Effects of the Glass-Forming Ability and Annealing Conditions on Cocrystallization Behaviors via Rapid Solvent Removal: A Case Study of Voriconazole


 and
and
Abstract
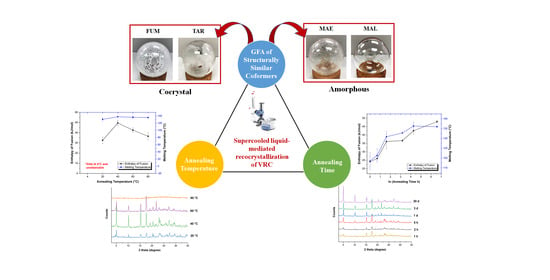
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Preparation of Cocrystals
2.3. Differential Scanning Calorimetry (DSC)
2.4. Powder X-ray Diffraction (PXRD)
2.5. Fourier-Transform Infrared Spectroscopy (FTIR)
2.6. High Performance Liquid Chromatography (HPLC)
2.7. Powder Dissolution Study
3. Results and Discussion
3.1. Role of C4 Diacid Coformers in Altering the Glass-Forming Ability and Crystallization Tendency of VRC
3.2. Effects of the Annealing Temperature and Annealing Time on the Supercooled Liquid-Mediated Re-Cocrystallization of the Amorphous VRC System: A Case Study of the VRC-TAR System
3.2.1. Annealing Temperature
3.2.2. Annealing Time
3.3. Physical Characterization of the VRC-TAR Cocrystal
3.4. Dissolution Performance
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Chow, S.F.; Shi, L.; Ng, W.W.; Leung, K.H.Y.; Nagapudi, K.; Sun, C.C.; Chow, A.H. Kinetic entrapment of a hidden curcumin cocrystal with phloroglucinol. Cryst. Growth Des. 2014, 14, 5079–5089. [Google Scholar] [CrossRef]
- Wong, S.N.; Hu, S.; Ng, W.W.; Xu, X.; Lai, K.L.; Lee, W.Y.T.; Chow, A.H.L.; Sun, C.C.; Chow, S.F. Cocrystallization of curcumin with benzenediols and benzenetriols via rapid solvent removal. Cryst. Growth Des. 2018, 18, 5534–5546. [Google Scholar] [CrossRef]
- Shi, X.; Song, S.; Ding, Z.; Fan, B.; Huang, W.; Xu, T. Improving the solubility, dissolution, and bioavailability of Ibrutinib by preparing it in a Coamorphous state with saccharin. J. Pharm. Sci. 2019, 108, 3020–3028. [Google Scholar] [CrossRef] [PubMed]
- Su, M.; Xia, Y.; Shen, Y.; Heng, W.; Wei, Y.; Zhang, L.; Gao, Y.; Zhang, J.; Qian, S. A novel drug–drug coamorphous system without molecular interactions: Improve the physicochemical properties of tadalafil and repaglinide. RSC Adv. 2020, 10, 565–583. [Google Scholar] [CrossRef] [Green Version]
- Ali, A.M.A.; Ali, A.A.; Maghrabi, I.A. Clozapine-carboxylic acid plasticized co-amorphous dispersions: Preparation, characterization and solution stability evaluation. Acta Pharm. 2015, 65, 133–146. [Google Scholar] [CrossRef] [Green Version]
- Xuan, B.; Wong, S.N.; Zhang, Y.; Weng, J.; Tong, H.H.; Wang, C.; Sun, C.C.; Chow, S.F. Extended release of highly water soluble isoniazid attained through cocrystallization with curcumin. Cryst. Growth Des. 2020, 20, 1951–1960. [Google Scholar] [CrossRef]
- Van Santen, R. The Ostwald step rule. J. Phys. Chem. 1984, 88, 5768–5769. [Google Scholar] [CrossRef]
- Chung, S.-Y.; Kim, Y.-M.; Kim, J.-G.; Kim, Y.-J. Multiphase transformation and Ostwald’s rule of stages during crystallization of a metal phosphate. Nat. Phys. 2009, 5, 68–73. [Google Scholar] [CrossRef]
- Laitinen, R.; Löbmann, K.; Strachan, C.J.; Grohganz, H.; Rades, T. Emerging trends in the stabilization of amorphous drugs. Int. J. Pharm. 2013, 453, 65–79. [Google Scholar] [CrossRef]
- Grohganz, H.; Priemel, P.A.; Löbmann, K.; Nielsen, L.H.; Laitinen, R.; Mullertz, A.; Van den Mooter, G.; Rades, T. Refining stability and dissolution rate of amorphous drug formulations. Expert Opin. Drug Deliv. 2014, 11, 977–989. [Google Scholar] [CrossRef]
- Dengale, S.J.; Grohganz, H.; Rades, T.; Löbmann, K. Recent advances in co-amorphous drug formulations. Adv. Drug Deliv. Rev. 2016, 100, 116–125. [Google Scholar] [CrossRef] [PubMed]
- Prestidge, C.A.; Barnes, T.J.; Lau, C.-H.; Barnett, C.; Loni, A.; Canham, L. Mesoporous silicon: A platform for the delivery of therapeutics. Expert Opin. Drug Deliv. 2007, 4, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Löbmann, K.; Laitinen, R.; Grohganz, H.; Gordon, K.C.; Strachan, C.; Rades, T. Coamorphous drug systems: Enhanced physical stability and dissolution rate of indomethacin and naproxen. Mol. Pharm. 2011, 8, 1919–1928. [Google Scholar] [CrossRef] [PubMed]
- Chow, S.F.; Chen, M.; Shi, L.; Chow, A.H.L.; Sun, C.C. Simultaneously improving the mechanical properties, dissolution performance, and hygroscopicity of ibuprofen and flurbiprofen by cocrystallization with nicotinamide. Pharm. Res. 2012, 29, 1854–1865. [Google Scholar] [CrossRef] [PubMed]
- Mannhold, R.B.; Buschmann, H.; Holenz, J. Innovative Dosage Forms: Design and Development at Early Stage; John Wiley & Sons: Hoboken, NJ, USA, 2019. [Google Scholar]
- Alhalaweh, A.; Alzghoul, A.; Kaialy, W.; Mahlin, D.; Bergström, C.A. Computational predictions of glass-forming ability and crystallization tendency of drug molecules. Mol. Pharm. 2014, 11, 3123–3132. [Google Scholar] [CrossRef] [PubMed]
- Baird, J.A.; Van Eerdenbrugh, B.; Taylor, L.S. A classification system to assess the crystallization tendency of organic molecules from undercooled melts. J. Pharm. Sci. 2010, 99, 3787–3806. [Google Scholar] [CrossRef]
- Fung, M.; Bēziņš, K.; Suryanarayanan, R. Physical stability and dissolution behavior of ketoconazole–organic acid coamorphous systems. Mol. Pharm. 2018, 15, 1862–1869. [Google Scholar] [CrossRef]
- Wu, W.; Wang, Y.; Löbmann, K.; Grohganz, H.; Rades, T. Transformations between Co-Amorphous and Co-Crystal Systems and Their Influence on the Formation and Physical Stability of Co-Amorphous Systems. Mol. Pharm. 2019, 16, 1294–1304. [Google Scholar] [CrossRef]
- Van den Mooter, G.; Wuyts, M.; Blaton, N.; Busson, R.; Grobet, P.; Augustijns, P.; Kinget, R. Physical stabilisation of amorphous ketoconazole in solid dispersions with polyvinylpyrrolidone K25. Eur. J. Pharm. Sci. 2001, 12, 261–269. [Google Scholar] [CrossRef]
- Karagianni, A.; Kachrimanis, K.; Nikolakakis, I. Co-amorphous solid dispersions for solubility and absorption improvement of drugs: Composition, preparation, characterization and formulations for oral delivery. Pharmaceutics 2018, 10, 98. [Google Scholar] [CrossRef] [Green Version]
- Patterson, T.F.; Thompson, G.R., III; Denning, D.W.; Fishman, J.A.; Hadley, S.; Herbrecht, R.; Kontoyiannis, D.P.; Marr, K.A.; Morrison, V.A.; Nguyen, M.H. Practice guidelines for the diagnosis and management of aspergillosis: 2016 update by the Infectious Diseases Society of America. Clin. Infect. Dis. 2016, 63, e1–e60. [Google Scholar] [CrossRef] [PubMed]
- Tortorano, A.; Richardson, M.; Roilides, E.; Van Diepeningen, A.; Caira, M.; Munoz, P.; Johnson, E.; Meletiadis, J.; Pana, Z.D.; Lackner, M. ESCMID and ECMM joint guidelines on diagnosis and management of hyalohyphomycosis: Fusarium spp., Scedosporium spp. and others. Clin. Microbiol. Infect. 2014, 20, 27–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pappas, P.G.; Kauffman, C.A.; Andes, D.R.; Clancy, C.J.; Marr, K.A.; Ostrosky-Zeichner, L.; Reboli, A.C.; Schuster, M.G.; Vazquez, J.A.; Walsh, T.J. Clinical practice guideline for the management of candidiasis: 2016 update by the Infectious Diseases Society of America. Clin. Infect. Dis. 2016, 62, e1–e50. [Google Scholar] [CrossRef]
- Roffey, S.; Cole, S.; Comby, P.; Gibson, D.; Jezequel, S.; Nedderman, A.; Smith, D.; Walker, D.; Wood, N. The disposition of voriconazole in mouse, rat, rabbit, guinea pig, dog, and human. Drug Metab. Dispos. 2003, 31, 731–741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, L.; Papadopoulos, D.; Dunn, P.; Bentham, A.; Dawson, N.; Mitchell, J.; Snowden, M. Predictive milling of pharmaceutical materials using nanoindentation of single crystals. Org. Process Res. Dev. 2004, 8, 674–679. [Google Scholar] [CrossRef]
- Ramos, J.J.M.; Diogo, H.P. The slow relaxation dynamics in active pharmaceutical ingredients studied by DSC and TSDC: Voriconazole, miconazole and itraconazole. Int. J. Pharm. 2016, 501, 39–48. [Google Scholar] [CrossRef]
- Hassan, R.; El-Kadi, S.; Sand, M. Effect of some organic acids on some fungal growth and their toxins production. Int. J. Adv. Biol. 2015, 2, 1–11. [Google Scholar] [CrossRef]
- Liao, Q.; Yip, L.; Chow, M.Y.; Chow, S.F.; Chan, H.-K.; Kwok, P.C.; Lam, J.K. Porous and highly dispersible voriconazole dry powders produced by spray freeze drying for pulmonary delivery with efficient lung deposition. Int. J. Pharm. 2019, 560, 144–154. [Google Scholar] [CrossRef]
- Sanphui, P.; Mishra, M.K.; Ramamurty, U.; Desiraju, G.R. Tuning mechanical properties of pharmaceutical crystals with multicomponent crystals: Voriconazole as a case study. Mol. Pharm. 2015, 12, 889–897. [Google Scholar] [CrossRef]
- Mahlin, D.; Bergström, C.A. Early drug development predictions of glass-forming ability and physical stability of drugs. Eur. J. Pharm. Sci. 2013, 49, 323–332. [Google Scholar] [CrossRef]
- Alie, J.; Lacabanne, C.; Menegotto, J.; Cardon, P.; Duplaa, H.; Caron, A.; Bauer, M. Dielectric study of the molecular mobility and the isothermal crystallization kinetics of an amorphous pharmaceutical drug substance. J. Pharm. Sci. 2004, 93, 218–233. [Google Scholar] [CrossRef] [PubMed]
- Hancock, B.C.; Shamblin, S.L.; Zografi, G. Molecular mobility of amorphous pharmaceutical solids below their glass transition temperatures. Pharm. Res. 1995, 12, 799–806. [Google Scholar] [CrossRef] [PubMed]
- Jameel, F.; Hershenson, S. Formulation and Process Development Strategies for Manufacturing Biopharmaceuticals; John Wiley & Sons: Hoboken, NJ, USA, 2010. [Google Scholar]
- Heuer, A.; Spiess, H.W. Universality of the glass transition temperature. J. Non-Cryst. Solids 1994, 176, 294–298. [Google Scholar] [CrossRef]
- Kanno, H. A simple derivation of the empirical rule TGTM = 23. J. Non-Cryst. Solids 1981, 44, 409–413. [Google Scholar] [CrossRef]
- Lin, Y.; Cogdill, R.P.; Wildfong, P.L. Informatic calibration of a materials properties database for predictive assessment of mechanically activated disordering potential for small molecule organic solids. J. Pharm. Sci. 2009, 98, 2696–2708. [Google Scholar] [CrossRef] [PubMed]
- Kerc, J.; Srcic, S. Thermal analysis of glassy pharmaceuticals. Thermochim. Acta 1995, 248, 81–95. [Google Scholar] [CrossRef]
- Fukuoka, E.; Makita, M.; Yamamura, S. Glassy state of pharmaceuticals. III.: Thermal properties and stability of glassy pharmaceuticals and their binary glass systems. Chem. Pharm. Bull. 1989, 37, 1047–1050. [Google Scholar] [CrossRef] [Green Version]
- Alhalaweh, A.; Alzghoul, A.; Mahlin, D.; Bergström, C.A. Physical stability of drugs after storage above and below the glass transition temperature: Relationship to glass-forming ability. Int. J. Pharm. 2015, 495, 312–317. [Google Scholar] [CrossRef]
- Curtin, V.; Amharar, Y.; Hu, Y.; Erxleben, A.; McArdle, P.; Caron, V.; Tajber, L.; Corrigan, O.I.; Healy, A.M. Investigation of the capacity of low glass transition temperature excipients to minimize amorphization of sulfadimidine on comilling. Mol. Pharm. 2013, 10, 386–396. [Google Scholar] [CrossRef] [Green Version]
- Amharar, Y.; Curtin, V.; Gallagher, K.H.; O’Siochru, E.; O’Connell, P.; Healy, A.-M. Mitigating unwanted amorphisation: A screening method for the selection of suitable excipients. Eur. J. Pharm. Sci. 2016, 81, 181–188. [Google Scholar] [CrossRef]
- Healy, A.M.; Worku, Z.A.; Kumar, D.; Madi, A.M. Pharmaceutical solvates, hydrates and amorphous forms: A special emphasis on cocrystals. Adv. Drug Deliv. Rev. 2017, 117, 25–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.; Lee, T.W.; Chow, A.H. Thermodynamic and kinetic evaluation of the impact of polymer excipients on storage stability of amorphous itraconazole. Int. J. Pharm. 2019, 555, 394–403. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Li, H.; Lang, B.; O’Donnell, K.; Zhang, H.; Wang, Z.; Dong, Y.; Wu, C.; Williams, R.O., III. Formulation and delivery of improved amorphous fenofibrate solid dispersions prepared by thin film freezing. Eur. J. Pharm. Biopharm. 2012, 82, 534–544. [Google Scholar] [CrossRef] [PubMed]
- Storey, R.A.; Ymén, I. Solid State Characterization of Pharmaceuticals; John Wiley & Sons: Hoboken, NJ, USA, 2011. [Google Scholar]
- Shevchenko, A.; Miroshnyk, I.; Pietilä, L.-O.; Haarala, J.; Salmia, J.; Sinervo, K.; Mirza, S.; van Veen, B.; Kolehmainen, E.; Yliruusi, J. Diversity in itraconazole cocrystals with aliphatic dicarboxylic acids of varying chain length. Cryst. Growth Des. 2013, 13, 4877–4884. [Google Scholar] [CrossRef]
- Weng, J.; Wong, S.N.; Xu, X.; Xuan, B.; Wang, C.; Chen, R.; Sun, C.C.; Lakerveld, R.; Kwok, P.C.L.; Chow, S.F. Cocrystal engineering of itraconazole with suberic acid via rotary evaporation and spray drying. Cryst. Growth Des. 2019, 19, 2736–2745. [Google Scholar] [CrossRef]
- Sarceviča, I.; Kons, A.; Orola, L. Isoniazid cocrystallisation with dicarboxylic acids: Vapochemical, mechanochemical and thermal methods. CrystEngComm 2016, 18, 1625–1635. [Google Scholar] [CrossRef]
- Espinosa-Lara, J.C.; Guzman-Villanueva, D.; Arenas-García, J.I.; Herrera-Ruiz, D.; Rivera-Islas, J.S.; Román-Bravo, P.; Morales-Rojas, H.; Höpfl, H. Cocrystals of Active Pharmaceutical Ingredients—Praziquantel in Combination with Oxalic, Malonic, Succinic, Maleic, Fumaric, Glutaric, Adipic, And Pimelic Acids. Cryst. Growth Des. 2013, 13, 169–185. [Google Scholar] [CrossRef]
- Trask, A.V.; Motherwell, W.S.; Jones, W. Pharmaceutical cocrystallization: Engineering a remedy for caffeine hydration. Cryst. Growth Des. 2005, 5, 1013–1021. [Google Scholar] [CrossRef]
- Lin, H.-L.; Wu, T.-K.; Lin, S.-Y. Screening and characterization of cocrystal formation of metaxalone with short-chain dicarboxylic acids induced by solvent-assisted grinding approach. Thermochim. Acta 2014, 575, 313–321. [Google Scholar] [CrossRef]
- Luo, Y.-H.; Sun, B.-W. Co-crystallization of pyridine-2-carboxamide with a series of alkyl dicarboxylic acids with different carbon chain: Crystal structure, spectroscopy and Hirshfeld analysis. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2014, 120, 228–236. [Google Scholar] [CrossRef]
- Vishweshwar, P.; Nangia, A.; Lynch, V.M. Molecular complexes of homologous alkanedicarboxylic acids with isonicotinamide: X-ray crystal structures, hydrogen bond synthons, and melting point alternation. Cryst. Growth Des. 2003, 3, 783–790. [Google Scholar] [CrossRef]
- Good, D.J.; Rodriguez-Hornedo, N. Solubility advantage of pharmaceutical cocrystals. Cryst. Growth Des. 2009, 9, 2252–2264. [Google Scholar] [CrossRef]
- Martin, F.A.; Pop, M.M.; Borodi, G.; Filip, X.; Kacso, I. Ketoconazole salt and co-crystals with enhanced aqueous solubility. Cryst. Growth Des. 2013, 13, 4295–4304. [Google Scholar] [CrossRef]
- Ahn, B.; Kim, S.; Kim, Y.; Yang, J. Synthesis and characterization of the biodegradable copolymers from succinic acid and adipic acid with 1, 4-butanediol. J. Appl. Polym. Sci. 2001, 82, 2808–2826. [Google Scholar] [CrossRef]
- Considine, D.M.; Considine, G.D. Van Nostrand’s Scientific Encyclopedia; Springer Science & Business Media: New York, NY, USA, 2013. [Google Scholar]
- Fox, T.G.; Flory, P.J. The glass temperature and related properties of polystyrene. Influence of molecular weight. J. Polym. Sci. 1954, 14, 315–319. [Google Scholar] [CrossRef]
- Brostow, W.; Chiu, R.; Kalogeras, I.M.; Vassilikou-Dova, A. Prediction of glass transition temperatures: Binary blends and copolymers. Mater. Lett. 2008, 62, 3152–3155. [Google Scholar] [CrossRef]
- Lu, X.; Weiss, R. Relationship between the glass transition temperature and the interaction parameter of miscible binary polymer blends. Macromolecules 1992, 25, 3242–3246. [Google Scholar] [CrossRef]
- Nair, R.; Nyamweya, N.; Gönen, S.; Martínez-Miranda, L.; Hoag, S. Influence of various drugs on the glass transition temperature of poly (vinylpyrrolidone): A thermodynamic and spectroscopic investigation. Int. J. Pharm. 2001, 225, 83–96. [Google Scholar] [CrossRef]
- Hancock, B.C.; Zografi, G. The relationship between the glass transition temperature and the water content of amorphous pharmaceutical solids. Pharm. Res. 1994, 11, 471–477. [Google Scholar] [CrossRef]
- Uvarov, V. The influence of X-ray diffraction pattern angular range on Rietveld refinement results used for quantitative analysis, crystallite size calculation and unit-cell parameter refinement. J. Appl. Crystallogr. 2019, 52, 252–261. [Google Scholar] [CrossRef]
- Avrami, M. Kinetics of phase change. I. General theory. J. Chem. Phys. 1939, 7, 1103–1112. [Google Scholar] [CrossRef]
- Avrami, M. Kinetics of phase change. II transformation-time relations for random distribution of nuclei. J. Chem. Phys. 1940, 8, 212–224. [Google Scholar] [CrossRef]
- Avrami, M. Kinetics of phase change. III: Granulation, phase change and microstructure. J. Chem. Phys. 1941, 9, 177–184. [Google Scholar] [CrossRef]
- Blázquez, J.S.; Manchón-Gordón, A.F.; Ipus, J.J.; Conde, C.F.; Conde, A. On the Use of JMAK Theory to Describe Mechanical Amorphization: A Comparison between Experiments, Numerical Solutions and Simulations. Metals 2018, 8, 450. [Google Scholar] [CrossRef] [Green Version]
- Lin, F.; Zhang, Y.; Tao, N.; Pantleon, W.; Jensen, D.J. Effects of heterogeneity on recrystallization kinetics of nanocrystalline copper prepared by dynamic plastic deformation. Acta Mater. 2014, 72, 252–261. [Google Scholar] [CrossRef] [Green Version]
- Siafaka, P.I.; Üstündağ Okur, N.; Mone, M.; Giannakopoulou, S.; Er, S.; Pavlidou, E.; Karavas, E.; Bikiaris, D.N. Two different approaches for oral administration of voriconazole loaded formulations: Electrospun fibers versus β-cyclodextrin complexes. Int. J. Mol. Sci. 2016, 17, 282. [Google Scholar] [CrossRef]
- Khare, A.; Singh, I.; Pawar, P.; Grover, K. Design and evaluation of voriconazole loaded solid lipid nanoparticles for ophthalmic application. J. Drug Deliv. 2016, 2016. [Google Scholar] [CrossRef] [Green Version]
- Pfizer, I. Vfend (Voriconazole) Package Insert; Pfizer Inc.: New York, NY, USA, 2010. [Google Scholar]
- Hope, W.W.; Billaud, E.M.; Lestner, J.; Denning, D.W. Therapeutic drug monitoring for triazoles. Curr. Opin. Infect. Dis. 2008, 21, 580–586. [Google Scholar] [CrossRef]
- Dolton, M.J.; Mikus, G.; Weiss, J.; Ray, J.E.; McLachlan, A.J. Understanding variability with voriconazole using a population pharmacokinetic approach: Implications for optimal dosing. J. Antimicrob. Chemother. 2014, 69, 1633–1641. [Google Scholar] [CrossRef]
- Trifilio, S.; Ortiz, R.; Pennick, G.; Verma, A.; Pi, J.; Stosor, V.; Zembower, T.; Mehta, J. Voriconazole therapeutic drug monitoring in allogeneic hematopoietic stem cell transplant recipients. Bone Marrow Transplant. 2005, 35, 509–513. [Google Scholar] [CrossRef] [Green Version]
- Kostewicz, E.S.; Wunderlich, M.; Brauns, U.; Becker, R.; Bock, T.; Dressman, J.B. Predicting the precipitation of poorly soluble weak bases upon entry in the small intestine. J. Pharm. Pharmacol. 2004, 56, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Hens, B.; Brouwers, J.; Corsetti, M.; Augustijns, P. Supersaturation and precipitation of posaconazole upon entry in the upper small intestine in humans. J. Pharm. Sci. 2016, 105, 2677–2684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tartaric Acid (Compound). Available online: https://pubchem.ncbi.nlm.nih.gov/compound/tartaric_acid#section=EPA-CPDat-Classification (accessed on 9 July 2020).













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chemical Name | M.W. (g/mol) | M.P. (°C) | ΔHf (kJ/mol) | Tg (°C) |
|---|---|---|---|---|
| VRC | 349.3 | 131.4 | 32.4 | 11.9 1 |
| FUM | 116.1 | 294.4 | 56.0 | 105.2 2 |
| TAR | 150.1 | 173.1 | 36.0 | 16 3 |
| MAE | 116.1 | 143.4 | 30.9 | 4.6 2 |
| MAL | 134.1 | 101.1 | 22.1 | −20 3 |
| Annealing Temp. (°C) | M.P. (°C) | ΔHf (kJ/mol) |
|---|---|---|
| 4 | − 1 | − 1 |
| 20 | 130.6 ± 5.0 | 32.5 ± 3.2 |
| 40 | 137.6 ± 0.5 | 49.6 ± 2.6 |
| 60 | 136.3 ± 2.1 | 42.5 ± 2.2 |
| 80 | 135.5 ± 1.7 | 36.5 ± 3.8 |
| Annealing Time | PA[t] at 5.10° 2θ | %RC at 5.10° 2θ 1 | PA[t] at 10.21° 2θ | %RC at 10.21° 2θ 1 |
|---|---|---|---|---|
| 1 h | 3.1 | 9.0 | 3.3 | 15.7 |
| 2 h | 7.5 | 22.0 | 2.6 | 12.4 |
| 5 h | 12.0 | 35.0 | 5.9 | 28.3 |
| 1 d | 18.0 | 52.4 | 14.7 | 69.9 |
| 3 d | 25.9 | 75.4 | 17.5 | 83.1 |
| 30 d | 34.4 | 100 | 21.0 | 100 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wong, S.N.; Chan, S.W.S.; Peng, X.; Xuan, B.; Lee, H.W.; Tong, H.H.Y.; Chow, S.F. Effects of the Glass-Forming Ability and Annealing Conditions on Cocrystallization Behaviors via Rapid Solvent Removal: A Case Study of Voriconazole. Pharmaceutics 2020, 12, 1209. https://doi.org/10.3390/pharmaceutics12121209
Wong SN, Chan SWS, Peng X, Xuan B, Lee HW, Tong HHY, Chow SF. Effects of the Glass-Forming Ability and Annealing Conditions on Cocrystallization Behaviors via Rapid Solvent Removal: A Case Study of Voriconazole. Pharmaceutics. 2020; 12(12):1209. https://doi.org/10.3390/pharmaceutics12121209
Chicago/Turabian StyleWong, Si Nga, Susan Wing Sze Chan, Xuexin Peng, Bianfei Xuan, Hok Wai Lee, Henry H. Y. Tong, and Shing Fung Chow. 2020. "Effects of the Glass-Forming Ability and Annealing Conditions on Cocrystallization Behaviors via Rapid Solvent Removal: A Case Study of Voriconazole" Pharmaceutics 12, no. 12: 1209. https://doi.org/10.3390/pharmaceutics12121209