Influence of Inter- and Intra-Batch Variability on the Sample Size Required for Demonstration of Equivalent Microstructure of Semisolid Dosage Forms


 , , and
, , and
Abstract
:
1. Introduction
2. Materials and Methods
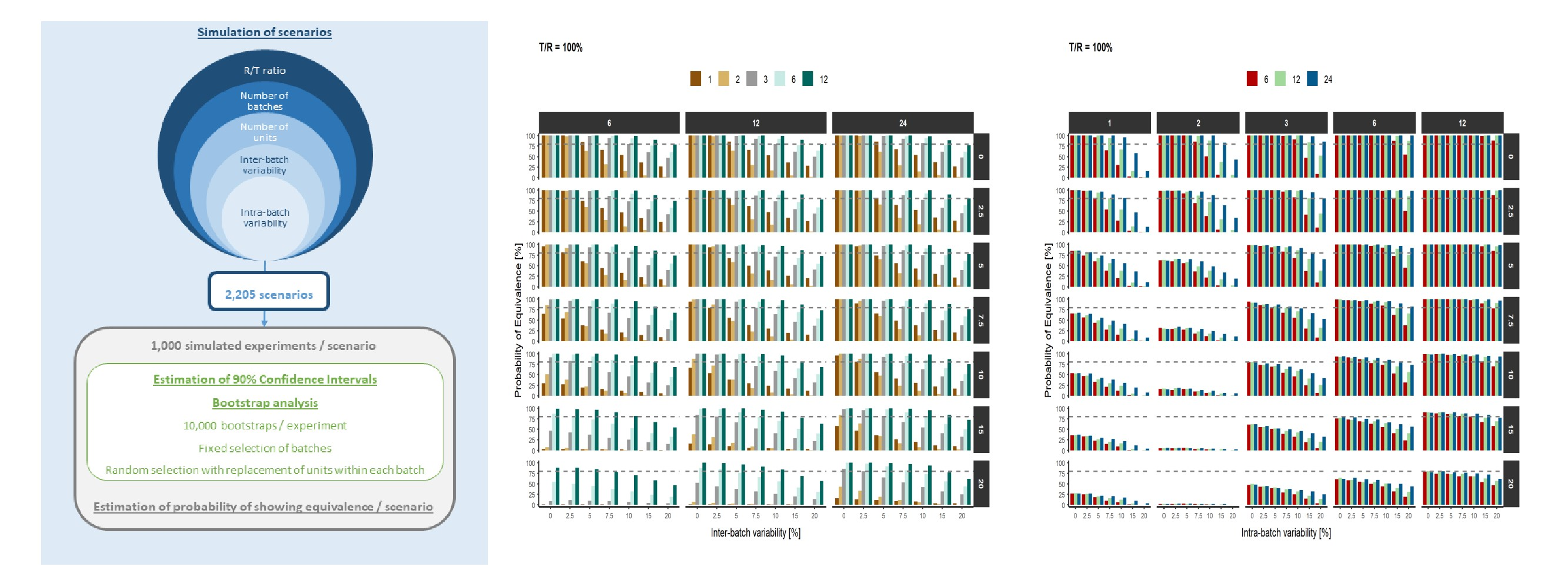
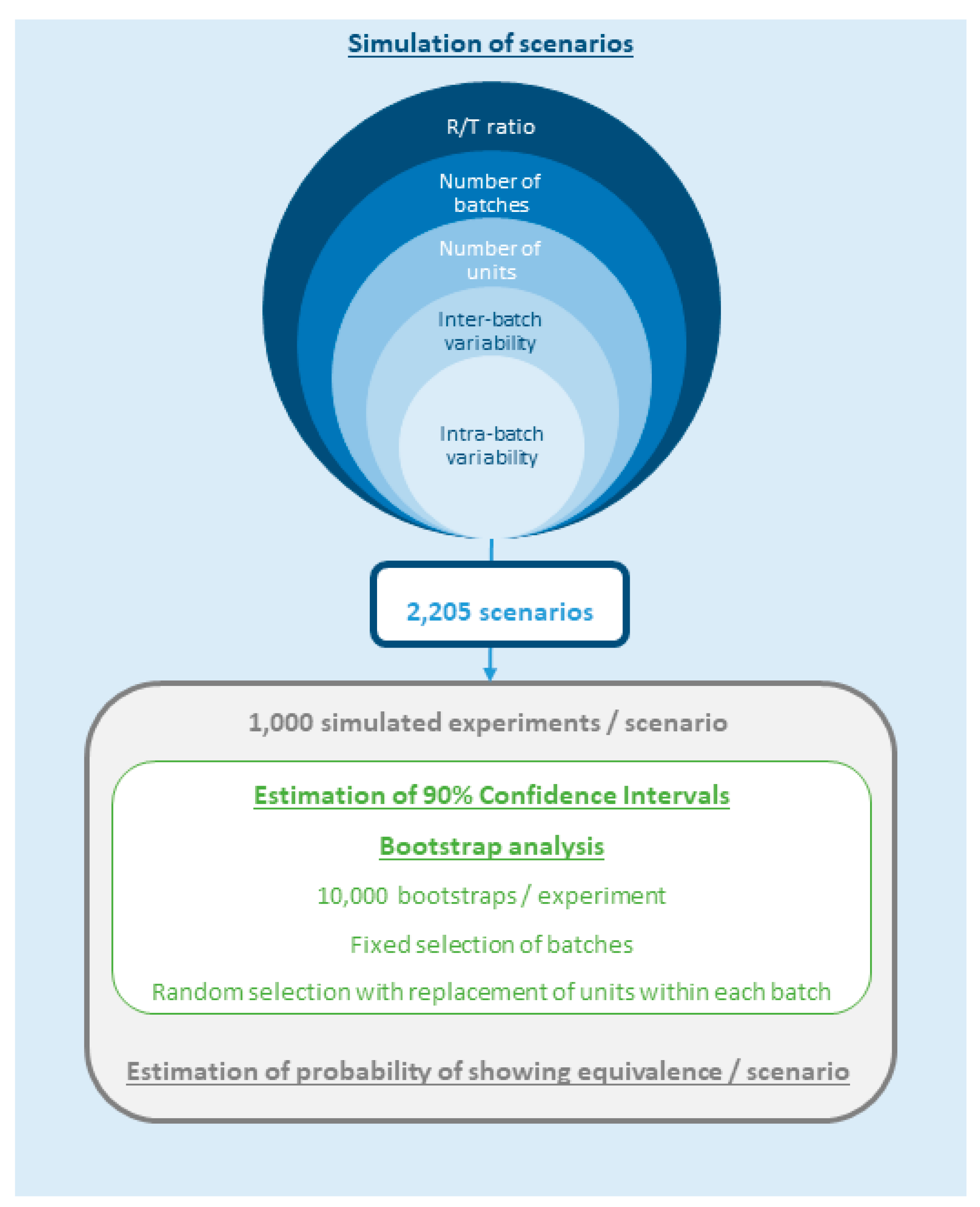
2.1. Study Design
- T/R: Test/Reference ratio (%): 100 (no difference), 97.5 (2.5% difference) and 95 (5% difference)
- NB: Number of batches per product: 1, 2, 3, 6 and 12
- NU: Number of units/replicates per batch: 6, 12 and 24
- IBV: Inter-batch variability (%): 0, 2.5, 5, 7.5, 10, 15 and 20
- ABV: Intra-batch variability (%): 0, 2.5, 5, 7.5, 10, 15 and 20
2.2. Data Simulation
2.3. Bootstrap Analysis
2.4. Software
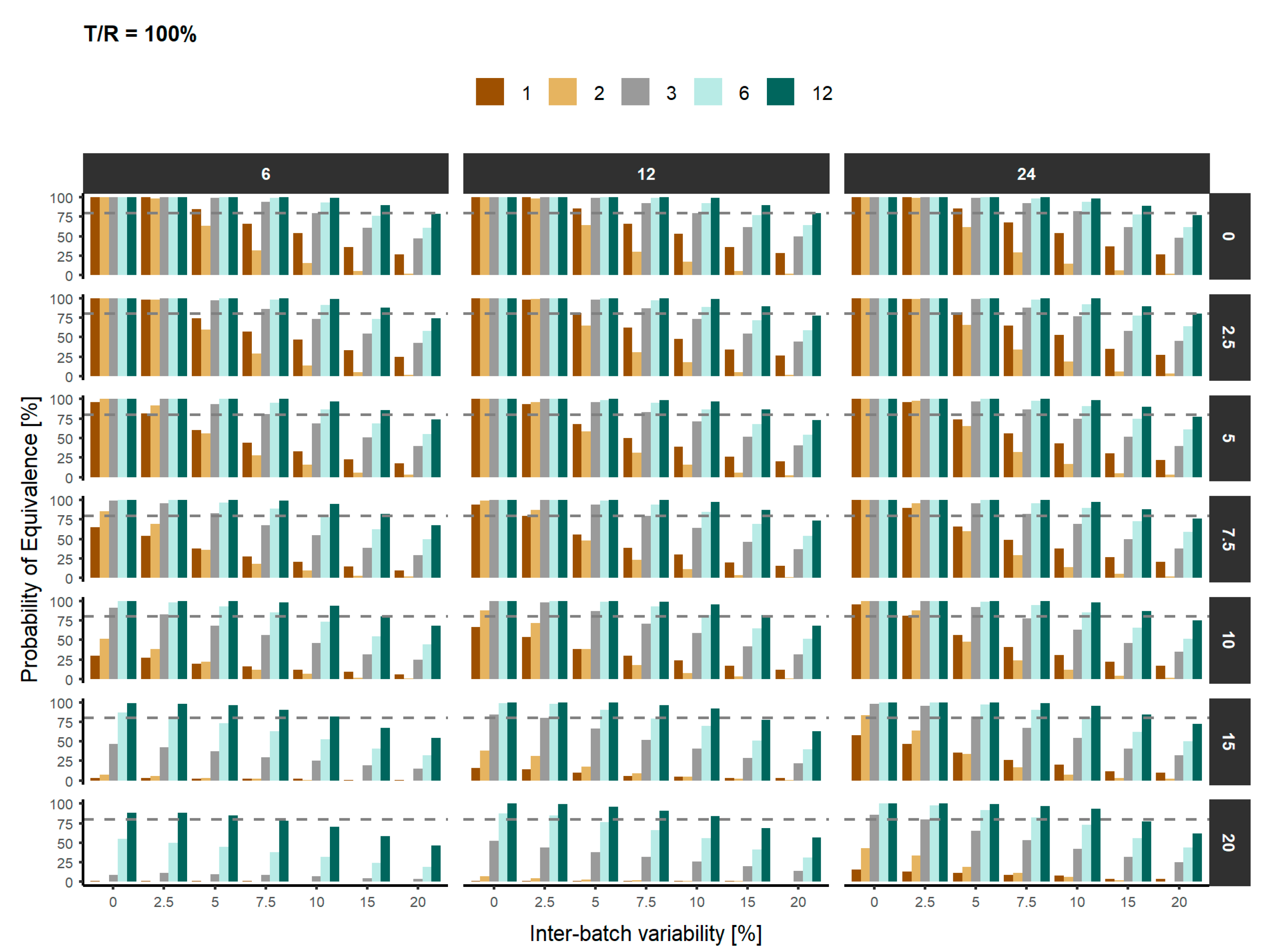
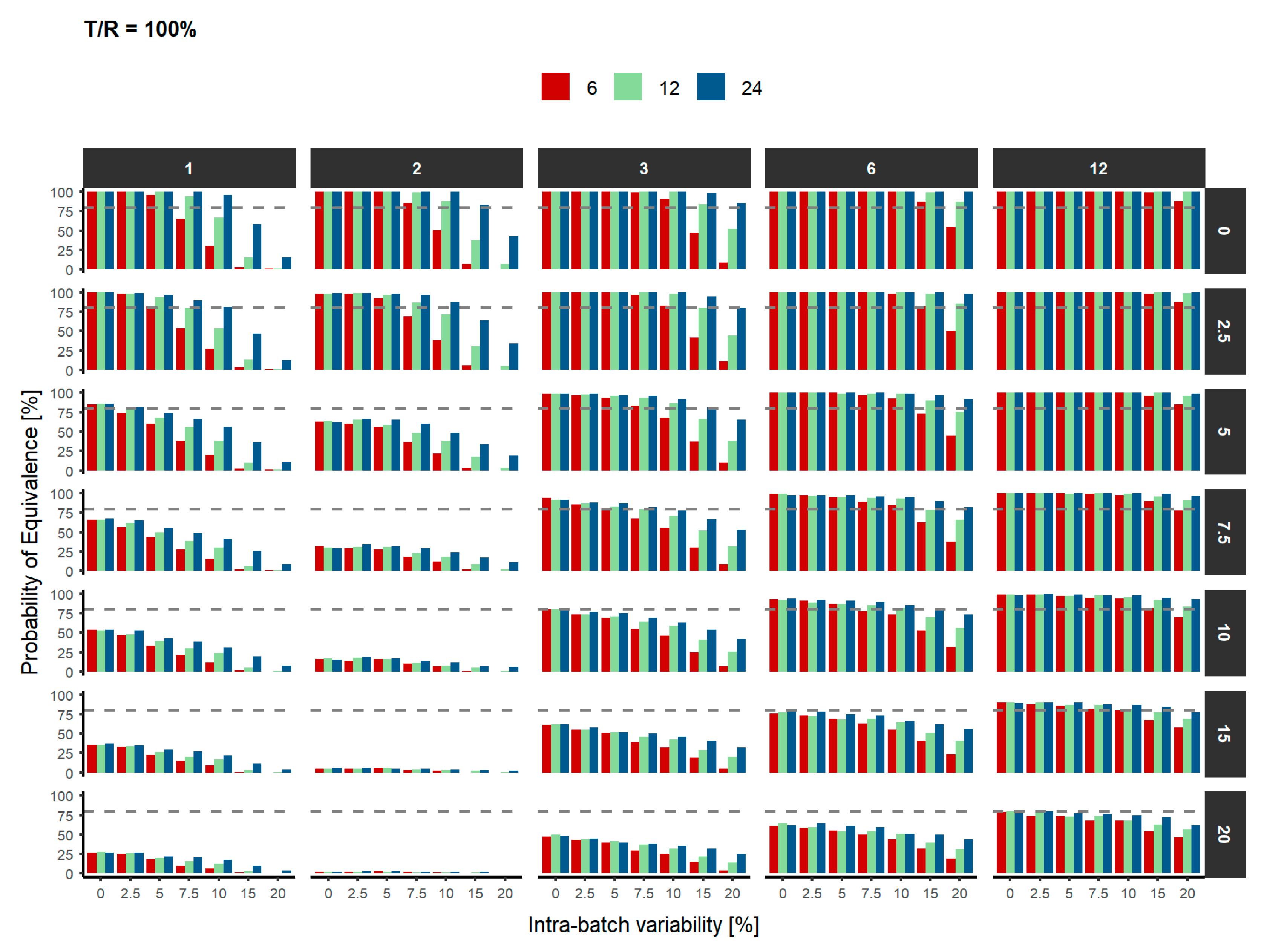
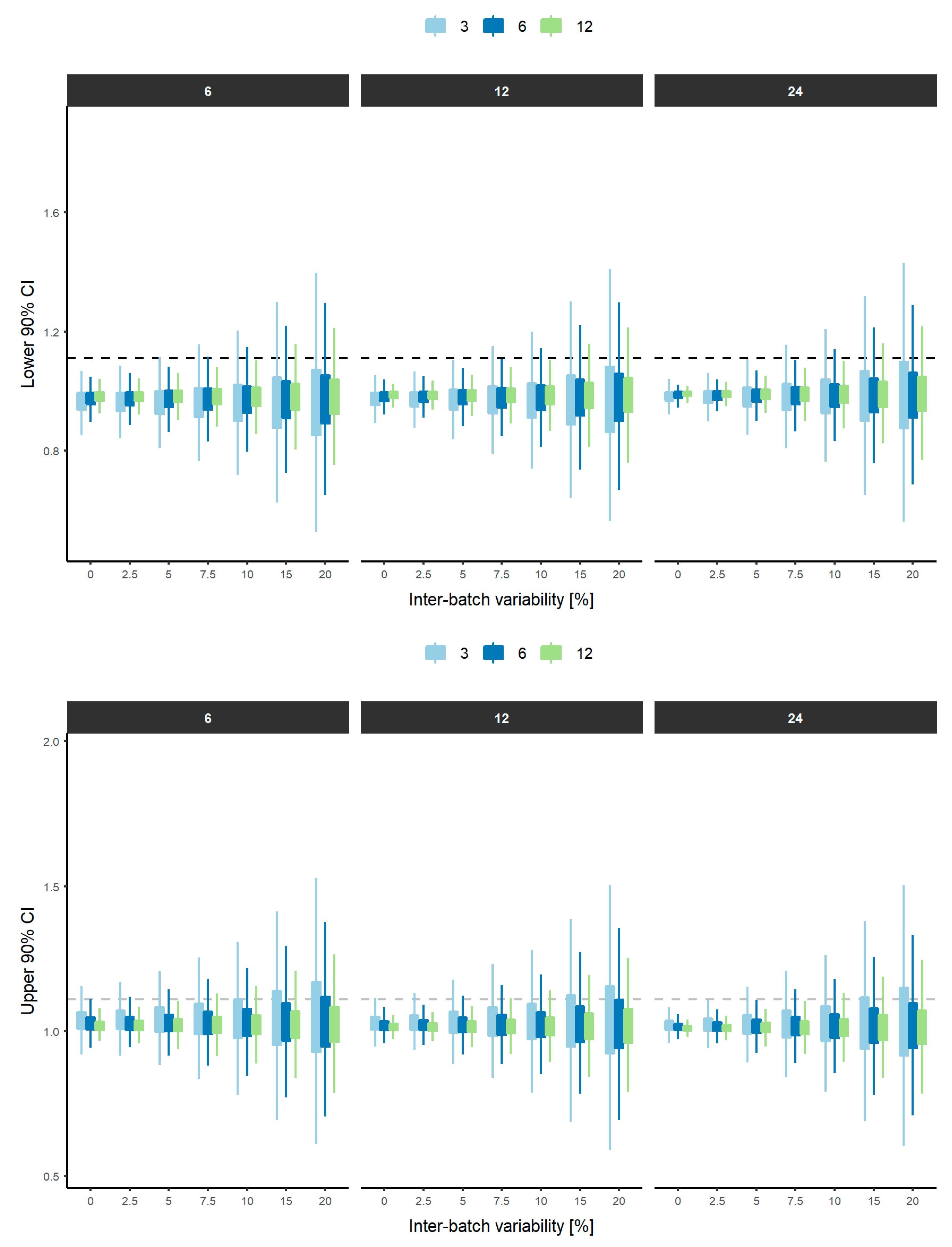
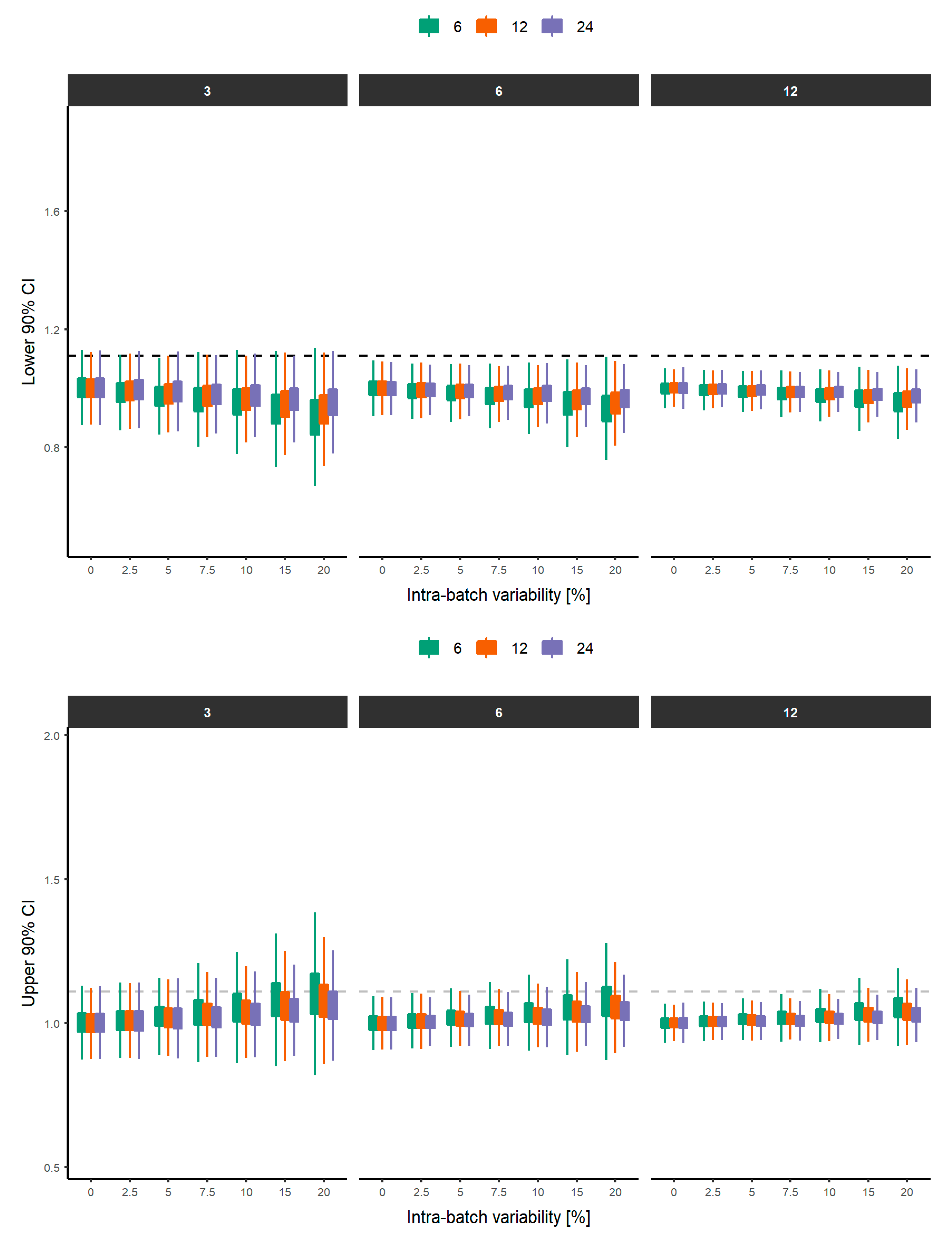
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Committee for Medicinal Products for Human Use (CHMP). Draft Guideline on Quality and Equivalence of Topical Product. CHMP/QWP/708282/2018. Available online: https://www.ema.europa.eu/en/quality-equivalence-topical-products (accessed on 25 September 2020).
- Food and Drug Administration. Draft Guidance on Acyclovir. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/psg/PSG_018604.pdf (accessed on 28 October 2020).
- Food and Drug Administration. Draft Guidance on Dapsone. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/psg/PSG_207154.pdf (accessed on 28 October 2020).
- Food and Drug Administration. Draft Guidance on Silver Sulfadiazine. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/psg/Silver%20sulfadiazine_topical%20cream_NDA%20017381_RC05-17.pdf (accessed on 28 October 2020).
- Food and Drug Administration. Draft Guidance on Benzyl alcohol. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/psg/Benzyl_alcohol_lot_022129_RC12-14.pdf (accessed on 28 October 2020).
- Food and Drug Administration. Draft Guidance on Ivermectin. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/psg/Ivermectin%20Topical%20Cream%201%20NDA%20206255%20PSG%20Page%20RV%20May%202019.pdf (accessed on 28 October 2020).
- Food and Drug Administration. Draft Guidance on Propofol. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/psg/Propofol_injectable%20injection_RLD%2019627_RC06-16.pdf (accessed on 28 October 2020).
- Food and Drug Administration. Draft Guidance on Budesonide. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/psg/PSG_020746.pdf (accessed on 28 October 2020).
- Committee for Medicinal Products for Human Use (CHMP); EMA. Guideline on Equivalence Studies for the Demonstration of Therapeutic Equivalence for Locally Applied, Locally Acting Products in the Gastrointestinal Tract. CPMP/EWP/239/95 Rev. 1, Corr.1. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-equivalence-studies-demonstration-therapeutic-equivalence-locally-applied-locally-acting_en.pdf (accessed on 28 October 2020).
- Committee for Medicinal Products for Human Use (CHMP). Guideline on the Requirements for Clinical Documentation for Orally Inhaled Products (OIP) Including the Requirements for Demonstration of Therapeutic Equivalence between Two Inhaled Products for Use in the Treatment of Asthma and Chronic Obstructive Pulmonary Disease (COPD) in Adults and for Use in the Treatment of Asthma in Children and Adolescents. CPMP/EWP/4151/00 Rev. 1. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-requirements-clinical-documentation-orally-inhaled-products-oip-including-requirements_en.pdf (accessed on 28 October 2020).
- Hertzog, M.A. Considerations in Determining Sample Size for Pilot Studies. Res. Nurs. Health 2008, 31, 180–191. [Google Scholar] [CrossRef] [PubMed]
- Browne, R.H. On the Use of a Pilot Sample for Sample Size Determination. Stat. Med. 1995, 14, 1933–1940. [Google Scholar] [CrossRef] [PubMed]
- Chang, R.-K.; Raw, A.; Lionberger, R.; Yu, L. Generic Development of Topical Dermatologic Products, Part II: Quality by Design for Topical Semisolid Products. AAPS J. 2013, 15, 674–683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mangas-Sanjuán, V.; Pleguezuelos-Villa, M.; Merino-Sanjuán, M.; Hernández, M.J.; Nácher, A.; García-Arieta, A.; Peris, D.; Hidalgo, I.; Soler, L.; Sallan, M.; et al. Assessment of the Inter-Batch Variability of Microstructure Parameters in Topical Semisolids and Impact on the Demonstration of Equivalence. Pharmaceutics 2019, 11, 503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ocaña, J.; Monleón-Getino, T.; Merino, V.; Peris, D.; Soler, L. Statistical Methods for Quality Equivalence of Topical Products. 0.5 Mg/g Betamethasone Ointment as a Case-Study. Pharmaceutics 2020, 12, 318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kryscio, D.R.; Sathe, P.M.; Lionberger, R.; Yu, L.; Bell, M.A.; Jay, M.; Hilt, J.Z. Spreadability Measurements to Assess Structural Equivalence (Q3) of Topical Formulations--a Technical Note. AAPS PharmSciTech 2008, 9, 84–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simões, A.; Miranda, M.; Cardoso, C.; Veiga, F.; Vitorino, C. Rheology by Design: A Regulatory Tutorial for Analytical Method Validation. Pharmaceutics 2020, 12, 820. [Google Scholar] [CrossRef] [PubMed]
- Miranda, M.; Cova, T.; Augusto, C.; Pais, A.A.C.C.; Cardoso, C.; Vitorino, C. Diving into Batch-to-Batch Variability of Topical Products-a Regulatory Bottleneck. Pharm. Res. 2020, 37, 218. [Google Scholar] [CrossRef] [PubMed]
- Pleguezuelos-Villa, M.; Merino-Sanjuán, M.; Hernández, M.J.; Nácher, A.; Peris, D.; Hidalgo, I.; Soler, L.; Sallan, M.; Merino, V. Relationship between Rheological Properties, in Vitro Release and in Vivo Equivalency of Topical Formulations of Diclofenac. Int. J. Pharm. 2019, 572, 118755. [Google Scholar] [CrossRef] [PubMed]
- Committee for Medicinal Products for Human Use (CHMP). Guideline on the Investigation of Bioequivalence. CPMP/EWP/QWP/1401/98 Rev. 1/ Corr **. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-investigation-bioequivalence-rev1_en.pdf (accessed on 28 October 2020).
- Stefan, W. Chapter 1. Introduction. In Testing Statistical Hypothesis of Equivalence; Chapman & Hall/CRC: Boca Raton, FL, USA, 2003; pp. 12–24. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| T/R = 100% | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Number of Units | ||||||||||||||||||||||||
| 6 | 12 | 24 | ||||||||||||||||||||||
| Intra-Batch Variability (%) | ||||||||||||||||||||||||
| 0 | 2.5 | 5 | 7.5 | 10 | 15 | 20 | 0 | 2.5 | 5 | 7.5 | 10 | 15 | 20 | 0 | 2.5 | 5 | 7.5 | 10 | 15 | 20 | ||||
| Number of batches | 1 | Inter-batch variability (%) | 0 | 100 | 100 | 96 | 65 | 30 | 3 | 1 | 100 | 100 | 100 | 94 | 67 | 16 | 1 | 100 | 100 | 100 | 100 | 96 | 58 | 16 |
| 2.5 | 100 | 98 | 82 | 54 | 27 | 3 | 1 | 100 | 98 | 94 | 80 | 54 | 14 | 1 | 100 | 99 | 96 | 90 | 81 | 47 | 13 | |||
| 5 | 85 | 74 | 60 | 38 | 20 | 2 | 1 | 86 | 81 | 68 | 56 | 38 | 10 | 1 | 86 | 82 | 74 | 66 | 56 | 36 | 11 | |||
| 7.5 | 66 | 57 | 44 | 28 | 16 | 2 | 1 | 66 | 62 | 50 | 39 | 30 | 6 | 1 | 68 | 65 | 56 | 49 | 41 | 26 | 9 | |||
| 10 | 54 | 47 | 33 | 21 | 12 | 2 | 0 | 53 | 48 | 39 | 30 | 24 | 5 | 1 | 54 | 53 | 43 | 38 | 31 | 20 | 8 | |||
| 15 | 36 | 33 | 23 | 15 | 9 | 1 | 0 | 36 | 34 | 26 | 20 | 17 | 3 | 1 | 37 | 35 | 30 | 27 | 22 | 12 | 4 | |||
| 20 | 27 | 25 | 18 | 10 | 6 | 1 | 0 | 28 | 26 | 20 | 16 | 12 | 3 | 0 | 27 | 27 | 22 | 21 | 17 | 10 | 4 | |||
| 2 | 0 | 100 | 100 | 100 | 86 | 51 | 7 | 0 | 100 | 100 | 100 | 99 | 88 | 38 | 7 | 100 | 100 | 100 | 100 | 100 | 83 | 43 | ||
| 2.5 | 98 | 98 | 92 | 69 | 38 | 6 | 0 | 98 | 99 | 96 | 87 | 72 | 31 | 5 | 99 | 99 | 98 | 96 | 88 | 64 | 34 | |||
| 5 | 63 | 60 | 56 | 36 | 22 | 3 | 0 | 64 | 65 | 59 | 48 | 38 | 18 | 3 | 62 | 66 | 65 | 60 | 48 | 34 | 19 | |||
| 7.5 | 32 | 29 | 28 | 18 | 12 | 2 | 0 | 30 | 31 | 31 | 23 | 18 | 9 | 2 | 29 | 34 | 32 | 29 | 24 | 17 | 11 | |||
| 10 | 16 | 14 | 16 | 10 | 7 | 1 | 0 | 17 | 18 | 16 | 11 | 8 | 5 | 1 | 15 | 19 | 17 | 14 | 12 | 7 | 6 | |||
| 15 | 5 | 5 | 6 | 3 | 2 | 0 | 0 | 5 | 5 | 6 | 4 | 3 | 2 | 1 | 6 | 6 | 5 | 5 | 4 | 3 | 2 | |||
| 20 | 2 | 2 | 3 | 2 | 1 | 0 | 0 | 2 | 2 | 2 | 1 | 1 | 1 | 0 | 2 | 3 | 3 | 2 | 2 | 2 | 0 | |||
| 3 | 0 | 100 | 100 | 100 | 99 | 91 | 47 | 9 | 100 | 100 | 100 | 100 | 100 | 84 | 52 | 100 | 100 | 100 | 100 | 100 | 98 | 86 | ||
| 2.5 | 100 | 100 | 100 | 96 | 83 | 42 | 11 | 100 | 100 | 100 | 100 | 98 | 80 | 44 | 100 | 100 | 100 | 100 | 100 | 95 | 80 | |||
| 5 | 99 | 97 | 94 | 83 | 68 | 37 | 10 | 99 | 98 | 96 | 94 | 87 | 66 | 38 | 99 | 99 | 97 | 96 | 92 | 82 | 65 | |||
| 7.5 | 94 | 86 | 81 | 68 | 56 | 30 | 9 | 92 | 87 | 83 | 80 | 71 | 52 | 32 | 92 | 88 | 87 | 82 | 78 | 67 | 53 | |||
| 10 | 80 | 73 | 69 | 55 | 46 | 25 | 7 | 80 | 73 | 71 | 64 | 59 | 41 | 26 | 82 | 77 | 75 | 69 | 63 | 54 | 42 | |||
| 15 | 61 | 55 | 51 | 39 | 32 | 19 | 5 | 62 | 55 | 52 | 46 | 42 | 29 | 20 | 62 | 58 | 52 | 50 | 46 | 41 | 32 | |||
| 20 | 47 | 43 | 40 | 29 | 25 | 15 | 4 | 50 | 44 | 41 | 37 | 32 | 22 | 14 | 48 | 45 | 40 | 38 | 35 | 32 | 25 | |||
| 6 | 0 | 100 | 100 | 100 | 100 | 100 | 87 | 55 | 100 | 100 | 100 | 100 | 100 | 99 | 87 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | ||
| 2.5 | 100 | 100 | 100 | 100 | 98 | 82 | 50 | 100 | 100 | 100 | 100 | 100 | 98 | 85 | 100 | 100 | 100 | 100 | 100 | 100 | 98 | |||
| 5 | 100 | 100 | 100 | 97 | 93 | 73 | 45 | 100 | 100 | 99 | 99 | 99 | 90 | 76 | 100 | 100 | 100 | 100 | 99 | 97 | 92 | |||
| 7.5 | 99 | 98 | 95 | 89 | 85 | 63 | 38 | 99 | 97 | 95 | 94 | 93 | 79 | 66 | 98 | 98 | 98 | 96 | 95 | 90 | 82 | |||
| 10 | 93 | 91 | 87 | 78 | 73 | 53 | 32 | 92 | 89 | 87 | 85 | 82 | 70 | 56 | 94 | 92 | 91 | 90 | 85 | 80 | 73 | |||
| 15 | 76 | 73 | 69 | 63 | 55 | 41 | 24 | 77 | 72 | 68 | 69 | 65 | 51 | 41 | 78 | 78 | 75 | 73 | 66 | 62 | 56 | |||
| 20 | 61 | 58 | 55 | 50 | 44 | 32 | 19 | 64 | 59 | 54 | 54 | 51 | 40 | 31 | 62 | 64 | 61 | 59 | 51 | 50 | 44 | |||
| 12 | 0 | 100 | 100 | 100 | 100 | 100 | 99 | 88 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | ||
| 2.5 | 100 | 100 | 100 | 100 | 100 | 98 | 88 | 100 | 100 | 100 | 100 | 100 | 100 | 99 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | |||
| 5 | 100 | 100 | 100 | 100 | 100 | 96 | 85 | 100 | 100 | 100 | 100 | 100 | 100 | 96 | 100 | 100 | 100 | 100 | 100 | 100 | 99 | |||
| 7.5 | 100 | 100 | 100 | 99 | 98 | 90 | 78 | 100 | 100 | 99 | 100 | 99 | 96 | 91 | 100 | 100 | 100 | 100 | 100 | 99 | 97 | |||
| 10 | 99 | 99 | 97 | 95 | 94 | 82 | 70 | 99 | 99 | 97 | 98 | 96 | 92 | 84 | 98 | 100 | 99 | 98 | 98 | 95 | 93 | |||
| 15 | 90 | 88 | 86 | 82 | 80 | 67 | 58 | 90 | 90 | 87 | 87 | 82 | 77 | 69 | 89 | 90 | 90 | 88 | 87 | 84 | 77 | |||
| 20 | 79 | 74 | 74 | 68 | 68 | 54 | 46 | 80 | 78 | 73 | 74 | 68 | 63 | 57 | 77 | 80 | 77 | 76 | 75 | 72 | 62 | |||
| T/R = 97.5% | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Number of Units | ||||||||||||||||||||||||
| 6 | 12 | 24 | ||||||||||||||||||||||
| Intra-Batch Variability (%) | ||||||||||||||||||||||||
| 0 | 2.5 | 5 | 7.5 | 10 | 15 | 20 | 0 | 2.5 | 5 | 7.5 | 10 | 15 | 20 | 0 | 2.5 | 5 | 7.5 | 10 | 15 | 20 | ||||
| Number of batches | 1 | Inter-batch variability (%) | 0 | 100 | 100 | 89 | 59 | 26 | 3 | 1 | 100 | 100 | 99 | 83 | 60 | 17 | 0 | 100 | 100 | 100 | 98 | 88 | 52 | 16 |
| 2.5 | 98 | 92 | 78 | 48 | 22 | 3 | 1 | 99 | 95 | 87 | 74 | 52 | 14 | 0 | 99 | 97 | 94 | 86 | 75 | 45 | 13 | |||
| 5 | 83 | 72 | 58 | 37 | 19 | 2 | 1 | 83 | 77 | 63 | 52 | 39 | 10 | 0 | 82 | 80 | 71 | 64 | 53 | 34 | 10 | |||
| 7.5 | 66 | 57 | 43 | 27 | 15 | 2 | 1 | 65 | 60 | 49 | 38 | 28 | 7 | 1 | 66 | 64 | 54 | 49 | 39 | 26 | 8 | |||
| 10 | 52 | 45 | 32 | 22 | 12 | 1 | 0 | 52 | 49 | 37 | 30 | 23 | 6 | 1 | 54 | 50 | 42 | 38 | 32 | 21 | 6 | |||
| 15 | 36 | 32 | 22 | 15 | 9 | 1 | 0 | 36 | 34 | 26 | 20 | 17 | 4 | 1 | 36 | 36 | 29 | 27 | 22 | 14 | 6 | |||
| 20 | 27 | 24 | 16 | 10 | 7 | 0 | 0 | 28 | 26 | 18 | 15 | 14 | 3 | 1 | 27 | 28 | 23 | 21 | 17 | 10 | 3 | |||
| 2 | 0 | 100 | 100 | 96 | 72 | 46 | 7 | 0 | 100 | 100 | 100 | 95 | 77 | 32 | 6 | 100 | 100 | 100 | 100 | 97 | 72 | 38 | ||
| 2.5 | 97 | 93 | 84 | 59 | 37 | 6 | 0 | 96 | 97 | 92 | 81 | 64 | 26 | 4 | 96 | 97 | 95 | 90 | 81 | 58 | 30 | |||
| 5 | 60 | 56 | 50 | 33 | 20 | 4 | 0 | 57 | 60 | 56 | 44 | 34 | 16 | 3 | 59 | 62 | 60 | 56 | 47 | 32 | 19 | |||
| 7.5 | 31 | 27 | 27 | 16 | 10 | 2 | 0 | 30 | 32 | 29 | 23 | 16 | 9 | 2 | 28 | 33 | 30 | 27 | 22 | 15 | 9 | |||
| 10 | 15 | 15 | 15 | 9 | 7 | 1 | 0 | 16 | 16 | 16 | 13 | 8 | 5 | 2 | 15 | 18 | 15 | 13 | 12 | 8 | 6 | |||
| 15 | 5 | 5 | 6 | 3 | 2 | 0 | 0 | 6 | 5 | 7 | 4 | 3 | 2 | 0 | 5 | 7 | 6 | 4 | 4 | 3 | 2 | |||
| 20 | 2 | 2 | 3 | 2 | 2 | 0 | 0 | 2 | 2 | 3 | 1 | 1 | 1 | 0 | 3 | 3 | 2 | 1 | 1 | 2 | 1 | |||
| 3 | 0 | 100 | 100 | 100 | 96 | 82 | 43 | 9 | 100 | 100 | 100 | 100 | 97 | 75 | 44 | 100 | 100 | 100 | 100 | 100 | 94 | 76 | ||
| 2.5 | 100 | 100 | 99 | 91 | 74 | 37 | 10 | 100 | 100 | 100 | 98 | 92 | 70 | 41 | 100 | 100 | 100 | 99 | 97 | 89 | 69 | |||
| 5 | 98 | 94 | 91 | 78 | 64 | 33 | 8 | 98 | 95 | 92 | 88 | 81 | 58 | 35 | 98 | 96 | 94 | 92 | 87 | 77 | 61 | |||
| 7.5 | 88 | 83 | 79 | 65 | 54 | 29 | 8 | 89 | 84 | 80 | 74 | 68 | 48 | 30 | 90 | 86 | 84 | 79 | 73 | 63 | 49 | |||
| 10 | 78 | 73 | 67 | 54 | 44 | 25 | 7 | 80 | 73 | 69 | 62 | 57 | 38 | 26 | 80 | 75 | 71 | 66 | 62 | 52 | 42 | |||
| 15 | 60 | 54 | 51 | 38 | 33 | 20 | 6 | 61 | 55 | 52 | 44 | 41 | 27 | 19 | 61 | 57 | 52 | 48 | 45 | 39 | 31 | |||
| 20 | 48 | 42 | 40 | 29 | 24 | 15 | 4 | 49 | 42 | 41 | 35 | 32 | 22 | 14 | 48 | 44 | 38 | 38 | 34 | 31 | 25 | |||
| 6 | 0 | 100 | 100 | 100 | 100 | 98 | 77 | 49 | 100 | 100 | 100 | 100 | 100 | 94 | 78 | 100 | 100 | 100 | 100 | 100 | 100 | 96 | ||
| 2.5 | 100 | 100 | 100 | 99 | 94 | 73 | 44 | 100 | 100 | 100 | 100 | 100 | 92 | 77 | 100 | 100 | 100 | 100 | 100 | 98 | 92 | |||
| 5 | 100 | 99 | 98 | 93 | 87 | 67 | 41 | 100 | 99 | 98 | 97 | 96 | 85 | 70 | 99 | 99 | 99 | 99 | 98 | 94 | 86 | |||
| 7.5 | 97 | 96 | 92 | 87 | 78 | 59 | 36 | 97 | 94 | 92 | 90 | 89 | 76 | 62 | 97 | 97 | 94 | 94 | 91 | 85 | 78 | |||
| 10 | 90 | 88 | 83 | 78 | 71 | 53 | 32 | 91 | 87 | 82 | 83 | 81 | 65 | 54 | 91 | 90 | 88 | 87 | 83 | 76 | 68 | |||
| 15 | 74 | 72 | 65 | 61 | 53 | 40 | 24 | 75 | 72 | 67 | 66 | 63 | 52 | 41 | 76 | 76 | 73 | 72 | 63 | 60 | 54 | |||
| 20 | 62 | 58 | 52 | 49 | 42 | 31 | 18 | 62 | 58 | 53 | 54 | 50 | 40 | 33 | 63 | 64 | 60 | 58 | 50 | 47 | 42 | |||
| 12 | 0 | 100 | 100 | 100 | 100 | 100 | 95 | 80 | 100 | 100 | 100 | 100 | 100 | 100 | 97 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | ||
| 2.5 | 100 | 100 | 100 | 100 | 100 | 93 | 77 | 100 | 100 | 100 | 100 | 100 | 100 | 96 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | |||
| 5 | 100 | 100 | 100 | 99 | 98 | 88 | 74 | 100 | 100 | 100 | 100 | 100 | 98 | 91 | 100 | 100 | 100 | 100 | 100 | 99 | 97 | |||
| 7.5 | 100 | 100 | 99 | 97 | 94 | 83 | 70 | 100 | 99 | 98 | 98 | 96 | 92 | 86 | 99 | 100 | 99 | 100 | 99 | 97 | 93 | |||
| 10 | 97 | 96 | 94 | 92 | 88 | 77 | 64 | 97 | 97 | 96 | 94 | 92 | 87 | 79 | 97 | 96 | 97 | 97 | 95 | 92 | 87 | |||
| 15 | 88 | 85 | 83 | 79 | 76 | 64 | 54 | 89 | 86 | 84 | 84 | 80 | 76 | 66 | 88 | 87 | 86 | 85 | 83 | 82 | 73 | |||
| 20 | 78 | 74 | 71 | 68 | 65 | 52 | 45 | 77 | 75 | 71 | 72 | 66 | 63 | 56 | 76 | 76 | 74 | 73 | 72 | 71 | 61 | |||
| T/R = 95% | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Number of Units | ||||||||||||||||||||||||
| 6 | 12 | 24 | ||||||||||||||||||||||
| Intra-Batch Variability (%) | ||||||||||||||||||||||||
| 0 | 2.5 | 5 | 7.5 | 10 | 15 | 20 | 0 | 2.5 | 5 | 7.5 | 10 | 15 | 20 | 0 | 2.5 | 5 | 7.5 | 10 | 15 | 20 | ||||
| Number of batches | 1 | Inter-batch variability (%) | 0 | 100 | 99 | 68 | 42 | 19 | 2 | 0 | 100 | 100 | 89 | 59 | 42 | 12 | 1 | 100 | 100 | 99 | 82 | 63 | 37 | 13 |
| 2.5 | 93 | 79 | 62 | 38 | 21 | 3 | 1 | 94 | 87 | 70 | 55 | 42 | 10 | 1 | 94 | 90 | 80 | 71 | 57 | 35 | 11 | |||
| 5 | 76 | 65 | 49 | 32 | 16 | 2 | 0 | 76 | 69 | 57 | 46 | 34 | 10 | 1 | 76 | 72 | 64 | 58 | 47 | 29 | 9 | |||
| 7.5 | 61 | 52 | 39 | 25 | 14 | 1 | 0 | 63 | 57 | 44 | 37 | 27 | 8 | 0 | 62 | 58 | 48 | 45 | 37 | 25 | 7 | |||
| 10 | 49 | 43 | 31 | 20 | 12 | 2 | 0 | 52 | 47 | 36 | 29 | 22 | 6 | 0 | 50 | 49 | 40 | 37 | 30 | 20 | 7 | |||
| 15 | 35 | 30 | 22 | 15 | 8 | 1 | 0 | 37 | 33 | 25 | 21 | 16 | 5 | 0 | 35 | 36 | 28 | 25 | 22 | 15 | 5 | |||
| 20 | 26 | 24 | 16 | 11 | 7 | 1 | 0 | 28 | 26 | 19 | 16 | 13 | 3 | 0 | 27 | 27 | 22 | 21 | 17 | 11 | 4 | |||
| 2 | 0 | 100 | 100 | 77 | 48 | 30 | 6 | 0 | 100 | 100 | 96 | 72 | 51 | 22 | 4 | 100 | 100 | 100 | 94 | 78 | 45 | 26 | ||
| 2.5 | 84 | 78 | 61 | 42 | 24 | 3 | 0 | 83 | 83 | 70 | 56 | 44 | 17 | 3 | 82 | 84 | 80 | 71 | 59 | 39 | 20 | |||
| 5 | 50 | 46 | 42 | 27 | 16 | 2 | 0 | 46 | 50 | 45 | 37 | 28 | 13 | 2 | 46 | 51 | 46 | 42 | 37 | 24 | 13 | |||
| 7.5 | 26 | 25 | 24 | 15 | 10 | 1 | 0 | 26 | 27 | 27 | 21 | 16 | 8 | 1 | 27 | 28 | 26 | 23 | 18 | 14 | 7 | |||
| 10 | 14 | 12 | 13 | 8 | 6 | 1 | 0 | 15 | 14 | 15 | 11 | 9 | 5 | 1 | 13 | 16 | 13 | 13 | 10 | 7 | 5 | |||
| 15 | 5 | 5 | 5 | 3 | 2 | 0 | 0 | 6 | 5 | 7 | 4 | 4 | 2 | 0 | 5 | 7 | 5 | 4 | 3 | 3 | 2 | |||
| 20 | 2 | 3 | 3 | 1 | 2 | 0 | 0 | 3 | 2 | 3 | 2 | 2 | 1 | 0 | 3 | 3 | 2 | 2 | 1 | 1 | 1 | |||
| 3 | 0 | 100 | 100 | 97 | 78 | 56 | 28 | 9 | 100 | 100 | 100 | 94 | 79 | 48 | 30 | 100 | 100 | 100 | 100 | 95 | 73 | 52 | ||
| 2.5 | 100 | 98 | 89 | 71 | 52 | 28 | 8 | 100 | 98 | 94 | 85 | 73 | 49 | 31 | 100 | 99 | 97 | 94 | 85 | 68 | 50 | |||
| 5 | 90 | 85 | 78 | 63 | 50 | 28 | 7 | 92 | 85 | 82 | 72 | 65 | 47 | 29 | 92 | 88 | 84 | 80 | 73 | 60 | 48 | |||
| 7.5 | 80 | 76 | 70 | 57 | 47 | 26 | 7 | 82 | 77 | 72 | 64 | 57 | 42 | 26 | 81 | 78 | 72 | 71 | 64 | 54 | 41 | |||
| 10 | 72 | 67 | 62 | 50 | 41 | 24 | 6 | 74 | 69 | 64 | 56 | 50 | 36 | 24 | 72 | 68 | 64 | 61 | 55 | 47 | 37 | |||
| 15 | 58 | 52 | 50 | 38 | 31 | 19 | 4 | 58 | 52 | 49 | 43 | 38 | 26 | 18 | 57 | 53 | 49 | 46 | 42 | 36 | 30 | |||
| 20 | 46 | 41 | 41 | 29 | 22 | 14 | 4 | 46 | 43 | 39 | 33 | 30 | 20 | 14 | 46 | 44 | 38 | 37 | 34 | 30 | 23 | |||
| 6 | 0 | 100 | 100 | 100 | 95 | 83 | 52 | 34 | 100 | 100 | 100 | 100 | 96 | 74 | 54 | 100 | 100 | 100 | 100 | 100 | 94 | 77 | ||
| 2.5 | 100 | 100 | 98 | 88 | 75 | 55 | 33 | 100 | 100 | 99 | 96 | 92 | 72 | 54 | 100 | 100 | 100 | 99 | 98 | 88 | 70 | |||
| 5 | 97 | 95 | 90 | 81 | 70 | 51 | 32 | 98 | 95 | 91 | 88 | 82 | 68 | 52 | 97 | 97 | 94 | 92 | 88 | 79 | 64 | |||
| 7.5 | 89 | 86 | 81 | 74 | 63 | 48 | 32 | 91 | 87 | 80 | 80 | 75 | 62 | 50 | 90 | 90 | 86 | 84 | 78 | 71 | 60 | |||
| 10 | 81 | 79 | 73 | 67 | 58 | 44 | 30 | 84 | 80 | 74 | 72 | 69 | 56 | 46 | 83 | 83 | 80 | 76 | 71 | 65 | 55 | |||
| 15 | 70 | 66 | 59 | 54 | 49 | 37 | 24 | 69 | 66 | 62 | 60 | 57 | 45 | 37 | 72 | 69 | 68 | 65 | 58 | 53 | 47 | |||
| 20 | 59 | 56 | 49 | 46 | 39 | 32 | 18 | 59 | 56 | 52 | 51 | 48 | 37 | 29 | 61 | 59 | 56 | 56 | 47 | 45 | 39 | |||
| 12 | 0 | 100 | 100 | 100 | 100 | 96 | 77 | 58 | 100 | 100 | 100 | 100 | 100 | 94 | 78 | 100 | 100 | 100 | 100 | 100 | 100 | 96 | ||
| 2.5 | 100 | 100 | 100 | 99 | 94 | 72 | 53 | 100 | 100 | 100 | 100 | 99 | 90 | 76 | 100 | 100 | 100 | 100 | 100 | 99 | 92 | |||
| 5 | 100 | 99 | 97 | 93 | 86 | 70 | 52 | 100 | 99 | 98 | 98 | 94 | 85 | 72 | 99 | 100 | 99 | 99 | 98 | 94 | 85 | |||
| 7.5 | 96 | 94 | 91 | 86 | 79 | 66 | 52 | 96 | 95 | 92 | 90 | 86 | 78 | 68 | 96 | 95 | 94 | 94 | 91 | 88 | 78 | |||
| 10 | 91 | 87 | 84 | 81 | 74 | 62 | 50 | 92 | 88 | 86 | 84 | 80 | 73 | 63 | 91 | 88 | 88 | 87 | 84 | 82 | 72 | |||
| 15 | 81 | 78 | 75 | 71 | 65 | 54 | 45 | 81 | 77 | 75 | 74 | 69 | 64 | 56 | 80 | 78 | 76 | 76 | 74 | 71 | 63 | |||
| 20 | 71 | 68 | 65 | 62 | 56 | 47 | 40 | 71 | 67 | 66 | 66 | 61 | 56 | 50 | 70 | 70 | 68 | 68 | 66 | 62 | 54 | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, Z.; Mangas-Sanjuán, V.; Merino-Sanjuán, M.; Merino, V.; García-Arieta, A. Influence of Inter- and Intra-Batch Variability on the Sample Size Required for Demonstration of Equivalent Microstructure of Semisolid Dosage Forms. Pharmaceutics 2020, 12, 1159. https://doi.org/10.3390/pharmaceutics12121159
Xu Z, Mangas-Sanjuán V, Merino-Sanjuán M, Merino V, García-Arieta A. Influence of Inter- and Intra-Batch Variability on the Sample Size Required for Demonstration of Equivalent Microstructure of Semisolid Dosage Forms. Pharmaceutics. 2020; 12(12):1159. https://doi.org/10.3390/pharmaceutics12121159
Chicago/Turabian StyleXu, Zhengguo, Víctor Mangas-Sanjuán, Matilde Merino-Sanjuán, Virginia Merino, and Alfredo García-Arieta. 2020. "Influence of Inter- and Intra-Batch Variability on the Sample Size Required for Demonstration of Equivalent Microstructure of Semisolid Dosage Forms" Pharmaceutics 12, no. 12: 1159. https://doi.org/10.3390/pharmaceutics12121159