Glycyrrhetinic Acid-Functionalized Mesoporous Silica Nanoparticles for the Co-Delivery of DOX/CPT-PEG for Targeting HepG2 Cells


Abstract
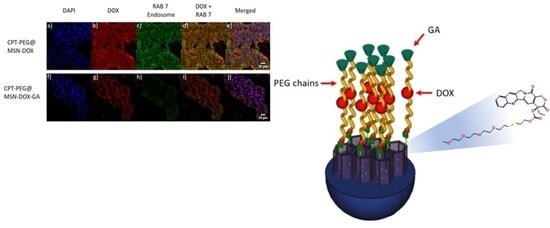
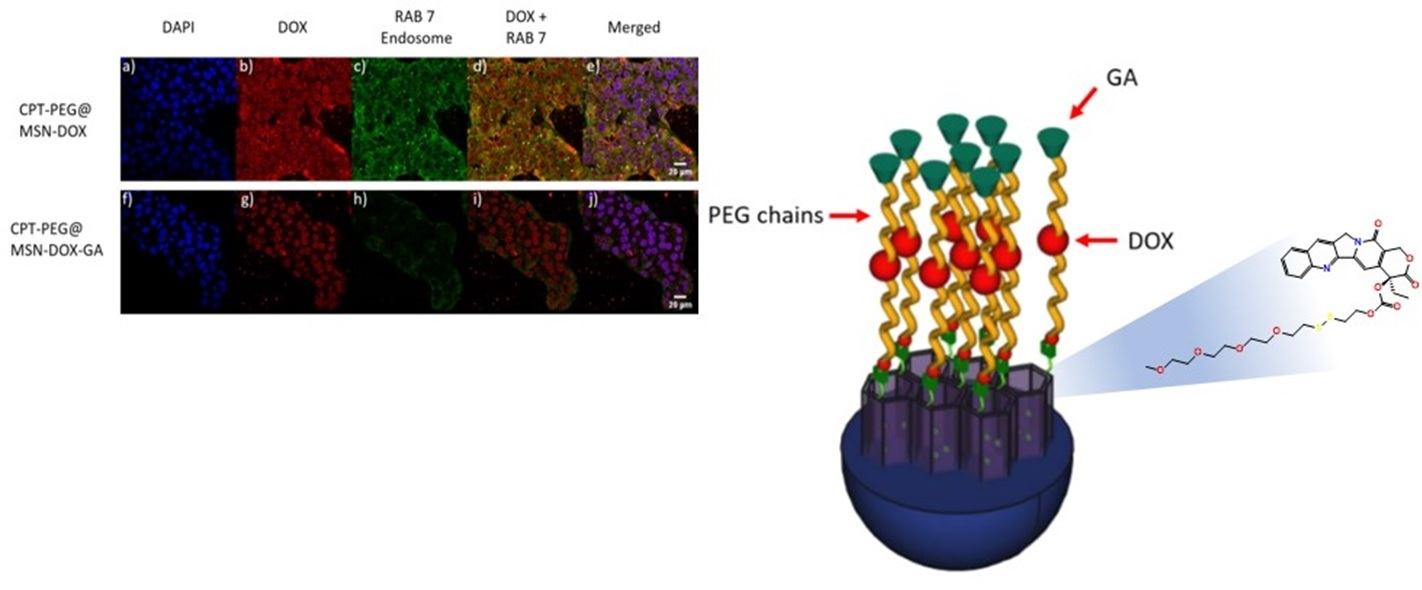
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Materials and Measurements
2.2. Instruments
2.3. Synthesis of 2-(Pyridyl-Disulfanyl)ethanol (4)
2.4. Synthesis of Camptothecin-(4-Pyridyldisulfanyl)ethyl Carbonate (2)
2.5. Synthesis of (S)-4-Ethyl-3,14-Dioxo-3,4,12,14-Tetrahydro-1H Pyrano [3′,4′:6,7] Indolizino [1,2-b]Quinolin-4-yl (2,5,8,11-Tetraoxa-14,15-Dithiaheptadecan-17-yl) Carbonate (1)
2.6. Synthesis of 4-(2-(2-(2-(2-Hydroxyethoxy) Ethoxy) Ethoxy) Ethoxy) Benzaldehyde (5)
2.7. Synthesis of 10-((3-Carboxypropanoyl)oxy)-2,4a,6a,6b,9,9,12a-Heptamethyl-13-oxo-1,2,3,4,4a,5,6,6a,6b,7,8,8a,9,10,11,12,12a,12b,13,14b-Icosahydropicene-2-Carboxylic Acid (6)
2.8. Synthesis of 10-((1-(4-Formylphenoxy)-13-oxo-3,6,9,12-Tetraoxahexadecan-16-oyl)oxy)-2,4a,6a,6b,9,9,12a-Heptamethyl-13-oxo-1,2,3,4,4a,5,6,6a,6b,7,8,8a,9,10,11,12,12a,12b,13,14b-Icosahydropicene-2-Carboxylic Acid (3)
2.9. Synthesis of MSN-(NH2) CTAB
2.10. Synthesis of MSN-(NH2)i(CHO)o
2.11. Synthesis of CPT-PEG@MSN-DOX
2.12. Conjugates CPT-PEG@MSN-DOX-OH and CPT-PEG@MSN-DOX-GA
2.13. Release Experiments of CPT-PEG@MSN-DOX-GA
2.14. In Vitro Cytotoxicity
2.15. Confocal Microscopy for Cellular Internalization
2.16. Flow Cytometry Analysis
3. Results and Discussion
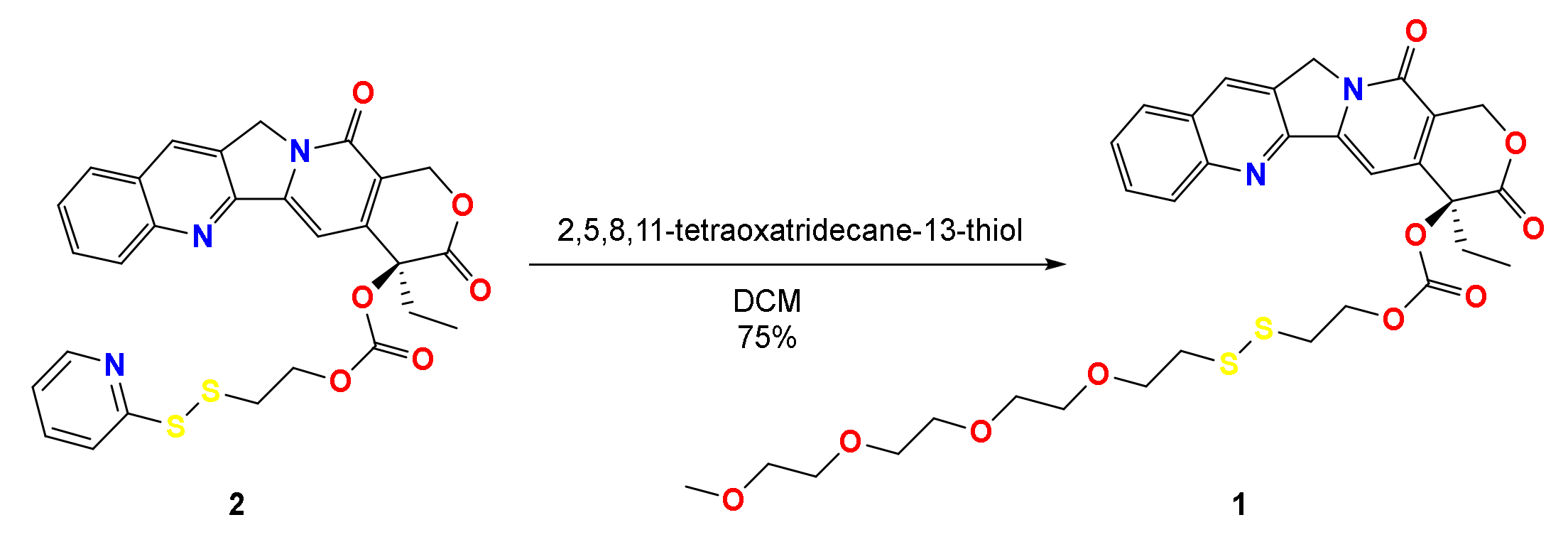
3.1. Synthesis of CPT Prodrug and Cleavage
3.2. Construction of the System CPT-PEG@MSN-DOX
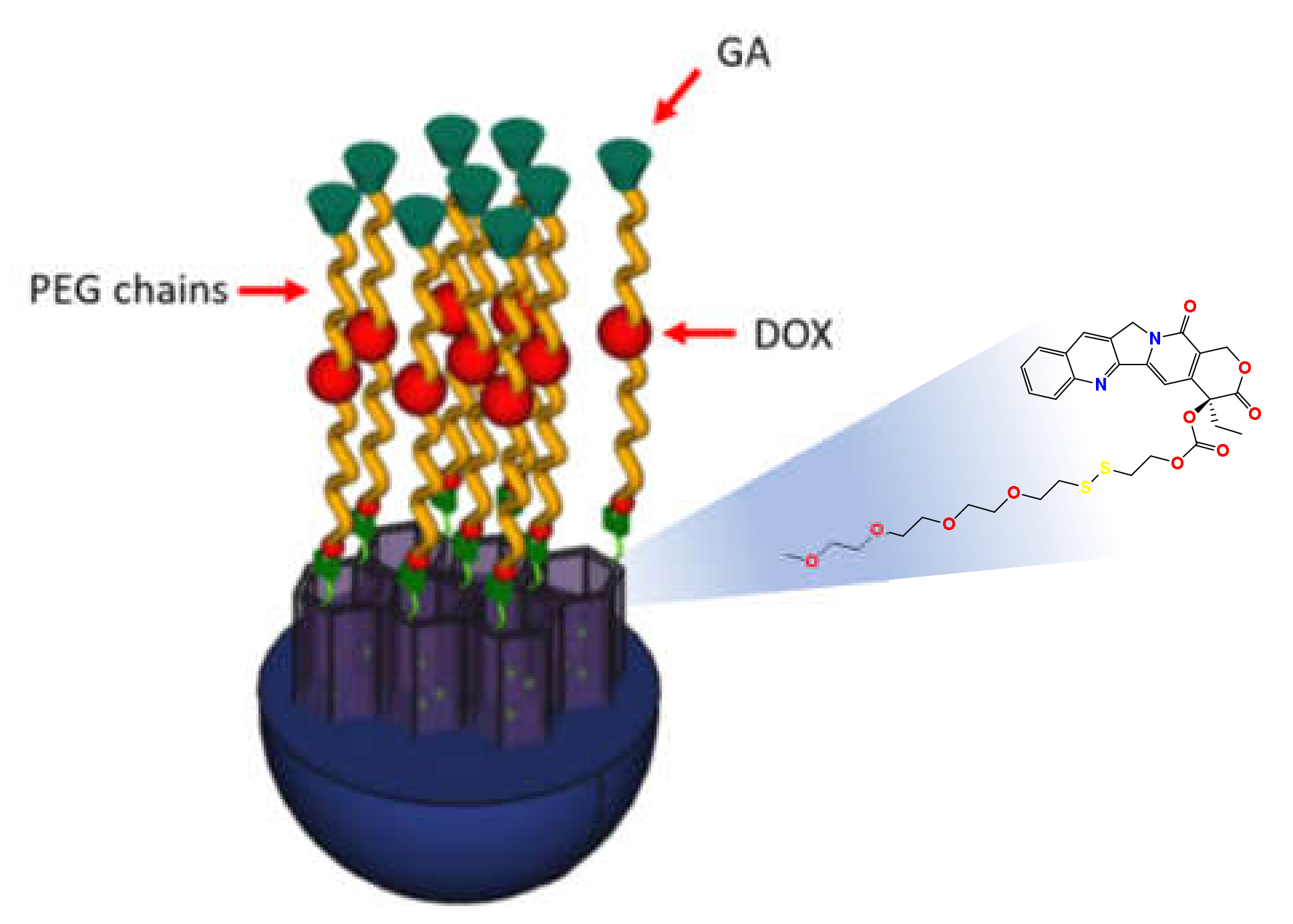
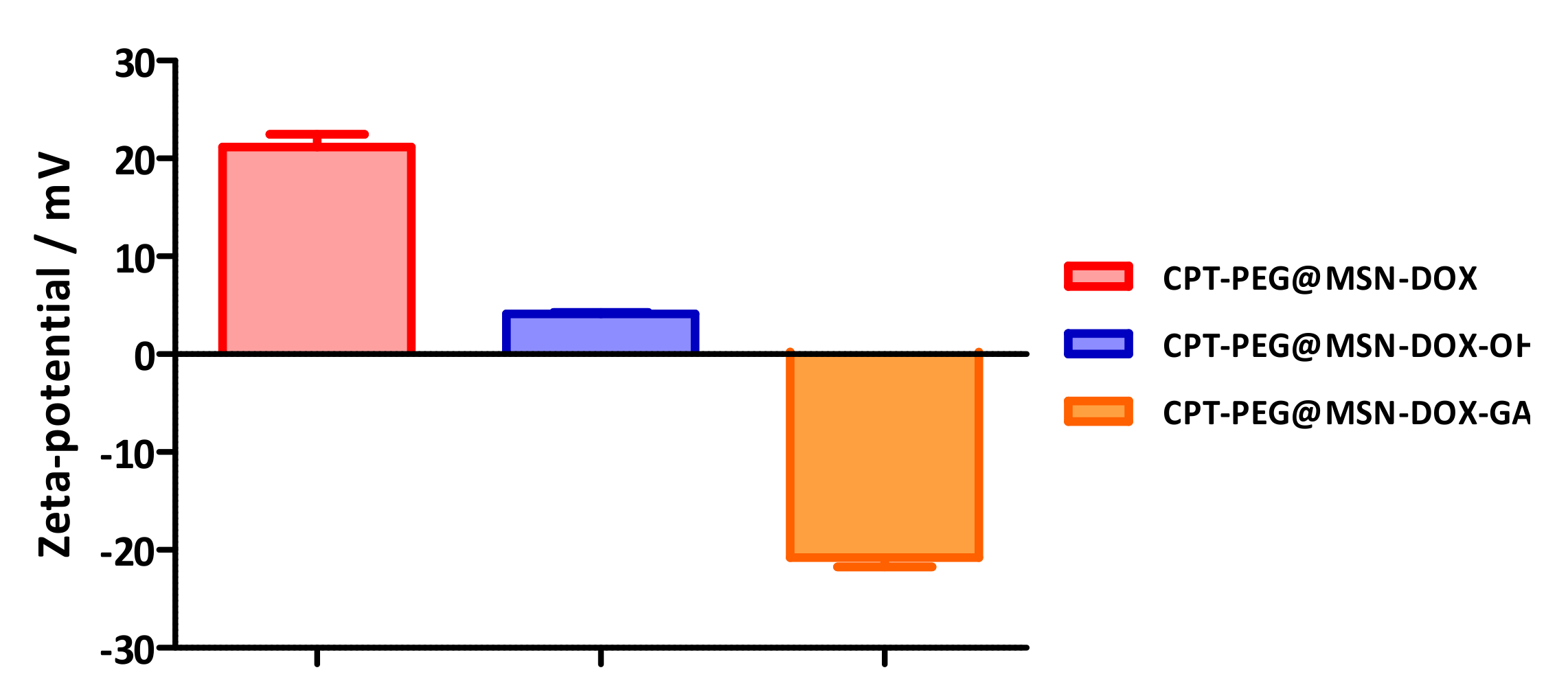
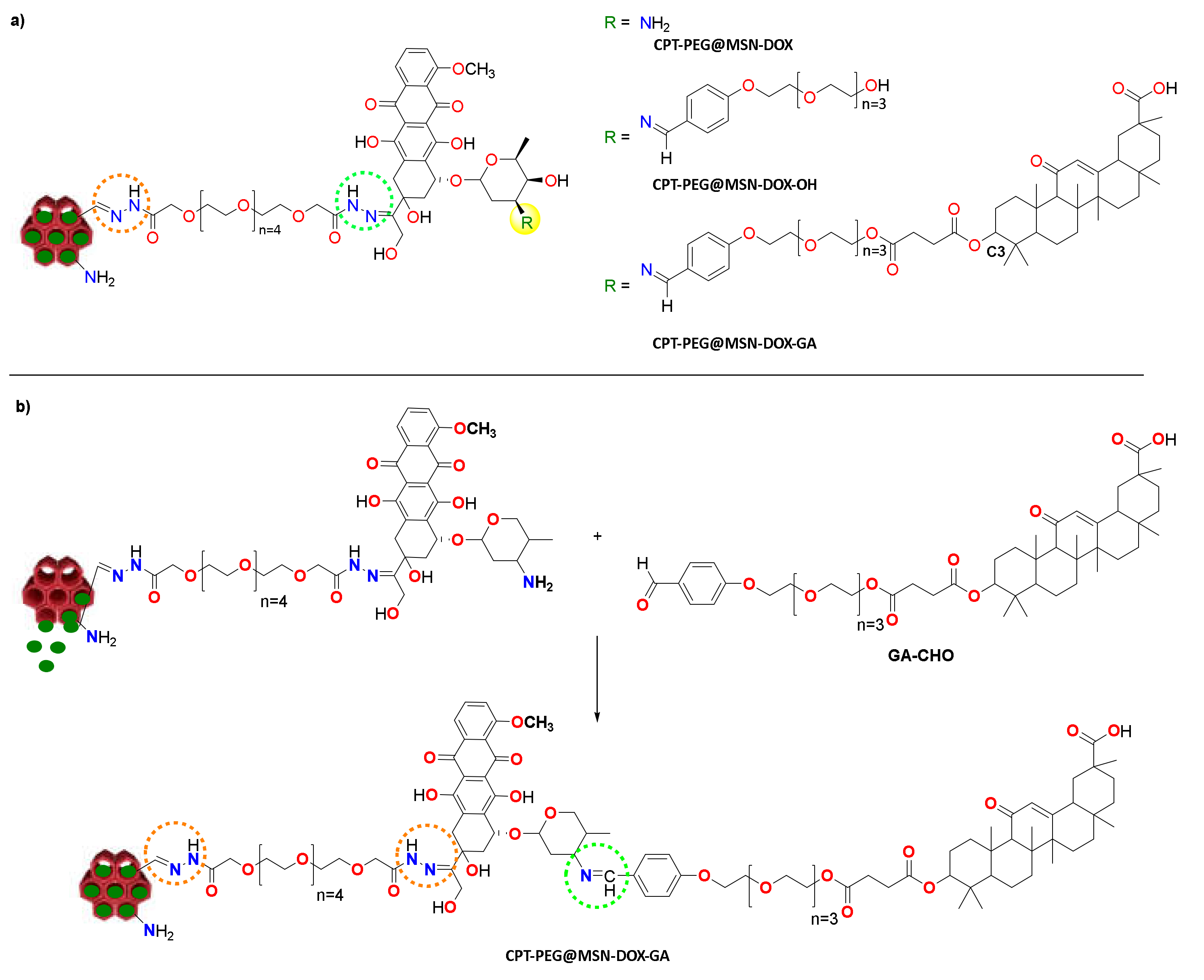
3.3. Construction of the DDS (CPT-PEG@MSN-DOX-GA)
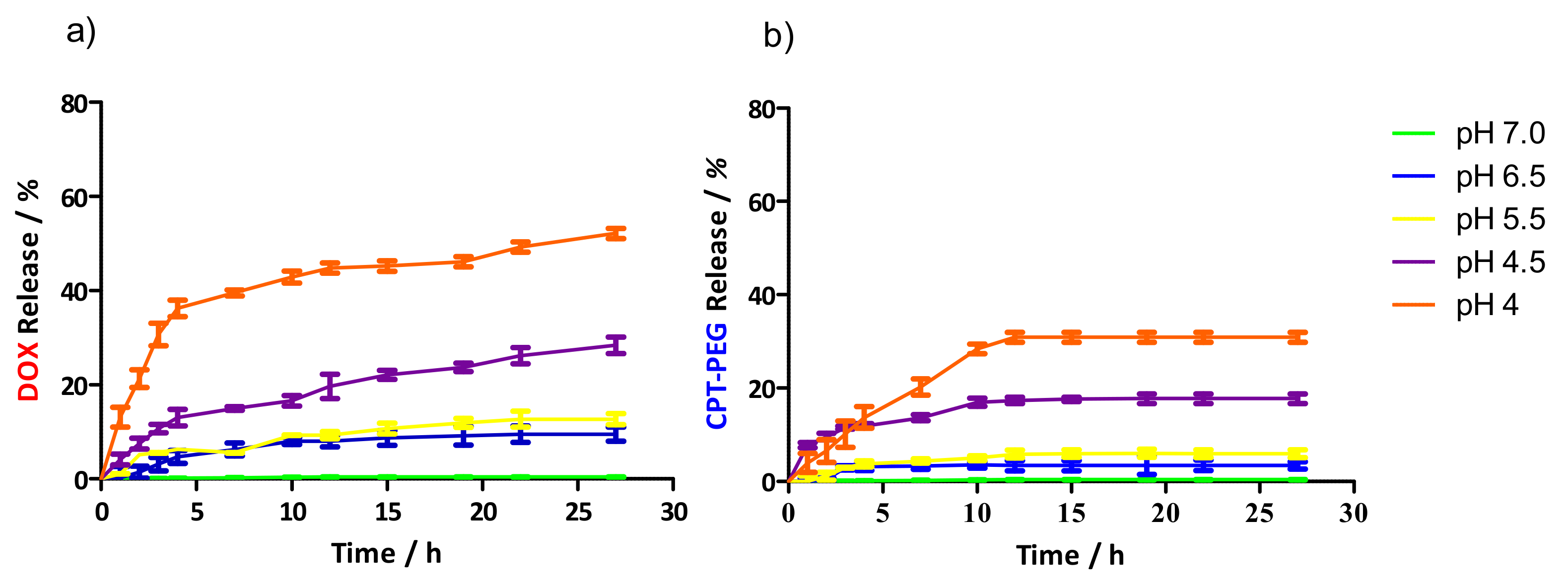
3.4. Controlled Drug Release
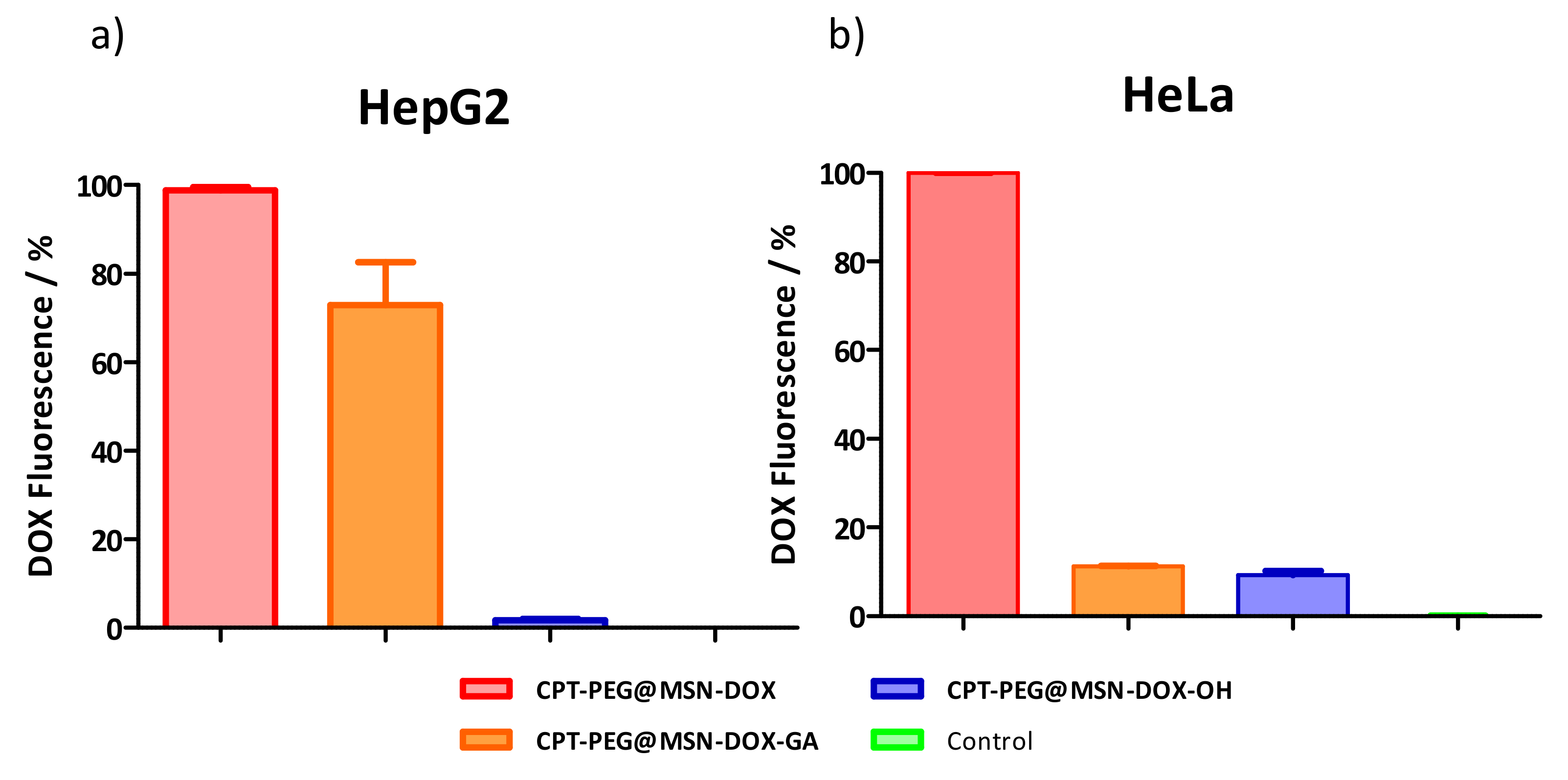
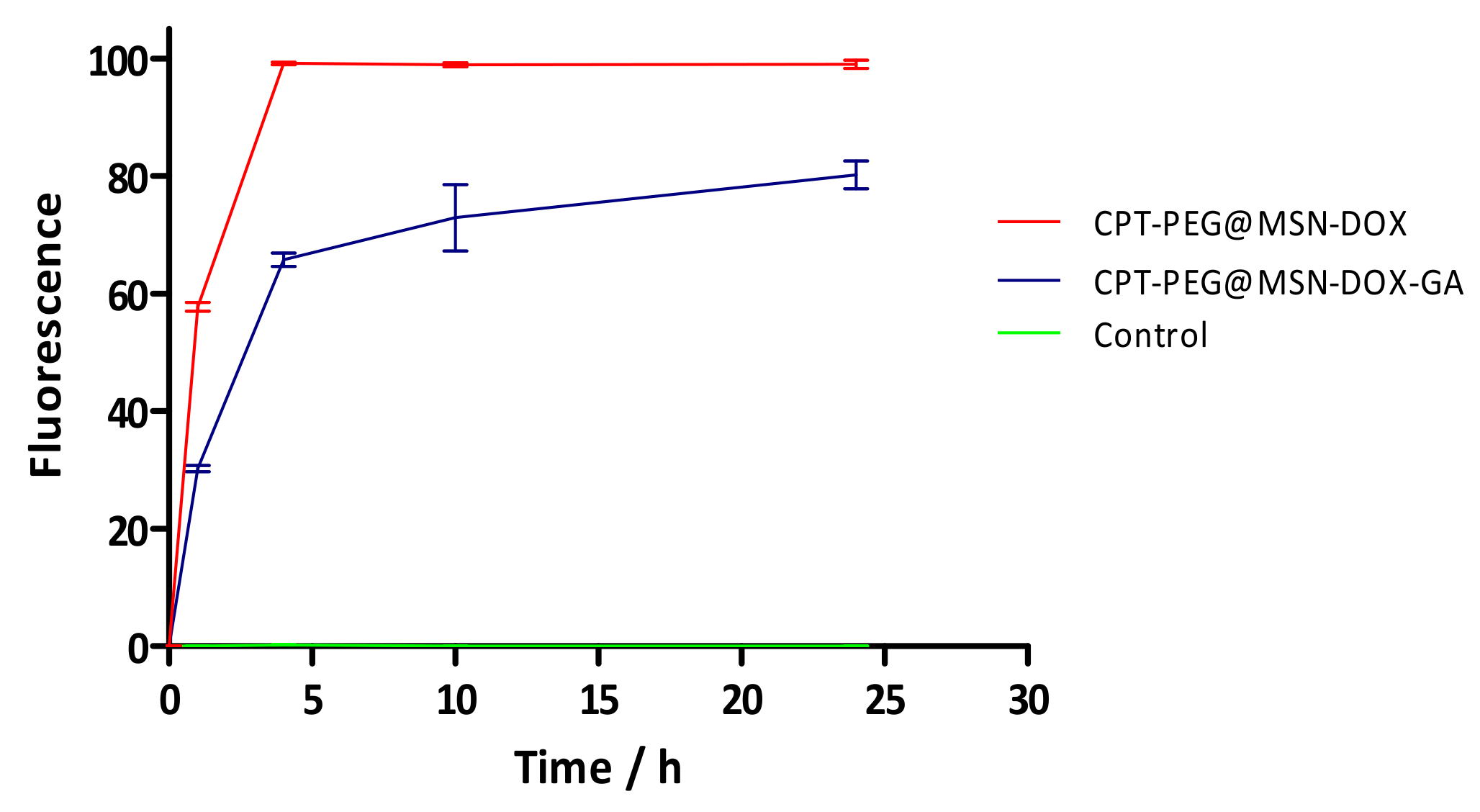
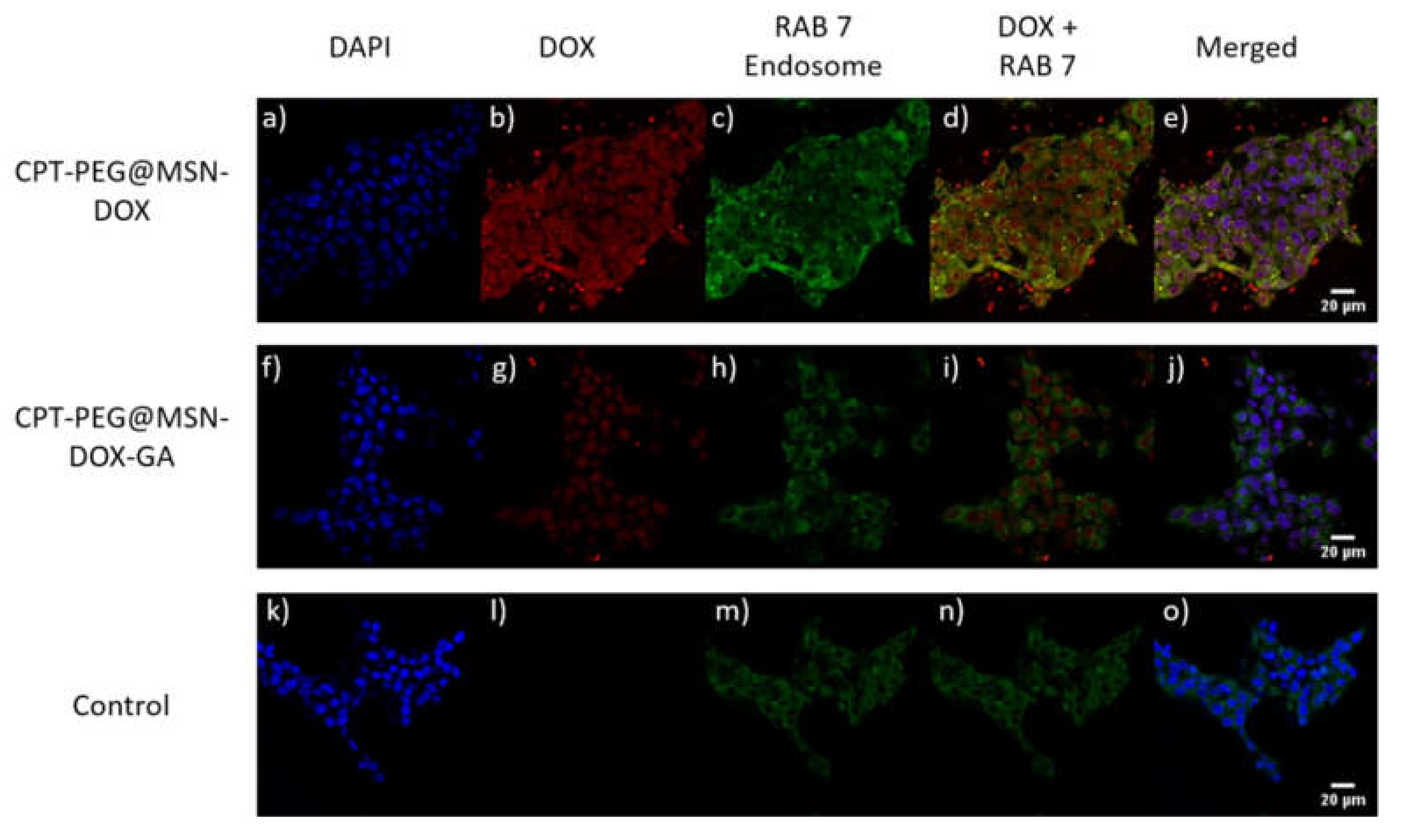
3.5. Selectivity in the Cellular Uptake
3.6. Cytotoxicity
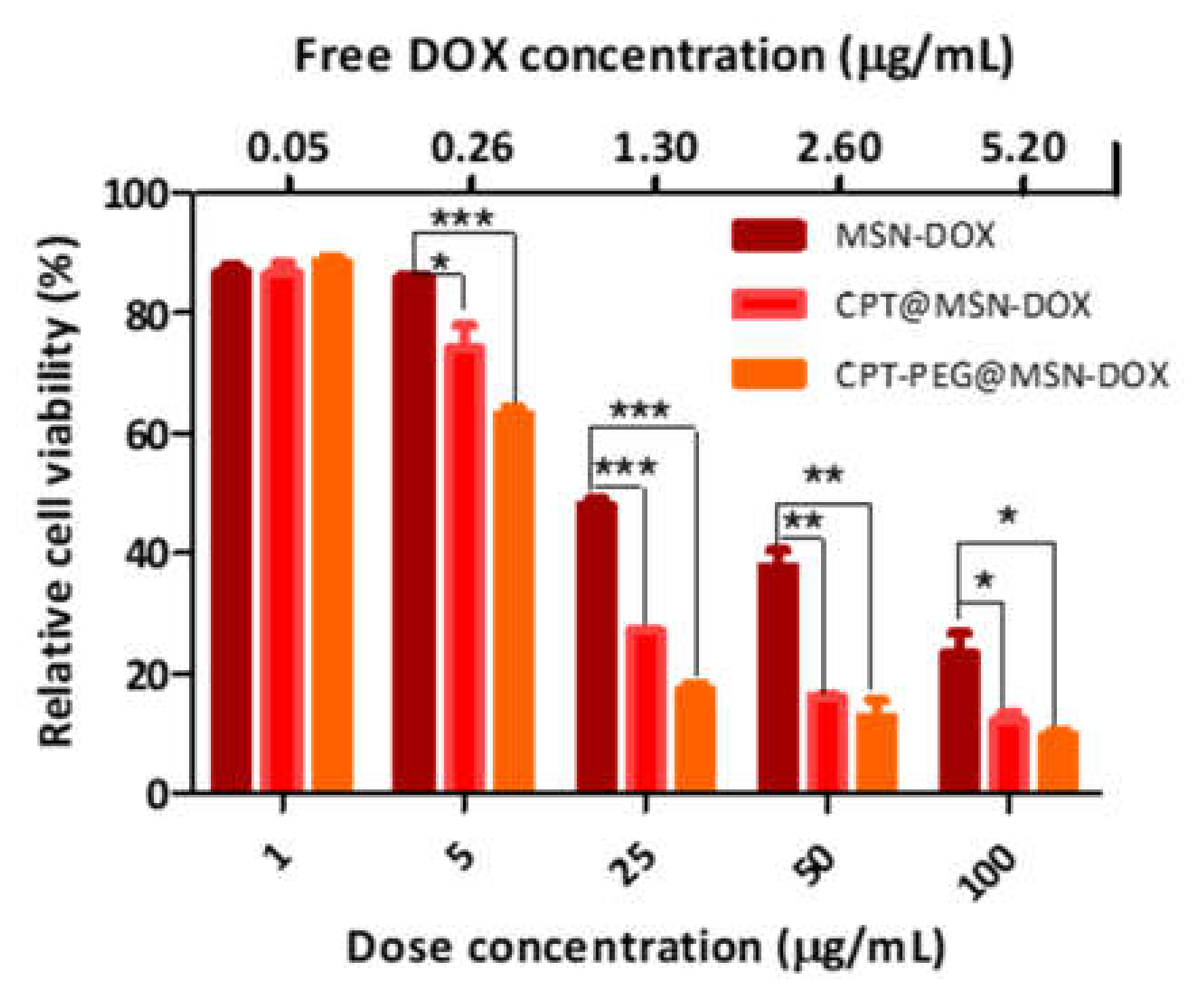
3.6.1. Assessment of the Cytotoxic Effect of CPT vs. CPT-PEG in Combination with DOX toward HepG2 Cells
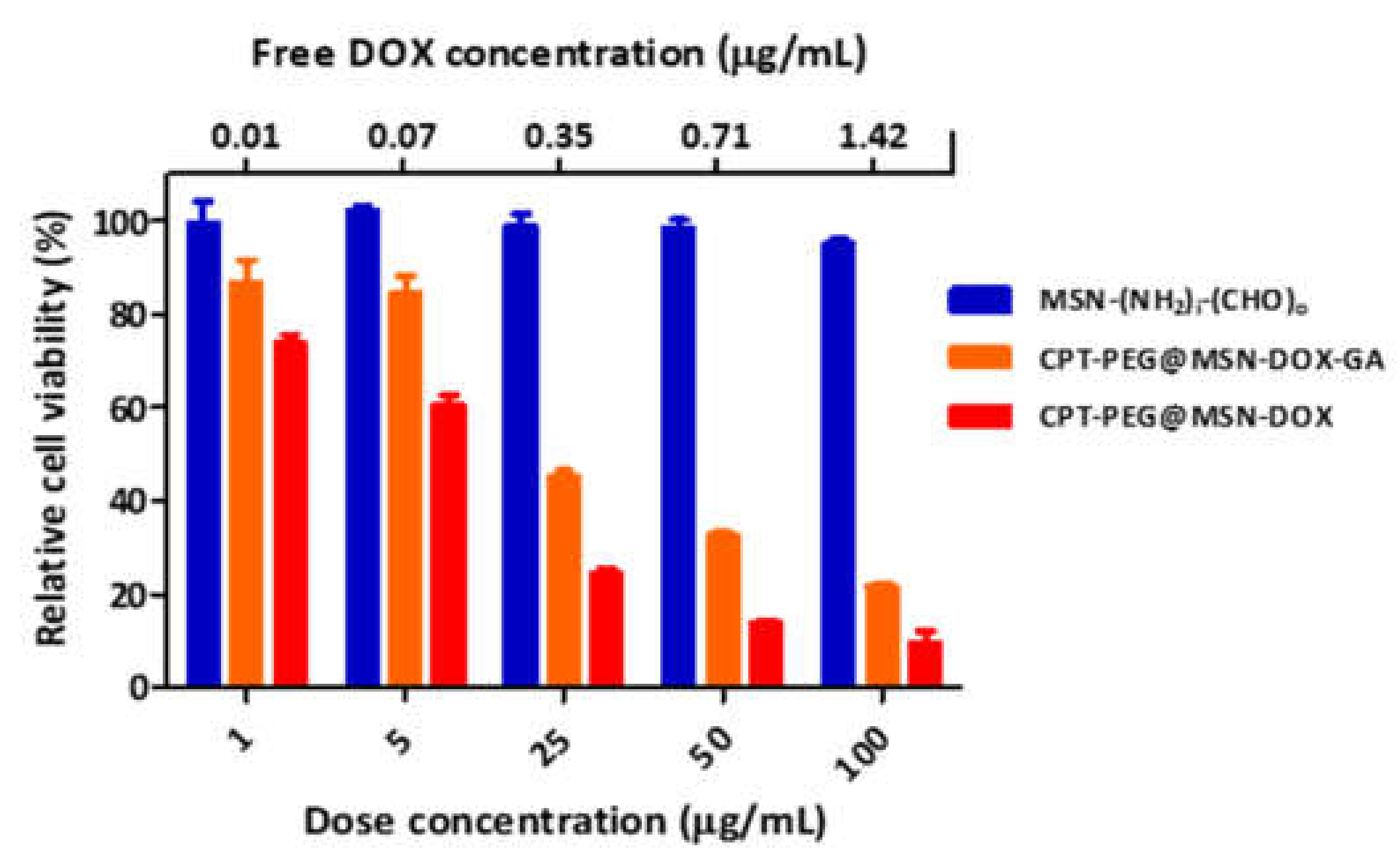
3.6.2. Cytotoxicity of CPT-PEG@MSN-DOX-GA toward HepG2 Cells
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Tunissiolli, N.M.; Castanhole-Nunes, M.; Biselli-Chicote, P.M.; Pavarino, É.C.; Da Silva, R.F.; Silva, R.D.C.M.A.D.; Goloni-Bertollo, E.M. Hepatocellular Carcinoma: A Comprehensive Review of Biomarkers, Clinical Aspects, and Therapy. Asian Pac. J. Cancer Prev. 2017, 18, 863–872. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.D.; Roberts, L.R. Hepatocellular carcinoma: A global view. Nat. Rev. Gastroenterol. Hepatol. 2010, 7, 448–458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, H.; Lim, J.-Y.; Sharma, A.; Yoon, D.W.; Kim, J.H.; Yang, Z.; Qu, J.; Kim, J.; Lee, S.G.; Kim, J.S. A pH-Responsive Glycyrrhetinic-Acid-Modified Small-Molecule Conjugate for NIR Imaging of Hepatocellular Carcinoma (HCC). ChemBioChem 2019, 20, 614–620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, Q.; Zhang, C.N.; Wang, X.H.; Wang, W.; Huang, W.; Cha, R.T.; Wang, C.H.; Yuan, Z.; Liu, M.; Wan, H.Y.; et al. Glycyrrhetinic acid-modified chitosan/poly(ethylene glycol) nanoparticles for liver-targeted delivery. Biomaterials 2010, 31, 4748–4756. [Google Scholar] [CrossRef] [PubMed]
- Llinàs, M.C.; Martínez-Edo, G.; Cascante, A.; Porcar, I.; Borros, S.; Sánchez-García, D. Preparation of a mesoporous silica-based nano-vehicle for dual DOX/CPT pH-triggered delivery. Drug Deliv. 2018, 25, 1137–1146. [Google Scholar] [CrossRef] [PubMed]
- Iturrioz-Rodriguez, N.; Correa-Duarte, M.A.; Fanarraga, M.L. Controlled drug delivery systems for cancer based on mesoporous silica nanoparticles. Int. J. Nanomed. 2019, 14, 3389–3401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, T.; Shi, S.; Goel, S.; Shen, X.; Xie, X.; Chen, Z.; Zhang, H.; Li, S.; Qin, X.; Yang, H.; et al. Recent Advancements in Mesoporous Silica Nanoparticles towards Therapeutic Applications for Cancer. Acta Biomater. 2019, 89, 1–13. [Google Scholar] [CrossRef]
- Narayan, R.; Nayak, U.Y.; Raichur, A.M.; Garg, S. Mesoporous Silica Nanoparticles: A Comprehensive Review on Synthesis and Recent Advances. Pharmaceutics 2018, 10, 118. [Google Scholar] [CrossRef] [Green Version]
- Castillo, R.R.; Colilla, M.; Vallet-Regi, M. Advances in mesoporous silica-based nanocarriers for co-delivery and combination therapy against cancer. Expert Opin. Drug Deliv. 2017, 14, 229–243. [Google Scholar] [CrossRef]
- Baeza, A.; Manzano, M.; Colilla, M.; Vallet-Regi, M. Recent advances in mesoporous silica nanoparticles for antitumor therapy: Our contribution. Biomater. Sci. 2016, 4, 803–813. [Google Scholar] [CrossRef] [Green Version]
- Martínez-Carmona, M.; Colilla, M.; Vallet-Regí, M. Smart Mesoporous Nanomaterials for Antitumor Therapy. Nanomaterials 2015, 5, 1906–1937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baeza, A.; Colilla, M.; Vallet-Regi, M. Advances in mesoporous silica nanoparticles for targeted stimuli-responsive drug delivery. Expert Opin. Drug Deliv. 2015, 12, 319–337. [Google Scholar] [CrossRef]
- Watermann, A.; Brieger, J. Mesoporous Silica Nanoparticles as Drug Delivery Vehicles in Cancer. Nanomaterials 2017, 7, 189. [Google Scholar] [CrossRef] [Green Version]
- Knežević, N.Ž.; Durand, J.-O. Targeted Treatment of Cancer with Nanotherapeutics Based on Mesoporous Silica Nanoparticles. ChemPlusChem 2015, 80, 26–36. [Google Scholar] [CrossRef]
- Mamaeva, V.; Sahlgren, C.; Linden, M. Mesoporous silica nanoparticles in medicine--recent advances. Adv. Drug. Deliv. Rev. 2013, 65, 689–702. [Google Scholar] [CrossRef] [PubMed]
- Coll, C.; Bernardos, A.; Martinez-Manez, R.; Sancenon, F. Gated silica mesoporous supports for controlled release and signaling applications. Acc. Chem. Res. 2013, 46, 339–349. [Google Scholar] [CrossRef]
- Qi, W.-W.; Yu, H.-Y.; Guo, H.; Lou, J.; Wang, Z.-M.; Liu, P.; Sapin-Minet, A.; Maincent, P.; Hong, X.-C.; Hu, X.-M.; et al. Doxorubicin-Loaded Glycyrrhetinic Acid Modified Recombinant Human Serum Albumin Nanoparticles for Targeting Liver Tumor Chemotherapy. Mol. Pharm. 2015, 12, 675–683. [Google Scholar] [CrossRef]
- Danhier, F.; Feron, O.; Préat, V. To exploit the tumor microenvironment: Passive and active tumor targeting of nanocarriers for anti-cancer drug delivery. J. Control. Release Off. J. Control. Release Soc. 2010, 148, 135–146. [Google Scholar] [CrossRef]
- Rosenholm, J.M.; Peuhu, E.; Eriksson, J.E.; Sahlgren, C.; Lindén, M. Targeted Intracellular Delivery of Hydrophobic Agents using Mesoporous Hybrid Silica Nanoparticles as Carrier Systems. Nano Lett. 2009, 9, 3308–3311. [Google Scholar] [CrossRef]
- Zhang, L.; Yao, J.; Zhou, J.; Wang, T.; Zhang, Q. Glycyrrhetinic acid-graft-hyaluronic acid conjugate as a carrier for synergistic targeted delivery of antitumor drugs. Int. J. Pharm. 2013, 441, 654–664. [Google Scholar] [CrossRef]
- Zhang, C.; Wang, W.; Liu, T.; Wu, Y.; Guo, H.; Wang, P.; Tian, Q.; Wang, Y.; Yuan, Z. Doxorubicin-loaded glycyrrhetinic acid-modified alginate nanoparticles for liver tumor chemotherapy. Biomaterials 2012, 33, 2187–2196. [Google Scholar] [CrossRef]
- Wang, X.; Gu, X.; Wang, H.; Sun, Y.; Wu, H.; Mao, S. Synthesis, characterization and liver targeting evaluation of self-assembled hyaluronic acid nanoparticles functionalized with glycyrrhetinic acid. Eur. J. Pharm. Sci. Off. J. Eur. Fed. Pharm. Sci. 2017, 96, 255–262. [Google Scholar] [CrossRef]
- Ma, P.; Sun, Y.; Chen, J.; Li, H.; Zhu, H.; Gao, X.; Bi, X.; Zhang, Y. Enhanced anti-hepatocarcinoma efficacy by GLUT1 targeting and cellular microenvironment-responsive PAMAM-camptothecin conjugate. Drug Deliv. 2018, 25, 153–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.-Y.; Liu, Y.; Wang, X.-Q.; Liu, L.-H.; Hu, J.-J.; Luo, G.-F.; Chen, W.-H.; Rong, L.; Zhang, X.-Z. One-Pot Construction of Functional Mesoporous Silica Nanoparticles for the Tumor-Acidity-Activated Synergistic Chemotherapy of Glioblastoma. ACS Appl. Mater. Interfaces 2013, 5, 7995–8001. [Google Scholar] [CrossRef] [PubMed]
- Camacho, K.M.; Kumar, S.; Menegatti, S.; Vogus, D.R.; Anselmo, A.C.; Mitragotri, S. Synergistic antitumor activity of camptothecin–doxorubicin combinations and their conjugates with hyaluronic acid. J. Control. Release 2015, 210, 198–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, M.; Huang, P.; Wang, Y.; Su, Y.; Zhou, L.; Zhu, X.; Yan, D. Synergistic Combination Chemotherapy of Camptothecin and Floxuridine through Self-Assembly of Amphiphilic Drug–Drug Conjugate. Bioconjug. Chem. 2015, 26, 2497–2506. [Google Scholar] [CrossRef]
- Henne, W.A.; Doorneweerd, D.D.; Hilgenbrink, A.R.; Kularatne, S.A.; Low, P.S. Synthesis and activity of a folate peptide camptothecin prodrug. Bioorg. Med. Chem. Lett. 2006, 16, 5350–5355. [Google Scholar] [CrossRef]
- Slichenmyer, W.J.; Rowinsky, E.K.; Grochow, L.B.; Kaufmann, S.H.; Donehower, R.C. Camptothecin analogues: Studies from The Johns Hopkins Oncology Center. Cancer Chemother. Pharmacol. 1994, 34, S53–S57. [Google Scholar] [CrossRef]
- Xu, Z.; Wang, D.; Xu, S.; Liu, X.; Zhang, X.; Zhang, H. Preparation of a Camptothecin Prodrug with Glutathione-Responsive Disulfide Linker for Anticancer Drug Delivery. Chem. Asian J. 2014, 9, 199–205. [Google Scholar] [CrossRef]
- Zhao, Z.; Meng, H.; Wang, N.; Donovan, M.J.; Fu, T.; You, M.; Chen, Z.; Zhang, X.; Tan, W. A Controlled-Release Nanocarrier with Extracellular pH Value Driven Tumor Targeting and Translocation for Drug Delivery. Angew. Chem. Int. Ed. 2013, 52, 7487–7491. [Google Scholar] [CrossRef]
- Muniesa, C.; Vicente, V.; Quesada, M.; Sáez-Atiénzar, S.; Blesa, J.R.; Abasolo, I.; Fernández, Y.; Botella, P. Glutathione-sensitive nanoplatform for monitored intracellular delivery and controlled release of Camptothecin. RSC Adv. 2013, 3, 15121–15131. [Google Scholar] [CrossRef]
- Juríček, M.; Barnes, J.C.; Strutt, N.L.; Vermeulen, N.A.; Ghooray, K.C.; Dale, E.J.; McGonigal, P.R.; Blackburn, A.K.; Avestro, A.-J.; Stoddart, J.F. An ExBox [2]catenane. Chem. Sci. 2014, 5, 2724–2731. [Google Scholar] [CrossRef]
- Feng, R.; Deng, P.; Song, Z.; Chu, W.; Zhu, W.; Teng, F.; Zhou, F. Glycyrrhetinic acid-modified PEG-PCL copolymeric micelles for the delivery of curcumin. React. Funct. Polym. 2017, 111, 30–37. [Google Scholar] [CrossRef]
- Li, X.-Q.; Wen, H.-Y.; Dong, H.-Q.; Xue, W.-M.; Pauletti, G.M.; Cai, X.-J.; Xia, W.-J.; Shi, D.; Li, Y.-Y. Self-assembling nanomicelles of a novel camptothecin prodrug engineered with a redox-responsive release mechanism. Chem. Commun. 2011, 47, 8647–8649. [Google Scholar] [CrossRef] [PubMed]
- Banerji, S.; Wright, A.J.; Noble, M.; Mahoney, D.J.; Campbell, I.D.; Day, A.J.; Jackson, D.G. Structures of the Cd44–hyaluronan complex provide insight into a fundamental carbohydrate-protein interaction. Nat. Struct. Mol. Biol. 2007, 14, 234–239. [Google Scholar] [CrossRef]
- Neises, B.; Steglich, W. Simple Method for the Esterification of Carboxylic Acids. Angew. Chem. Int. Ed. Engl. 1978, 17, 522–524. [Google Scholar] [CrossRef]
- Nunes, S.S.; Fernandes, R.S.; Cavalcante, C.H.; da Costa César, I.; Leite, E.A.; Lopes, S.C.A.; Ferretti, A.; Rubello, D.; Townsend, D.M.; de Oliveira, M.C.; et al. Influence of PEG coating on the biodistribution and tumor accumulation of pH-sensitive liposomes. Drug Deliv. Transl. Res. 2019, 9, 123–130. [Google Scholar] [CrossRef]
- Zhou, S.; Sha, H.; Ke, X.; Liu, B.; Wang, X.; Du, X. Combination drug release of smart cyclodextrin-gated mesoporous silica nanovehicles. Chem. Commun. 2015, 51, 7203–7206. [Google Scholar] [CrossRef]
- Lee, C.-H.; Cheng, S.H.; Huang, I.P.; Souris, J.S.; Yang, C.S.; Mou, C.Y.; Lo, L.W. Intracellular pH-responsive mesoporous silica nanoparticles for the controlled release of anticancer chemotherapeutics. Angew. Chem. Int. Ed. Engl. 2010, 49, 8214–8219. [Google Scholar] [CrossRef]
- Gao, Y.; Yang, C.; Liu, X.; Ma, R.; Kong, D.; Shi, L. A Multifunctional Nanocarrier Based on Nanogated Mesoporous Silica for Enhanced Tumor-Specific Uptake and Intracellular Delivery. Macromol. Biosci. 2012, 12, 251–259. [Google Scholar] [CrossRef]
- Villegas, M.R.; Baeza, A.; Vallet-Regi, M. Nanotechnological Strategies for Protein Delivery. Molecules 2018, 23, 1008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slowing, I.; Trewyn, B.G.; Lin, V.S.Y. Effect of Surface Functionalization of MCM-41-Type Mesoporous Silica Nanoparticles on the Endocytosis by Human Cancer Cells. J. Am. Chem. Soc. 2006, 128, 14792–14793. [Google Scholar] [CrossRef] [Green Version]











Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martínez-Edo, G.; Fornaguera, C.; Borrós, S.; Sánchez-García, D. Glycyrrhetinic Acid-Functionalized Mesoporous Silica Nanoparticles for the Co-Delivery of DOX/CPT-PEG for Targeting HepG2 Cells. Pharmaceutics 2020, 12, 1048. https://doi.org/10.3390/pharmaceutics12111048
Martínez-Edo G, Fornaguera C, Borrós S, Sánchez-García D. Glycyrrhetinic Acid-Functionalized Mesoporous Silica Nanoparticles for the Co-Delivery of DOX/CPT-PEG for Targeting HepG2 Cells. Pharmaceutics. 2020; 12(11):1048. https://doi.org/10.3390/pharmaceutics12111048
Chicago/Turabian StyleMartínez-Edo, Gabriel, Cristina Fornaguera, Salvador Borrós, and David Sánchez-García. 2020. "Glycyrrhetinic Acid-Functionalized Mesoporous Silica Nanoparticles for the Co-Delivery of DOX/CPT-PEG for Targeting HepG2 Cells" Pharmaceutics 12, no. 11: 1048. https://doi.org/10.3390/pharmaceutics12111048