [Google Scholar] [CrossRef]
- Ransohoff, R.M.; Kivisäkk, P.; Kidd, G. Three or more routes for leukocyte migration into the central nervous system. Nat. Rev. Immunol. 2003, 3, 569–581. [Google Scholar] [CrossRef]
- Greenhalgh, A.D.; David, S.; Bennett, F.C. Immune cell regulation of glia during CNS injury and disease. Nat. Rev. Neurosci. 2020, 21, 139–152. [Google Scholar] [CrossRef] [PubMed]
- Engelhardt, B.; Vajkoczy, P.; Weller, R.O. The movers and shapers in immune privilege of the CNS. Nat. Immunol. 2017, 18, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Wilson, E.H.; Weninger, W.; Hunter, C.A. Trafficking of immune cells in the central nervous system. J. Clin. Investig. 2010, 120, 1368–1379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engelhardt, B.; Ransohoff, R.M. The ins and outs of T-lymphocyte trafficking to the CNS: Anatomical sites and molecular mechanisms. Trends Immunol. 2005, 26, 485–495. [Google Scholar] [CrossRef] [PubMed]
- Marchetti, L.; Engelhardt, B. Immune cell trafficking across the blood-brain barrier in the absence and presence of neuroinflammation. Vasc. Biol. 2020, 2, H1–H18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carrithers, M.D.; Visintin, I.; Kang, S.J.; Janeway, C.A., Jr. Differential adhesion molecule requirements for immune surveillance and inflammatory recruitment. Brain 2000, 123 Pt 6, 1092–1101. [Google Scholar] [CrossRef] [Green Version]
- Steiner, O.; Coisne, C.; Cecchelli, R.; Boscacci, R.; Deutsch, U.; Engelhardt, B.; Lyck, R. Differential roles for endothelial ICAM-1, ICAM-2, and VCAM-1 in shear-resistant T cell arrest, polarization, and directed crawling on blood-brain barrier endothelium. J. Immunol. 2010, 185, 4846–4855. [Google Scholar] [CrossRef]
- Abadier, M.; Haghayegh Jahromi, N.; Cardoso Alves, L.; Boscacci, R.; Vestweber, D.; Barnum, S.; Deutsch, U.; Engelhardt, B.; Lyck, R. Cell surface levels of endothelial ICAM-1 influence the transcellular or paracellular T-cell diapedesis across the blood-brain barrier. Eur. J. Immunol. 2015, 45, 1043–1058. [Google Scholar] [CrossRef]
- Gorina, R.; Lyck, R.; Vestweber, D.; Engelhardt, B. β2 integrin-mediated crawling on endothelial ICAM-1 and ICAM-2 is a prerequisite for transcellular neutrophil diapedesis across the inflamed blood-brain barrier. J. Immunol. 2014, 192, 324–337. [Google Scholar] [CrossRef] [Green Version]
- Marcos-Contreras, O.A.; Brenner, J.S.; Kiseleva, R.Y.; Zuluaga-Ramirez, V.; Greineder, C.F.; Villa, C.H.; Hood, E.D.; Myerson, J.W.; Muro, S.; Persidsky, Y.; et al. Combining vascular targeting and the local first pass provides 100-fold higher uptake of ICAM-1-targeted vs untargeted nanocarriers in the inflamed brain. J. Control. Release 2019, 301, 54–61. [Google Scholar] [CrossRef]
- Hsu, J.; Rappaport, J.; Muro, S. Specific binding, uptake, and transport of ICAM-1-targeted nanocarriers across endothelial and subendothelial cell components of the blood-brain barrier. Pharm. Res. 2014, 31, 1855–1866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marcos-Contreras, O.A.; Greineder, C.F.; Kiseleva, R.Y.; Parhiz, H.; Walsh, L.R.; Zuluaga-Ramirez, V.; Myerson, J.W.; Hood, E.D.; Villa, C.H.; Tombacz, I.; et al. Selective targeting of nanomedicine to inflamed cerebral vasculature to enhance the blood-brain barrier. Proc. Natl. Acad. Sci. USA 2020, 117, 3405–3414. [Google Scholar] [CrossRef] [PubMed]
- Manthe, R.L.; Loeck, M.; Bhowmick, T.; Solomon, M.; Muro, S. Intertwined mechanisms define transport of anti-ICAM nanocarriers across the endothelium and brain delivery of a therapeutic enzyme. J. Control. Release 2020, 324, 181–193. [Google Scholar] [CrossRef] [PubMed]
- Arima, Y.; Kamimura, D.; Sabharwal, L.; Yamada, M.; Bando, H.; Ogura, H.; Atsumi, T.; Murakami, M. Regulation of immune cell infiltration into the CNS by regional neural inputs explained by the gate theory. Mediators Inflamm. 2013, 2013, 898165. [Google Scholar] [CrossRef]
- Arima, Y.; Harada, M.; Kamimura, D.; Park, J.H.; Kawano, F.; Yull, F.E.; Kawamoto, T.; Iwakura, Y.; Betz, U.A.; Márquez, G.; et al. Regional neural activation defines a gateway for autoreactive T cells to cross the blood-brain barrier. Cell 2012, 148, 447–457. [Google Scholar] [CrossRef] [Green Version]
- Kamimura, D.; Yamada, M.; Harada, M.; Sabharwal, L.; Meng, J.; Bando, H.; Ogura, H.; Atsumi, T.; Arima, Y.; Murakami, M. The gateway theory: Bridging neural and immune interactions in the CNS. Front. Neurosci. 2013, 7, 204. [Google Scholar] [CrossRef] [Green Version]
- Ogura, H.; Arima, Y.; Kamimura, D.; Murakami, M. The gateway theory: How regional neural activation creates a gateway for immune cells via an inflammation amplifier. Biomed. J. 2013, 36, 269–273. [Google Scholar] [CrossRef]
- Kamimura, D.; Murakami, M. Neural stimulations regulate the infiltration of immune cells into the CNS. J. Intern Med. 2019, 286, 259–267. [Google Scholar] [CrossRef] [Green Version]
- Sabharwal, L.; Kamimura, D.; Meng, J.; Bando, H.; Ogura, H.; Nakayama, C.; Jiang, J.J.; Kumai, N.; Suzuki, H.; Atsumi, T.; et al. The Gateway Reflex, which is mediated by the inflammation amplifier, directs pathogenic immune cells into the CNS. J. Biochem. 2014, 156, 299–304. [Google Scholar] [CrossRef] [Green Version]
- Stofkova, A.; Murakami, M. Neural activity regulates autoimmune diseases through the gateway reflex. Bioelectron. Med. 2019, 5, 14. [Google Scholar] [CrossRef]
- Tanaka, Y.; Arima, Y.; Kamimura, D.; Murakami, M. The Gateway Reflex, a Novel Neuro-Immune Interaction for the Regulation of Regional Vessels. Front. Immunol. 2017, 8, 1321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herrmann, A.K.; Grimm, D. High-Throughput Dissection of AAV-Host Interactions: The Fast and the Curious. J. Mol. Biol. 2018, 430, 2626–2640. [Google Scholar] [CrossRef] [PubMed]
- Deverman, B.E.; Pravdo, P.L.; Simpson, B.P.; Kumar, S.R.; Chan, K.Y.; Banerjee, A.; Wu, W.L.; Yang, B.; Huber, N.; Pasca, S.P.; et al. Cre-dependent selection yields AAV variants for widespread gene transfer to the adult brain. Nat. Biotechnol. 2016, 34, 204–209. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaki, Y.; Konno, A.; Mochizuki, R.; Shinohara, Y.; Nitta, K.; Okada, Y.; Hirai, H. Intravenous administration of the adeno-associated virus-PHP.B capsid fails to upregulate transduction efficiency in the marmoset brain. Neurosci. Lett. 2018, 665, 182–188. [Google Scholar] [CrossRef]
- Liguore, W.A.; Domire, J.S.; Button, D.; Wang, Y.; Dufour, B.D.; Srinivasan, S.; McBride, J.L. AAV-PHP.B Administration Results in a Differential Pattern of CNS Biodistribution in Non-human Primates Compared with Mice. Mol. Ther. 2019, 27, 2018–2037. [Google Scholar] [CrossRef]
- Huang, Q.; Chan, K.Y.; Tobey, I.G.; Chan, Y.A.; Poterba, T.; Boutros, C.L.; Balazs, A.B.; Daneman, R.; Bloom, J.M.; Seed, C.; et al. Delivering genes across the blood-brain barrier: LY6A, a novel cellular receptor for AAV-PHP.B capsids. PLoS ONE 2019, 14, e0225206. [Google Scholar] [CrossRef] [Green Version]
- Batista, A.R.; King, O.D.; Reardon, C.P.; Davis, C.; Shankaracharya; Philip, V.; Gray-Edwards, H.; Aronin, N.; Lutz, C.; Landers, J.; et al. Ly6a Differential Expression in Blood-Brain Barrier Is Responsible for Strain Specific Central Nervous System Transduction Profile of AAV-PHP.B. Hum. Gene Ther. 2020, 31, 90–102. [Google Scholar] [CrossRef] [Green Version]
- Hordeaux, J.; Yuan, Y.; Clark, P.M.; Wang, Q.; Martino, R.A.; Sims, J.J.; Bell, P.; Raymond, A.; Stanford, W.L.; Wilson, J.M. The GPI-Linked Protein LY6A Drives AAV-PHP.B Transport across the Blood-Brain Barrier. Mol. Ther. 2019, 27, 912–921. [Google Scholar] [CrossRef]
- Yamazaki, Y.; Kanekiyo, T. Blood-Brain Barrier Dysfunction and the Pathogenesis of Alzheimer’s Disease. Int. J. Mol. Sci. 2017, 18, 1965. [Google Scholar] [CrossRef] [Green Version]
- Oku, N. Innovations in Liposomal DDS Technology and Its Application for the Treatment of Various Diseases. Biol. Pharm. Bull. 2017, 40, 119–127. [Google Scholar] [CrossRef] [Green Version]
- Deane, R.; Wu, Z.; Sagare, A.; Davis, J.; Du Yan, S.; Hamm, K.; Xu, F.; Parisi, M.; LaRue, B.; Hu, H.W.; et al. LRP/amyloid beta-peptide interaction mediates differential brain efflux of Abeta isoforms. Neuron 2004, 43, 333–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vogelgesang, S.; Cascorbi, I.; Schroeder, E.; Pahnke, J.; Kroemer, H.K.; Siegmund, W.; Kunert-Keil, C.; Walker, L.C.; Warzok, R.W. Deposition of Alzheimer’s beta-amyloid is inversely correlated with P-glycoprotein expression in the brains of elderly non-demented humans. Pharmacogenetics 2002, 12, 535–541. [Google Scholar] [CrossRef] [PubMed]
- Carrano, A.; Hoozemans, J.J.; van der Vies, S.M.; van Horssen, J.; de Vries, H.E.; Rozemuller, A.J. Neuroinflammation and blood-brain barrier changes in capillary amyloid angiopathy. Neurodegener. Dis. 2012, 10, 329–331. [Google Scholar] [CrossRef] [PubMed]
- Waubant, E. Biomarkers indicative of blood-brain barrier disruption in multiple sclerosis. Dis. Markers 2006, 22, 235–244. [Google Scholar] [CrossRef]
- Chen, X.; Lan, X.; Roche, I.; Liu, R.; Geiger, J.D. Caffeine protects against MPTP-induced blood-brain barrier dysfunction in mouse striatum. J. Neurochem. 2008, 107, 1147–1157. [Google Scholar] [CrossRef] [Green Version]
- Gu, X.L.; Long, C.X.; Sun, L.; Xie, C.; Lin, X.; Cai, H. Astrocytic expression of Parkinson’s disease-related A53T alpha-synuclein causes neurodegeneration in mice. Mol. Brain 2010, 3, 12. [Google Scholar] [CrossRef] [Green Version]
- McCall, A.L.; Van Bueren, A.M.; Nipper, V.; Moholt-Siebert, M.; Downes, H.; Lessov, N. Forebrain ischemia increases GLUT1 protein in brain microvessels and parenchyma. J. Cereb. Blood Flow Metab. 1996, 16, 69–76. [Google Scholar] [CrossRef] [Green Version]
- Haley, M.J.; Lawrence, C.B. The blood-brain barrier after stroke: Structural studies and the role of transcytotic vesicles. J. Cereb. Blood Flow Metab. 2017, 37, 456–470. [Google Scholar] [CrossRef] [Green Version]
- Knowland, D.; Arac, A.; Sekiguchi, K.J.; Hsu, M.; Lutz, S.E.; Perrino, J.; Steinberg, G.K.; Barres, B.A.; Nimmerjahn, A.; Agalliu, D. Stepwise recruitment of transcellular and paracellular pathways underlies blood-brain barrier breakdown in stroke. Neuron 2014, 82, 603–617. [Google Scholar] [CrossRef] [Green Version]
- van Tellingen, O.; Yetkin-Arik, B.; de Gooijer, M.C.; Wesseling, P.; Wurdinger, T.; de Vries, H.E. Overcoming the blood-brain tumor barrier for effective glioblastoma treatment. Drug Resist. Updat. 2015, 19, 1–12. [Google Scholar] [CrossRef]
- Aronica, E.; Gorter, J.A.; Redeker, S.; van Vliet, E.A.; Ramkema, M.; Scheffer, G.L.; Scheper, R.J.; van der Valk, P.; Leenstra, S.; Baayen, J.C.; et al. Localization of breast cancer resistance protein (BCRP) in microvessel endothelium of human control and epileptic brain. Epilepsia 2005, 46, 849–857. [Google Scholar] [CrossRef] [PubMed]
- Grimm, D.; Zolotukhin, S. E Pluribus Unum: 50 Years of Research, Millions of Viruses, and One Goal--Tailored Acceleration of AAV Evolution. Mol. Ther. 2015, 23, 1819–1831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, E.J.; Guenther, C.M.; Suh, J. Adeno-Associated Virus (AAV) Vectors: Rational Design Strategies for Capsid Engineering. Curr. Opin. Biomed. Eng. 2018, 7, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Foust, K.D.; Nurre, E.; Montgomery, C.L.; Hernandez, A.; Chan, C.M.; Kaspar, B.K. Intravascular AAV9 preferentially targets neonatal neurons and adult astrocytes. Nat. Biotechnol. 2009, 27, 59–65. [Google Scholar] [CrossRef] [Green Version]
- Iida, A.; Takino, N.; Miyauchi, H.; Shimazaki, K.; Muramatsu, S. Systemic delivery of tyrosine-mutant AAV vectors results in robust transduction of neurons in adult mice. Biomed. Res. Int. 2013, 2013, 974819. [Google Scholar] [CrossRef] [Green Version]
- Choudhury, S.R.; Fitzpatrick, Z.; Harris, A.F.; Maitland, S.A.; Ferreira, J.S.; Zhang, Y.; Ma, S.; Sharma, R.B.; Gray-Edwards, H.L.; Johnson, J.A.; et al. In Vivo Selection Yields AAV-B1 Capsid for Central Nervous System and Muscle Gene Therapy. Mol. Ther. 2016, 24, 1247–1257. [Google Scholar] [CrossRef] [Green Version]
- Choudhury, S.R.; Harris, A.F.; Cabral, D.J.; Keeler, A.M.; Sapp, E.; Ferreira, J.S.; Gray-Edwards, H.L.; Johnson, J.A.; Johnson, A.K.; Su, Q.; et al. Widespread Central Nervous System Gene Transfer and Silencing After Systemic Delivery of Novel AAV-AS Vector. Mol. Ther. 2016, 24, 726–735. [Google Scholar] [CrossRef] [Green Version]
- Saraiva, J.; Nobre, R.J.; Pereira de Almeida, L. Gene therapy for the CNS using AAVs: The impact of systemic delivery by AAV9. J. Control. Release 2016, 241, 94–109. [Google Scholar] [CrossRef]
- Murlidharan, G.; Samulski, R.J.; Asokan, A. Biology of adeno-associated viral vectors in the central nervous system. Front. Mol. Neurosci. 2014, 7, 76. [Google Scholar] [CrossRef] [Green Version]
- Duque, S.; Joussemet, B.; Riviere, C.; Marais, T.; Dubreil, L.; Douar, A.M.; Fyfe, J.; Moullier, P.; Colle, M.A.; Barkats, M. Intravenous administration of self-complementary AAV9 enables transgene delivery to adult motor neurons. Mol. Ther. 2009, 17, 1187–1196. [Google Scholar] [CrossRef]
- Körbelin, J.; Dogbevia, G.; Michelfelder, S.; Ridder, D.A.; Hunger, A.; Wenzel, J.; Seismann, H.; Lampe, M.; Bannach, J.; Pasparakis, M.; et al. A brain microvasculature endothelial cell-specific viral vector with the potential to treat neurovascular and neurological diseases. EMBO Mol. Med. 2016, 8, 609–625. [Google Scholar] [CrossRef] [PubMed]
- Griffin, J.M.; Fackelmeier, B.; Fong, D.M.; Mouravlev, A.; Young, D.; O’Carroll, S.J. Astrocyte-selective AAV gene therapy through the endogenous GFAP promoter results in robust transduction in the rat spinal cord following injury. Gene Ther. 2019, 26, 198–210. [Google Scholar] [CrossRef] [PubMed]
- Vagner, T.; Dvorzhak, A.; Wójtowicz, A.M.; Harms, C.; Grantyn, R. Systemic application of AAV vectors targeting GFAP-expressing astrocytes in Z-Q175-KI Huntington’s disease mice. Mol. Cell Neurosci. 2016, 77, 76–86. [Google Scholar] [CrossRef] [PubMed]
- Ravindra Kumar, S.; Miles, T.F.; Chen, X.; Brown, D.; Dobreva, T.; Huang, Q.; Ding, X.; Luo, Y.; Einarsson, P.H.; Greenbaum, A.; et al. Multiplexed Cre-dependent selection yields systemic AAVs for targeting distinct brain cell types. Nat. Methods 2020, 17, 541–550. [Google Scholar] [CrossRef] [PubMed]
- D’Amico, A.; Mercuri, E.; Tiziano, F.D.; Bertini, E. Spinal muscular atrophy. Orphanet. J. Rare Dis. 2011, 6, 71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parente, V.; Corti, S. Advances in spinal muscular atrophy therapeutics. Ther. Adv. Neurol. Disord. 2018, 11. [Google Scholar] [CrossRef]
- Dabbous, O.; Maru, B.; Jansen, J.P.; Lorenzi, M.; Cloutier, M.; Guérin, A.; Pivneva, I.; Wu, E.Q.; Arjunji, R.; Feltner, D.; et al. Survival, Motor Function, and Motor Milestones: Comparison of AVXS-101 Relative to Nusinersen for the Treatment of Infants with Spinal Muscular Atrophy Type 1. Adv. Ther. 2019, 36, 1164–1176. [Google Scholar] [CrossRef] [Green Version]
- Hinderer, C.; Katz, N.; Buza, E.L.; Dyer, C.; Goode, T.; Bell, P.; Richman, L.K.; Wilson, J.M. Severe Toxicity in Nonhuman Primates and Piglets Following High-Dose Intravenous Administration of an Adeno-Associated Virus Vector Expressing Human SMN. Hum. Gene Ther. 2018, 29, 285–298. [Google Scholar] [CrossRef] [Green Version]
- Note: Zolgensma data manipulation. Med. Lett. Drugs Ther. 2019, 61, 129.
- Parks, W.P.; Boucher, D.W.; Melnick, J.L.; Taber, L.H.; Yow, M.D. Seroepidemiological and ecological studies of the adenovirus-associated satellite viruses. Infect. Immun. 1970, 2, 716–722. [Google Scholar] [CrossRef] [Green Version]
- Mimuro, J.; Mizukami, H.; Shima, M.; Matsushita, T.; Taki, M.; Muto, S.; Higasa, S.; Sakai, M.; Ohmori, T.; Madoiwa, S.; et al. The prevalence of neutralizing antibodies against adeno-associated virus capsids is reduced in young Japanese individuals. J. Med. Virol. 2014, 86, 1990–1997. [Google Scholar] [CrossRef] [PubMed]
- Mingozzi, F.; Chen, Y.; Edmonson, S.C.; Zhou, S.; Thurlings, R.M.; Tak, P.P.; High, K.A.; Vervoordeldonk, M.J. Prevalence and pharmacological modulation of humoral immunity to AAV vectors in gene transfer to synovial tissue. Gene Ther. 2013, 20, 417–424. [Google Scholar] [CrossRef] [PubMed]
- Louis Jeune, V.; Joergensen, J.A.; Hajjar, R.J.; Weber, T. Pre-existing anti-adeno-associated virus antibodies as a challenge in AAV gene therapy. Hum. Gene Ther. Methods 2013, 24, 59–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manno, C.S.; Pierce, G.F.; Arruda, V.R.; Glader, B.; Ragni, M.; Rasko, J.J.; Ozelo, M.C.; Hoots, K.; Blatt, P.; Konkle, B.; et al. Successful transduction of liver in hemophilia by AAV-Factor IX and limitations imposed by the host immune response. Nat. Med. 2006, 12, 342–347. [Google Scholar] [CrossRef]
- Mimuro, J.; Mizukami, H.; Hishikawa, S.; Ikemoto, T.; Ishiwata, A.; Sakata, A.; Ohmori, T.; Madoiwa, S.; Ono, F.; Ozawa, K.; et al. Minimizing the inhibitory effect of neutralizing antibody for efficient gene expression in the liver with adeno-associated virus 8 vectors. Mol. Ther. 2013, 21, 318–323. [Google Scholar] [CrossRef] [Green Version]
- Duan, D. Micro-Dystrophin Gene Therapy Goes Systemic in Duchenne Muscular Dystrophy Patients. Hum. Gene Ther. 2018, 29, 733–736. [Google Scholar] [CrossRef]
- Shankar, R.; Joshi, M.; Pathak, K. Lipid Nanoparticles: A Novel Approach for Brain Targeting. Pharm. Nanotechnol. 2018, 6, 81–93. [Google Scholar] [CrossRef]
- Agrawal, M.; Saraf, S.; Dubey, S.K.; Puri, A.; Patel, R.J.; Ajazuddin; Ravichandiran, V.; Murty, U.S.; Alexander, A. Recent strategies and advances in the fabrication of nano lipid carriers and their application towards brain targeting. J. Control. Release 2020, 321, 372–415. [Google Scholar] [CrossRef]
- Lu, Y.; Jiang, C. Brain-Targeted Polymers for Gene Delivery in the Treatment of Brain Diseases. Top Curr. Chem. (Cham.) 2017, 375, 48. [Google Scholar] [CrossRef]
- Fu, C.; Xiang, Y.; Li, X.; Fu, A. Targeted transport of nanocarriers into brain for theranosis with rabies virus glycoprotein-derived peptide. Mater. Sci. Eng. C. Mater. Biol. Appl. 2018, 87, 155–166. [Google Scholar] [CrossRef]
- Oswald, M.; Geissler, S.; Goepferich, A. Targeting the Central Nervous System (CNS): A Review of Rabies Virus-Targeting Strategies. Mol. Pharm. 2017, 14, 2177–2196. [Google Scholar] [CrossRef] [PubMed]
- Fisher, C.R.; Streicker, D.G.; Schnell, M.J. The spread and evolution of rabies virus: Conquering new frontiers. Nat. Rev. Microbiol. 2018, 16, 241–255. [Google Scholar] [CrossRef] [PubMed]
- Schnell, M.J.; McGettigan, J.P.; Wirblich, C.; Papaneri, A. The cell biology of rabies virus: Using stealth to reach the brain. Nat. Rev. Microbiol. 2010, 8, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Al-Obaidi, M.M.J.; Bahadoran, A.; Wang, S.M.; Manikam, R.; Raju, C.S.; Sekaran, S.D. Disruption of the blood brain barrier is vital property of neurotropic viral infection of the central nervous system. Acta Virol. 2018, 62, 16–27. [Google Scholar] [CrossRef] [Green Version]
- Preuss, M.A.; Faber, M.L.; Tan, G.S.; Bette, M.; Dietzschold, B.; Weihe, E.; Schnell, M.J. Intravenous inoculation of a bat-associated rabies virus causes lethal encephalopathy in mice through invasion of the brain via neurosecretory hypothalamic fibers. PLoS Pathog. 2009, 5, e1000485. [Google Scholar] [CrossRef]
- Fu, A.; Wang, Y.; Zhan, L.; Zhou, R. Targeted delivery of proteins into the central nervous system mediated by rabies virus glycoprotein-derived peptide. Pharm. Res. 2012, 29, 1562–1569. [Google Scholar] [CrossRef]
- Fu, A.; Zhang, M.; Gao, F.; Xu, X.; Chen, Z. A novel peptide delivers plasmids across blood-brain barrier into neuronal cells as a single-component transfer vector. PLoS ONE 2013, 8, e59642. [Google Scholar] [CrossRef] [Green Version]
- Zou, Z.; Sun, Z.; Li, P.; Feng, T.; Wu, S. Cre Fused with RVG Peptide Mediates Targeted Genome Editing in Mouse Brain Cells In Vivo. Int. J. Mol. Sci. 2016, 17, 2104. [Google Scholar] [CrossRef] [Green Version]
- Capriotti, A.; Piovesana, S.; Zenezini Chiozzi, R.; Montone, C.M.; Bossi, A.M.; Laganà, A. Does the protein corona take over the selectivity of molecularly imprinted nanoparticles? The biological challenges to recognition. J. Proteom. 2020, 219, 103736. [Google Scholar] [CrossRef]
- Rampado, R.; Crotti, S.; Caliceti, P.; Pucciarelli, S.; Agostini, M. Recent Advances in Understanding the Protein Corona of Nanoparticles and in the Formulation of: “Stealthy” Nanomaterials. Front. Bioeng. Biotechnol. 2020, 8, 166. [Google Scholar] [CrossRef]
- Zhang, Y.; Wu, J.L.Y.; Lazarovits, J.; Chan, W.C.W. An Analysis of the Binding Function and Structural Organization of the Protein Corona. J. Am. Chem. Soc. 2020, 142, 8827–8836. [Google Scholar] [CrossRef]
- Chen, D.; Ganesh, S.; Wang, W.; Amiji, M. Protein Corona-Enabled Systemic Delivery and Targeting of Nanoparticles. AAPS J. 2020, 22, 83. [Google Scholar] [CrossRef]
- Kelly, P.M.; Åberg, C.; Polo, E.; O’Connell, A.; Cookman, J.; Fallon, J.; Krpetić, Ž.; Dawson, K.A. Mapping protein binding sites on the biomolecular corona of nanoparticles. Nat. Nanotechnol. 2015, 10, 472–479. [Google Scholar] [CrossRef]
- Mirshafiee, V.; Kim, R.; Park, S.; Mahmoudi, M.; Kraft, M.L. Impact of protein pre-coating on the protein corona composition and nanoparticle cellular uptake. Biomaterials 2016, 75, 295–304. [Google Scholar] [CrossRef]
- Cheng, Z.; Al Zaki, A.; Hui, J.Z.; Muzykantov, V.R.; Tsourkas, A. Multifunctional nanoparticles: Cost versus benefit of adding targeting and imaging capabilities. Science 2012, 338, 903–910. [Google Scholar] [CrossRef] [Green Version]
- Vyas, S.P.; Sihorkar, V. Endogenous carriers and ligands in non-immunogenic site-specific drug delivery. Adv. Drug Deliv. Rev. 2000, 43, 101–164. [Google Scholar] [CrossRef]
- Burgess, A.; Hynynen, K. Noninvasive and targeted drug delivery to the brain using focused ultrasound. ACS Chem. Neurosci. 2013, 4, 519–526. [Google Scholar] [CrossRef] [Green Version]
- Burgess, A.; Hynynen, K. Drug delivery across the blood-brain barrier using focused ultrasound. Expert Opin. Drug Deliv. 2014, 11, 711–721. [Google Scholar] [CrossRef] [Green Version]
- Burgess, A.; Shah, K.; Hough, O.; Hynynen, K. Focused ultrasound-mediated drug delivery through the blood-brain barrier. Expert Rev. Neurother. 2015, 15, 477–491. [Google Scholar] [CrossRef] [Green Version]
- Fisher, D.G.; Price, R.J. Recent Advances in the Use of Focused Ultrasound for Magnetic Resonance Image-Guided Therapeutic Nanoparticle Delivery to the Central Nervous System. Front. Pharmacol. 2019, 10, 1348. [Google Scholar] [CrossRef]
- Meairs, S. Facilitation of Drug Transport across the Blood-Brain Barrier with Ultrasound and Microbubbles. Pharmaceutics 2015, 7, 275–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Endo-Takahashi, Y.; Negishi, Y. Microbubbles and Nanobubbles with Ultrasound for Systemic Gene Delivery. Pharmaceutics 2020, 12, 964. [Google Scholar] [CrossRef] [PubMed]
- Fan, C.H.; Ting, C.Y.; Lin, C.Y.; Chan, H.L.; Chang, Y.C.; Chen, Y.Y.; Liu, H.L.; Yeh, C.K. Noninvasive, Targeted, and Non-Viral Ultrasound-Mediated GDNF-Plasmid Delivery for Treatment of Parkinson’s Disease. Sci. Rep. 2016, 6, 19579. [Google Scholar] [CrossRef] [PubMed]
- Fan, C.H.; Lin, C.Y.; Liu, H.L.; Yeh, C.K. Ultrasound targeted CNS gene delivery for Parkinson’s disease treatment. J. Control. Release 2017, 261, 246–262. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.Y.; Hsieh, H.Y.; Chen, C.M.; Wu, S.R.; Tsai, C.H.; Huang, C.Y.; Hua, M.Y.; Wei, K.C.; Yeh, C.K.; Liu, H.L. Non-invasive, neuron-specific gene therapy by focused ultrasound-induced blood-brain barrier opening in Parkinson’s disease mouse model. J. Control. Release 2016, 235, 72–81. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.Y.; Tsai, C.H.; Feng, L.Y.; Chai, W.Y.; Lin, C.J.; Huang, C.Y.; Wei, K.C.; Yeh, C.K.; Chen, C.M.; Liu, H.L. Focused ultrasound-induced blood brain-barrier opening enhanced vascular permeability for GDNF delivery in Huntington’s disease mouse model. Brain Stimul. 2019, 12, 1143–1150. [Google Scholar] [CrossRef] [PubMed]
- Mead, B.P.; Kim, N.; Miller, G.W.; Hodges, D.; Mastorakos, P.; Klibanov, A.L.; Mandell, J.W.; Hirsh, J.; Suk, J.S.; Hanes, J.; et al. Novel Focused Ultrasound Gene Therapy Approach Noninvasively Restores Dopaminergic Neuron Function in a Rat Parkinson’s Disease Model. Nano Lett. 2017, 17, 3533–3542. [Google Scholar] [CrossRef]
- Bilang-Bleuel, A.; Revah, F.; Colin, P.; Locquet, I.; Robert, J.J.; Mallet, J.; Horellou, P. Intrastriatal injection of an adenoviral vector expressing glial-cell-line-derived neurotrophic factor prevents dopaminergic neuron degeneration and behavioral impairment in a rat model of Parkinson disease. Proc. Natl. Acad. Sci. USA 1997, 94, 8818–8823. [Google Scholar] [CrossRef] [Green Version]
- Tomac, A.; Lindqvist, E.; Lin, L.F.; Ogren, S.O.; Young, D.; Hoffer, B.J.; Olson, L. Protection and repair of the nigrostriatal dopaminergic system by GDNF in vivo. Nature 1995, 373, 335–339. [Google Scholar] [CrossRef]
- Lin, L.F.; Doherty, D.H.; Lile, J.D.; Bektesh, S.; Collins, F. GDNF: A glial cell line-derived neurotrophic factor for midbrain dopaminergic neurons. Science 1993, 260, 1130–1132. [Google Scholar] [CrossRef]
- Gash, D.M.; Zhang, Z.; Ovadia, A.; Cass, W.A.; Yi, A.; Simmerman, L.; Russell, D.; Martin, D.; Lapchak, P.A.; Collins, F.; et al. Functional recovery in parkinsonian monkeys treated with GDNF. Nature 1996, 380, 252–255. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Han, L.; Qin, J.; Lu, W.; Wang, J. The use of borneol as an enhancer for targeting aprotinin-conjugated PEG-PLGA nanoparticles to the brain. Pharm. Res. 2013, 30, 2560–2572. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Zhang, A.; Lu, H.; Cheng, Q. The Role and Mechanism of Borneol to Open the Blood-Brain Barrier. Integr. Cancer. Ther. 2018, 17, 806–812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garg, P.; Pandey, S.; Seonwoo, H.; Yeom, S.; Choung, Y.H.; Cho, C.S.; Choung, P.H.; Hoon Chung, J. Hyperosmotic polydixylitol for crossing the blood brain barrier and efficient nucleic acid delivery. Chem. Commun. (Camb.) 2015, 51, 3645–3648. [Google Scholar] [CrossRef] [PubMed]
- Bynoe, M.S.; Viret, C.; Yan, A.; Kim, D.G. Adenosine receptor signaling: A key to opening the blood-brain door. Fluids Barriers CNS 2015, 12, 20. [Google Scholar] [CrossRef] [Green Version]
- Carman, A.J.; Mills, J.H.; Krenz, A.; Kim, D.G.; Bynoe, M.S. Adenosine receptor signaling modulates permeability of the blood-brain barrier. J. Neurosci. 2011, 31, 13272–13280. [Google Scholar] [CrossRef]
- Gao, X.; Qian, J.; Zheng, S.; Changyi, Y.; Zhang, J.; Ju, S.; Zhu, J.; Li, C. Overcoming the blood-brain barrier for delivering drugs into the brain by using adenosine receptor nanoagonist. ACS Nano 2014, 8, 3678–3689. [Google Scholar] [CrossRef]
- Jacobson, K.A.; Gao, Z.G. Adenosine receptors as therapeutic targets. Nat. Rev. Drug Discov. 2006, 5, 247–264. [Google Scholar] [CrossRef] [Green Version]
- Salahuddin, T.S.; Johansson, B.B.; Kalimo, H.; Olsson, Y. Structural changes in the rat brain after carotid infusions of hyperosmolar solutions: A light microscopic and immunohistochemical study. Neuropathol. Appl. Neurobiol. 1988, 14, 467–482. [Google Scholar] [CrossRef]
- Bicker, J.; Fortuna, A.; Alves, G.; Falcão, A. Nose-to-brain Delivery of Natural Compounds for the Treatment of Central Nervous System Disorders. Curr. Pharm. Des. 2020, 26, 594–619. [Google Scholar] [CrossRef]
- Musumeci, T.; Serapide, M.F.; Pellitteri, R.; Dalpiaz, A.; Ferraro, L.; Dal Magro, R.; Bonaccorso, A.; Carbone, C.; Veiga, F.; Sancini, G.; et al. Oxcarbazepine free or loaded PLGA nanoparticles as effective intranasal approach to control epileptic seizures in rodents. Eur. J. Pharm. Biopharm. 2018, 133, 309–320. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N.N.; Lochhead, J.J.; Pizzo, M.E.; Nehra, G.; Boroumand, S.; Greene, G.; Thorne, R.G. Delivery of immunoglobulin G antibodies to the rat nervous system following intranasal administration: Distribution, dose-response, and mechanisms of delivery. J. Control. Release 2018, 286, 467–484. [Google Scholar] [CrossRef] [PubMed]
- Lochhead, J.J.; Kellohen, K.L.; Ronaldson, P.T.; Davis, T.P. Distribution of insulin in trigeminal nerve and brain after intranasal administration. Sci. Rep. 2019, 9, 2621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez, M.; Lapierre, J.; Ojha, C.R.; Kaushik, A.; Batrakova, E.; Kashanchi, F.; Dever, S.M.; Nair, M.; El-Hage, N. Intranasal drug delivery of small interfering RNA targeting Beclin1 encapsulated with polyethylenimine (PEI) in mouse brain to achieve HIV attenuation. Sci. Rep. 2017, 7, 1862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belur, L.R.; Temme, A.; Podetz-Pedersen, K.M.; Riedl, M.; Vulchanova, L.; Robinson, N.; Hanson, L.R.; Kozarsky, K.F.; Orchard, P.J.; Frey, W.H., 2nd; et al. Intranasal Adeno-Associated Virus Mediated Gene Delivery and Expression of Human Iduronidase in the Central Nervous System: A Noninvasive and Effective Approach for Prevention of Neurologic Disease in Mucopolysaccharidosis Type I. Hum. Gene Ther. 2017, 28, 576–587. [Google Scholar] [CrossRef] [PubMed]
- Aly, A.E.; Harmon, B.; Padegimas, L.; Sesenoglu-Laird, O.; Cooper, M.J.; Yurek, D.M.; Waszczak, B.L. Intranasal delivery of hGDNF plasmid DNA nanoparticles results in long-term and widespread transfection of perivascular cells in rat brain. Nanomedicine 2019, 16, 20–33. [Google Scholar] [CrossRef]
- Alarcón-Arís, D.; Recasens, A.; Galofré, M.; Carballo-Carbajal, I.; Zacchi, N.; Ruiz-Bronchal, E.; Pavia-Collado, R.; Chica, R.; Ferrés-Coy, A.; Santos, M.; et al. Selective α-Synuclein Knockdown in Monoamine Neurons by Intranasal Oligonucleotide Delivery: Potential Therapy for Parkinson’s Disease. Mol. Ther. 2018, 26, 550–567. [Google Scholar] [CrossRef] [Green Version]
- Aly, A.E.; Waszczak, B.L. Intranasal gene delivery for treating Parkinson’s disease: Overcoming the blood-brain barrier. Expert Opin. Drug Deliv. 2015, 12, 1923–1941. [Google Scholar] [CrossRef]
- Kim, I.D.; Shin, J.H.; Kim, S.W.; Choi, S.; Ahn, J.; Han, P.L.; Park, J.S.; Lee, J.K. Intranasal delivery of HMGB1 siRNA confers target gene knockdown and robust neuroprotection in the postischemic brain. Mol. Ther. 2012, 20, 829–839. [Google Scholar] [CrossRef] [Green Version]
- Lochhead, J.J.; Thorne, R.G. Intranasal delivery of biologics to the central nervous system. Adv. Drug Deliv. Rev. 2012, 64, 614–628. [Google Scholar] [CrossRef]
- Podolska, K.; Stachurska, A.; Hajdukiewicz, K.; Małecki, M. Gene therapy prospects--intranasal delivery of therapeutic genes. Adv. Clin. Exp. Med. 2012, 21, 525–534. [Google Scholar] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Product (Company) | Vector | Delivery Gene | Target Disease | Approved Country (Year) | Reference |
|---|---|---|---|---|---|
| Gendicine (Shenzhen SiBiono GeneTech, Shenzhen, China) | Adenovirus (in vivo, IT) | Human wild-type p53 | Head and neck squamous cell carcinoma | China (2003) | |
| Oncorine (Shanghai Sunway Biotech, Shanghai, China) | Oncolytic adenovirus (in vivo, IT) | Head and neck cancer | China (2006) | [26] | |
| Rexin G (Epeius Biotechnology, Monrovia, CA, USA) | Retrovirus (in vivo, IV) | Mutant cyclin G1 | Chemoresistant solid tumor | Philippines (2007) | |
| Neovagulgen (Human Stem Cell Institute, Moscow, Russia) | Plasmid (in vivo, IM) | Vascular endothelial growth factor (VEGF) | Peripheral artery disease | Russia (2011) Ukraine (2013) | |
| Glybera (Uniqure, Amsterdam, Netherlands) Discontinued | AAV1 (in vivo IM) | Lipoprotein lipase | Lipoprotein lipase deficiency | Europe (2012) | [27] |
| Imlygic (Amgen, Thousand Oaks, CA, USA) | Oncolytic HSV1 (in vivo, IT) | Granulocyte macrophage colony-stimulating factor (GM-CSF) | Malignant melanoma | America (2015) Europe (2015) | [28] |
| Strimveils (GSK, Brentford, United Kingdam) | Retrovirus (ex vivo) | Adenosine deaminase (ADA) | ADA deficiency | Europe (2016) | [29] |
| Zalmoxis (MolMed S.p.A, Milano, Italy) | Retrovirus (ex vivo) | Herpesvirus thymidine kinase | Graft versus host disease | Europe (2016) | |
| Kymriah (Novartis, Basel, Switzerland) | Retrovirus (ex vivo) | Chimeric antigen receptor (CAR) against CD19 | Acute lymphoblastic leukemia | America (2017) Europe (2018) Japan (2019) | [30] |
| Yescarta (Kite Pharma, Santa Monica, CA, USA) | Retrovirus (ex vivo) | CAR against CD19 | Large B cell lymphoma | America (2017) Europe (2018) | [31] |
| Luxturna (Spark Therapeutics, Philadelphia, PA, USA) | AAV2 (in vivo, SR) | RPE65 | Retinal dystrophy | America (2017) Europe (2018) | |
| Zynteglo (Bluebird Bio, Cambridge, MA, USA) | Lentivirus (ex vivo) | Beta-globin | Beta thalassemia | Europe (2019) | [32] |
| Zolgensma (Novartis, Basel, Switzerland) | AAV9 (in vivo, IV) | SMN1 | Spinal mascular atrophy (SMA) | America (2019) Europe (2019) | [25] |
| Collategene (Mitsubishi Tanabe Pharma Corporation, Osaka, Japan) | Plasmid (in vivo, IM) | HGF | Critical limb ischemia | Japan (2019) |
| Disease | Target Gene (Protein) | Mechanism | Reference |
|---|---|---|---|
| PD | AADC | Replace | [16] |
| GAD | Replace | [17] | |
| GBA1 | Replace | [33] | |
| SNCA | Silence | [34] | |
| AD | APOE4 | Silence/immunotherapy | [20] |
| APOE2 | Replace | [35] | |
| APP (amyloid-beta) | Silence | [22] | |
| MAPT (tau) | Silence/immunotherapy | [36] | |
| PQBP1 | Replace | [23] | |
| ALS | SOD1 | Silence | [37] |
| C9orf72 | Silence | [38] | |
| TARDBP | Silence | [39] | |
| SMA | SMN1 | Replace | [24] |
| CNS Disease | The Type of BBB Breakdown and Dysfunction | Reference |
|---|---|---|
| AD | Down-regulation of glucose transporter 1 (GLUT1) | [201] |
| Down-regulation of low density lipoprotein receptor (LDLR) | [203] | |
| Decreased expression and functionality of P-gp | [204] | |
| Low expression of tight junction (TJ) proteins | [205] | |
| Multiple sclerosis (MS) | Abundance of chemokines and cytokines, leading to damage on TJ proteins | [206] |
| PD | Decrease in TJ protein expression, leaky BBB | [207] |
| Abnormal distribution of GLUT1 | [208] | |
| Stroke | Up-regulation of GLUT1 Increased BBB permeability | [209] |
| [210] | ||
| [211] | ||
| Glioblastoma | Induction of blood-brain tumor barrier | [212] |
| Overexpression of certain receptor-mediated transcytosis (RMT) | [213] |
| Serotype | Relative Efficiency (vs. AAV9) | Notable Features Capsid Mutation Pattern | Reference |
|---|---|---|---|
| AAV9 | 1 | Neonatal mice→neurons | [216] |
| Adult mice→astrocytes | |||
| Used as the vector of Zolgensma | |||
| AAV.GTX | AAV9 capsid VP1; Y446F and Y731F→inhibit ubiquitination | [217] | |
| AAV-PHP.B | <40 | AAV9 capsid; Q588-TLAVPFK-A589→AAV-PHP.B | [195] |
| AAV-PHP.eB | AAV-PHP.B capsid; A587D, Q588G→AAV-PHP.eB | ||
| AAV.B1 | ~10 | DNA shuffling, Less reactive to neutralizing antibodies than AAV9 | [218] |
| AAV.AS | ~15 | AAV9 4 amino acids; S414N, G453D, K557E, T582I→AAV9.47 | [219] |
| AAV9.47 VP2; +19 alanine |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kimura, S.; Harashima, H. Current Status and Challenges Associated with CNS-Targeted Gene Delivery across the BBB. Pharmaceutics 2020, 12, 1216. https://doi.org/10.3390/pharmaceutics12121216
Kimura S, Harashima H. Current Status and Challenges Associated with CNS-Targeted Gene Delivery across the BBB. Pharmaceutics. 2020; 12(12):1216. https://doi.org/10.3390/pharmaceutics12121216
Chicago/Turabian StyleKimura, Seigo, and Hideyoshi Harashima. 2020. "Current Status and Challenges Associated with CNS-Targeted Gene Delivery across the BBB" Pharmaceutics 12, no. 12: 1216. https://doi.org/10.3390/pharmaceutics12121216