Mechanochemical Synthesis and Structure of the Tetrahydrate and Mesoporous Anhydrous Metforminium(2+)-N,N′-1,4-Phenylenedioxalamic Acid (1:2) Salt: The Role of Hydrogen Bonding and n→π * Charge Assisted Interactions

 , , , and
, , , and
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials and Crystal Synthesis
2.2. Instrumental
2.3. X-ray Structure Determination
3. Results and Discussion
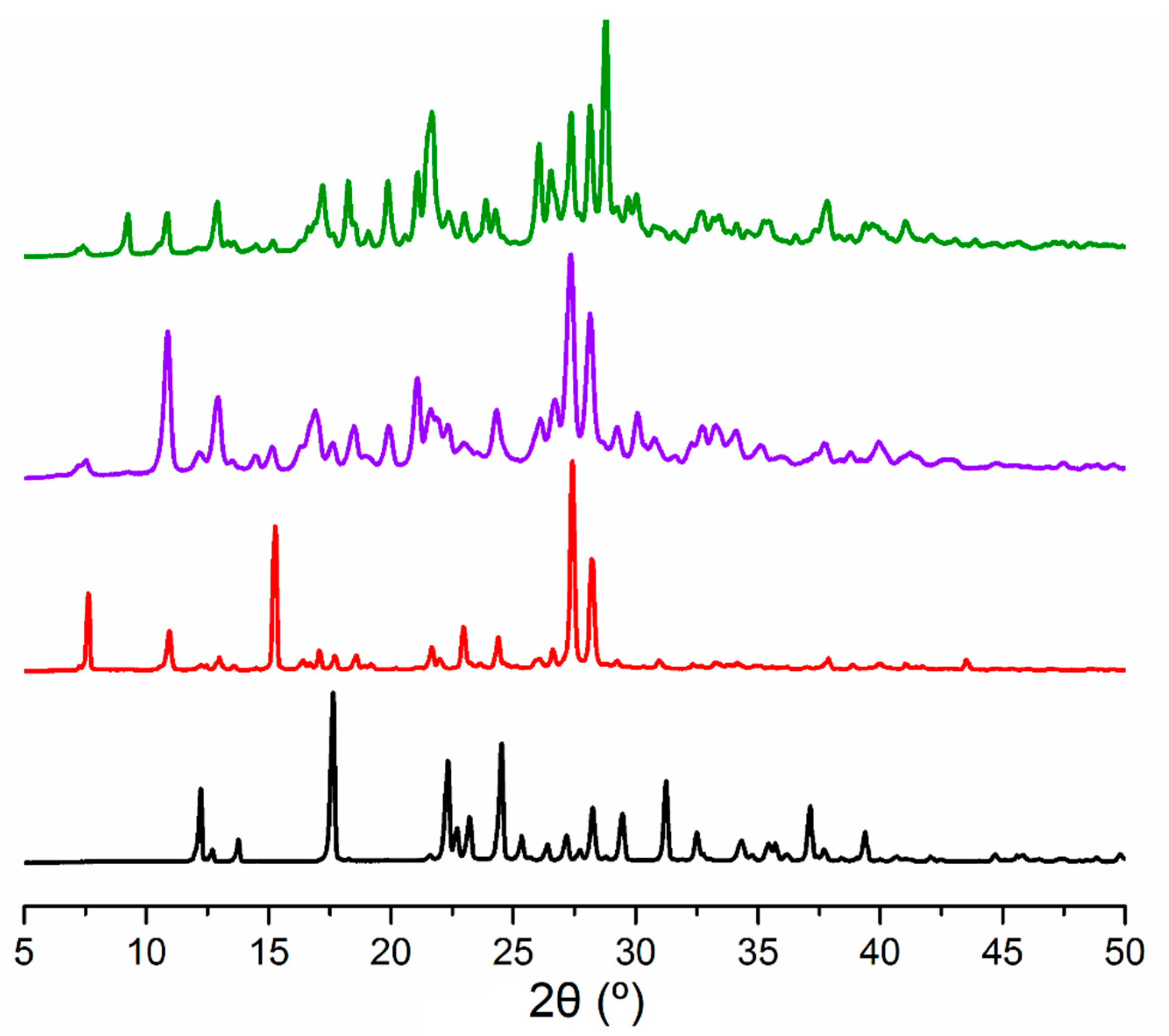
3.1. Synthesis
3.2. The Molecular and Supramolecular Structure of H2Mf(HpOXA)2∙4W
3.3. The Synthesis, Molecular and Supramolecular Structures of H2Mf(HpOXA)2 Anhydrate
3.4. The Nature and Structure of Dicationic Metfomin in H2Mf(HpOXA)2∙4W and H2Mf(HpOXA)2 Organic Salts
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Shan, N.; Zaworotko, M.J. The role of cocrystals in pharmaceutical science. Drug Discov. Today 2008, 13, 441–446. [Google Scholar] [CrossRef]
- Das, P.; Delost, M.D.; Qureshi, M.H.; Smith, D.T.; Njardarson, J.T. A Survey of the Structures of US FDA Approved Combination Drugs. J. Med. Chem. 2019, 62, 4265–4311. [Google Scholar] [CrossRef] [PubMed]
- Bian, X.; Jiang, L.; Gan, Z.; Zhang, L.; Cai, L.; Hu, X. A Glimepiride-Metformin Multidrug Crystal: Synthesis, Crystal Structure Analysis, and Physicochemical Properties. Molecules 2019, 24, 3786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhatt, J.A.; Bahl, D.; Morris, K.; Stevens, L.L.; Haware, R.V. Structure-mechanics and improved tabling performance of the drug-drug cocrystal metformin-salicylic acid. Eur. J. Pharm. Biopharm. 2020, 153, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Plata-Vargas, E.; de la Cruz-Hernández, C.; Dorazco-González, A.; Fuentes-Noriega, I.; Morales-Morales, D.; Germán-Acacio, J.M. Synthesis of Metforminium Succinate by Melting. Crystal Structure, Thermal, Spectroscopic and Dissolution Properties. J. Mex. Chem. Soc. 2017, 61, 197–204. [Google Scholar] [CrossRef]
- Kathuria, D.; Bankar, A.A.; Bharatam, P.V. What’s in a structure? The story of biguanides. Mol. Struct. 2018, 1152, 61–78. [Google Scholar] [CrossRef]
- Hariharan, M.; Rajan, S.S.; Srinivasan, R. Structure if metformin hydrochloride. Acta Cryst. 1989, C45, 911–913. [Google Scholar] [CrossRef]
- Childs, S.L.; Chyall, L.J.; Dunlap, J.T.; Coates, D.A.; Stahly, B.C.; Stahly, G.P. A Metastable Polymorph of Metformin Hydrochloride: Isolation and Characterization Using Capillary Crystallization and Thermal Microscopy Techniques. Cryst. Growth Des. 2004, 4, 441–449. [Google Scholar] [CrossRef]
- Nanubolu, J.B.; Sridhar, B.; Ravikumar, K.; Sawant, K.D.; Naik, T.A.; Patkar, L.N.; Cherukuvada, S.; Sreedhar, B. Polymorphism in metformin embonate salt-recurrence of dimeric and tetrameric guanidinium-carboxylate synthons. Cryst. Eng. Commun. 2013, 15, 4448–4464. [Google Scholar] [CrossRef]
- González-González, J.S.; Martínez-Martínez, F.J.; García-Báez, E.V.; Cruz, A.; Morín-Sánchez, L.M.; Rojas-Lima, S.; Padilla-Martínez, I.I. Molecular Complexes of Diethyl N,N′-1,3-Phenyldioxalamate and Resorcinols: Conformational Switching through Intramolecular Three-Centered Hydrogen-Bonding. Cryst. Growth Des. 2014, 14, 628–642. [Google Scholar] [CrossRef]
- Hall, C.M.; Wright, J.B. Antiallergic Phenylenedioxamic Acids. Ger. Offen DE 2362409 A1, 14 May 1980. [Google Scholar]
- Wright, J.B.; Hall, C.M.; Johnson, H.G.J. N,N’-(phenylene)dioxamic acids and their esters as antiallergy agents. Med. Chem. 1978, 21, 930–935. [Google Scholar] [CrossRef] [PubMed]
- Petyunin, G.P. Synthesis and pharmacological activity of phenylenedioxamic acids. Khimiko-Farmatsevticheskii Zhurnal 1986, 20, 827–830. [Google Scholar]
- Oliveira, W.X.C.; Pinheiro, C.B.; da Costa, M.M.; Fontes, A.P.S.; Nunes, W.C.; Lloret, F.; Julve, M.; Pereira, C.L.M. Crystal Engineering Applied to Modulate the Structure and Magnetic Properties of Oxamate Complexes Containing the [Cu(bpca)]+ Cation. Cryst. Growth Des. 2016, 16, 4094–4107. [Google Scholar] [CrossRef]
- Lisnard, L.; Chamoreau, L.-M.; Li, Y.; Journaux, Y. Solvothermal Synthesis of Oxamate-Based Helicate: Temperature Dependence of the Hydrogen Bond Structuring in the Solid. Cryst. Growth Des. 2012, 12, 4955–4962. [Google Scholar] [CrossRef] [Green Version]
- Ramírez-Milanés, E.G.; Martínez-Martínez, F.J.; Magaña-Vergara, N.E.; Rojas Lima, S.; Avendaño-Jiménez, Y.A.; García-Báez, E.V.; Morín-Sánchez, L.M.; Padilla-Martínez, I.I. Positional isomerism and steric effects in the self-assemblies of phenylene bis-monothiooxalamides. Cryst. Growth Des. 2017, 17, 2513–2528. [Google Scholar] [CrossRef]
- Gómez-Castro, C.Z.; Padilla-Martínez, I.I.; García-Báez, E.V.; Castrejón-Flores, J.L.; Peraza-Campos, A.L.; Martínez-Martínez, F.J. Solid state structure and solution thermodynamics of the three-centered hydrogen bond (O∙∙∙H∙∙∙O) using N-(2-benzoylphenyl) oxalyl derivatives as model compounds. Molecules 2014, 19, 14446–14460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cabrera-Pérez, L.C.; García-Báez, E.V.; Franco-Hernández, M.O.; Martínez-Martínez, F.J.; Padilla-Martínez, I.I. Carbonyl–carbonyl interactions and amide π-stacking as the directing motifs of the supramolecular assembly of ethyl N-(2-acetylphenyl)-oxalamate in a synperiplanar conformation. Acta Cryst. 2015, C71, 381–385. [Google Scholar]
- Martin, S.; Beitia, J.I.; Ugalde, M.; Vitoria, P.; Cortes, R. Diethyl N,N’-o-phenylenedioxamate. Acta Crystallogr. E Struct. Rep. Online 2002, 58, o913–o915. [Google Scholar] [CrossRef]
- James, S.L.; Adams, C.J.; Bolm, C.; Braga, D.; Collier, P.; Friščić, T.; Grepioni, F.; Harris, K.D.M.; Hyett, G.; Jones, W.; et al. Mechanochemistry: Opportunities for new and cleaner synthesis. Chem. Soc. Rev. 2012, 41, 413–447. [Google Scholar] [CrossRef] [Green Version]
- Agilent. CrysAlis PRO; Agilent Technologies: Oxfordshire, UK, 2012. [Google Scholar]
- Sheldrick, G.M. A short history of SHELX. Acta Cryst. 2008, A64, 112–122. [Google Scholar] [CrossRef] [Green Version]
- Farrugia, L.J. WinGX v2018.3 and ORTEP for windows: An update. J. Appl. Cryst. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELX. Acta Cryst. 2007, C71, 3–8. [Google Scholar]
- Spek, A.L. Single-crystal structure validation with the program PLATON. J. Appl. Cryst. 2003, 36, 7–13. [Google Scholar] [CrossRef] [Green Version]
- Macrae, C.F.; Sovago, I.; Cottrell, S.J.; Galek, P.T.A.; McCabe, P.; Pidcock, V.; Platings, M.; Shields, G.P.; Stevens, J.S.; Towler, M.; et al. Mercury 4.0: From visualization to analysis, design and prediction. J. Appl. Cryst. 2020, 53, 226–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pardo, E.; Cangussu, D.; Lescouezec, R.; Journaux, Y.; Pasan, J.; Delgado, F.S.; Ruiz-Pérez, C.; Ruiz-García, R.; Cano, J.; Julve, M.; et al. Molecular-Programmed Self-Assembly of Homo- and Heterometallic Tetranuclear Coordination Compounds: Synthesis, Crystal Structures, and Magnetic Properties of Rack-Type CuII2MII2 Complexes (M = Cu and Ni) with Tetranucleating Phenylenedioxamato Bridging Ligands. Inorg. Chem. 2009, 48, 4661–4673. [Google Scholar] [PubMed]
- Devi, R.N.; Jelsch, C.; Israel, S.; Aubert, E.; Anzline, C.; Hosamani, A.A. Charge density analysis of metformin chloride, a biguanide anti-hyperglycemic agent. Acta Cryst. 2017, B73, 10–22. [Google Scholar]
- Allen, F.H.; Kennard, O.; Watson, D.G.; Brammer, L.; Orpen, A.G.; Taylor, R. Tables of bond lengths determined by X-ray and neutron diffraction. Part 1. Bond lengths in organic compounds. J. Chem. Soc. Perkin Trans. 2 1987, 12, S1–S19. [Google Scholar] [CrossRef]
- Lu, L.P.; Zhang, H.M.; Feng, S.S.; Zhu, M.L. Two N,N-dimethylbiguanidium salts displaying double hydrogen bonds to the counter-ions. Acta Cryst. 2004, C60, o740–o743. [Google Scholar] [CrossRef] [Green Version]
- Chernyshov, I.Y.; Ananyev, I.V.; Pidko, E.A. Revisiting van der Waals Radii: From Comprehensive Structural Analysis to Knowledge-Based Classification of Interatomic Contacts. Chem. Phys. Chem. 2020, 21, 370–376. [Google Scholar] [CrossRef]
- Allen, F.H.; Baalham, C.A.; Lommerse, J.P.M.; Raithby, P.R. Carbonyl-Carbonyl Interactions can be Competitive with Hydrogen Bonds. Acta Cryst. 1998, B54, 320–329. [Google Scholar] [CrossRef]
- Kashid, S.M.; Sayan, B. Experimental Determination of the Electrostatic Nature of Carbonyl Hydrogen-Bonding Interactions Using IR-NMR Correlations. J. Phys. Chem. Lett. 2014, 5, 3211–3215. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Araki, T.; Kitaoka, H.; Terada, K. Characterization of Non-stoichiometric Hydration and the Dehydration Behavior of Sitafloxacin Hydrate. Chem. Pharm. Bull. 2012, 60, 45–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thommes, M.; Kaneko, K.; Neimark, A.V.; Olivier, J.P.; Rodriguez-Reinoso, F.; Rouquerol, J.; Sing, K.S.W. Physisorption of gases, with special reference to the evaluation of surface area and pore size distribution (IUPAC Technical Report). Pure Appl. Chem. 2015, 87, 1051–1069. [Google Scholar] [CrossRef] [Green Version]
- Langmaier, J.; Pižl, M.; Samec, Z.; Záliž, S. Extreme Basicity of Biguanide Drugs in Aqueous Solutions: Ion Transfer Voltammetry and DFT Calculations. J. Phys. Chem. A 2016, 120, 7344–7350. [Google Scholar] [CrossRef]
- Cruz-Cabeza, A.J. Acid–base crystalline complexes and the pKa rule. Cryst. Eng. Commun. 2012, 14, 6362–6365. [Google Scholar] [CrossRef]
- Lemoine, P.; Tomas, A. Tétrachlorocuprate(II) de metformine. Acta Cryst. 1994, C50, 1437–1439. [Google Scholar] [CrossRef] [Green Version]
- Fridrichová, M.; Císařová, I.; Němec, I. 1,1-Dimethylbiguanidium(2+) dinitrate. Acta Cryst. 2012, E68, o18–o19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Şerb, M.D.; Kalf, I.; Englert, U. Biguanide and squaric acid as pH-dependent building blocks in crystal engineering. Cryst. Eng. Commun. 2014, 16, 10631–10639. [Google Scholar] [CrossRef]
- Guo, D.S.; Zhang, H.Q.; Ding, F.; Liu, Y. Thermodynamic origins of selective binding affinity between p-sulfonatocalix[4,5]arenes with biguanidiniums. Org. Biomol. Chem. 2012, 10, 1527–1536. [Google Scholar] [CrossRef]
- Bauzá, A.; Mooibroek, T.J.; Frontera, A. The Bright Future of Unconventional s/p-Hole Interactions. Chem. Phys. Chem. 2015, 16, 2496–2517. [Google Scholar] [CrossRef]
- Wang, C.; Hu, S.; Sun, C.C. Expedited development of a high dose orally disintegrating metformin tablet enabled by sweet salt formation with acesulfame. Int. J. Pharm. 2017, 532, 435–443. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.K.; Das, A. The n→π* interaction: A rapidly emerging non-covalent interaction. Phys. Chem. Chem. Phys. 2015, 17, 9596–9612. [Google Scholar] [CrossRef] [PubMed] [Green Version]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HpOXA− Moiety | |||
| Atoms | Bond Lengths/Å | Atoms | Bond Lengths/Å |
| C8–C9 | 1.534(3) | C18–C19 | 1.556(3) |
| C28–C29 | 1.542(3) | C38–C39 | 1.550(3) |
| Torsion Angles/° | Torsion Angles/° | ||
| C2–C1–N7–C8 | −16.7(4) | C3–C4–N17–C18 | −2.7(4) |
| O8–C8–N7–C1 | 1.5(4) | O8–C8–C9–O9 | −179.7(2) |
| O18–C18–N17–C4 | 1.1(4) | O18–C18–C19–O19 | 177.0(2) |
| C22–C21–N27–C28 | −11.1(4) | C23–C24–C25–C26 | −1.1(3) |
| O28–C28–N27–C21 | 2.4(4) | O28–C28–C29–O29 | −178.8(2) |
| O28–C28–N27–C21 | 2.4(4) | O28–C28–C29–O29 | −178.8(2) |
| O38–C38–N37–C24 | 4.8(4) | O38–C38–C39–O39 | −171.6(2) |
| H2Mf2+ Moiety | |||
| Atoms | Bond Lengths/Å | Atoms | Bond Lengths/Å |
| N50–C52 | 1.304(3) | N51–C52 | 1.308(3) |
| N53–C52 | 1.367(3) | N53–C54 | 1.378(3) |
| N55–C54 | 1.302(3) | N56–C54 | 1.316(3) |
| N56–C58 | 1.457(3) | N56–C57 | 1.452(4) |
| Torsion Angle/° | |||
| N56–C54–N53–C52 | 148.8(2) | ||
| C–O∙∙∙A | C–O/Å | O∙∙∙A/Å | C–O∙∙∙A/° |
|---|---|---|---|
| C38–O38∙∙∙C9 | 1.219(2) | 3.083(3) | 89.0(2) |
| C38–O38∙∙∙C8 | 1.219(2) | 3.075(3) | 102.1(2) |
| C29–O30∙∙∙C19 | 1.264(3) | 3.198(3) | 97.4(2) |
| C8–O8∙∙∙C54 | 1.222(2) | 3.142(3) | 133.4(2) |
| C8–O8∙∙∙N56 | 1.222(2) | 3.003(3) | 143.6(2) |
| C18–O18 vii∙∙∙C54 | 1.222(3) | 3.109(3) | 131.8(2) |
| D–H···A | D–H/Å | H···A/Å | D···A/Å | D–H···A/° |
|---|---|---|---|---|
| C5–H5···O1 i | 0.93 | 2.62 | 3.438 (3) | 147 |
| C6–H6···O3 ii | 0.93 | 2.48 | 3.217 (3) | 136 |
| N7–H7···O3 ii | 0.86 | 2.17 | 2.991 (2) | 159 |
| N17–H17···O1 i | 0.86 | 2.37 | 3.208 (3) | 166 |
| N27–H27···O2 iii | 0.86 | 2.58 | 3.154 (3) | 125 |
| N37–H37···O1 iv | 0.86 | 2.49 | 3.185 (3) | 139 |
| N50–H50B···O40 | 0.86 | 2.02 | 2.820 (3) | 154 |
| N50–H50A···O4 v | 0.86 | 2.27 | 2.999 (3) | 143 |
| N50–H50A···O9 vi | 0.86 | 2.37 | 2.982 (2) | 129 |
| N51–H51A···O4 v | 0.86 | 2.20 | 2.947 (3) | 145 |
| N51–H51B···O18 vii | 0.86 | 2.10 | 2.918 (2) | 159 |
| N53–H53···O38 | 0.86 | 2.00 | 2.843 (2) | 168 |
| N55–H55A···O2 | 0.86 | 1.92 | 2.730 (2) | 157 |
| N55—H55B···O28 vii | 0.86 | 2.05 | 2.832 (2) | 151 |
| O1—H1A···O3 vii | 0.85 | 2.15 | 2.985 (3) | 169 |
| O1—H1B···O19 i | 0.85 | 2.33 | 2.998 (3) | 136 |
| O2—H2A···O39 vi | 0.85 | 1.89 | 2.742 (2) | 177 |
| O2—H2B···O19 viii | 0.85 | 1.93 | 2.760 (2) | 166 |
| O3—H3A···O4 | 0.85 | 1.93 | 2.760 (3) | 167 |
| O3—H3B···O39 vi | 0.85 | 2.16 | 2.982 (3) | 161 |
| O4—H4A···O8 | 0.85 | 2.00 | 2.773 (3) | 151 |
| O4—H4A···O10 | 0.85 | 2.64 | 3.145 (2) | 120 |
| O4—H4B···O29 ix | 0.85 | 1.80 | 2.611 (3) | 160 |
| O10—H10···O40 vi | 0.82 | 1.72 | 2.5362 (19) | 176 |
| O30—H30···O20 x | 0.82 | 1.66 | 2.477 (2) | 177 |
| Bond Wavenumber (cm−1) | ||||
|---|---|---|---|---|
| Compounds | O–H | N–H | C=O | C=N |
| H2pOXA∙2W a | 3488 (br), 3349 (br) | 3335 (m), 3307 (m) | 1733 (m), 1714 (m), 1681 (s), 1657 (s) | |
| HMfCl | 3390 (sh), 3379 (m) 3298 (br), 3155 (m) 3095 (sh) | 1624 (m), 1584 (sh), 1585 (s), 1550 (s) | ||
| H2Mf(HpOXA)2∙4W | 3442 (br), 3102 (br) | 3432 (br), 3358 (br), 3334 (w), 3317 (w) | 1686 (vs), 1524 (vs, COO-) | 1672 (sh) |
| H2Mf(HpOXA)2 | 3045 (br, m), 3180 (vw) | 3445 (w), 3414 (w) 3372 (w), 3348 (w) 3329 (sh), 3309 (w) | 1707 (sh), 1681 (s), 1524 (vs, COO-) | 1650 (sh), 1524 (sh) 1500 (vs) |
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| Compounds | C1 | C2 | C3 | C8O | C9O | C52 | C54 | CH3 |
| H2pOXA a | 134.6 | 121.0 | 121.0 | 157.1 | 162.6 | |||
| HMfCl a | 158.9 | 159.6 | 37.9 | |||||
| HMfCl crystals | 159 | 162 | 42, 40 | |||||
| (H2Mf)(HpOXA)2∙4W | 136 b 134, 132 | 120 118 c | 121 b 120 c | 159 b 155 b | 164 d | 159 | 160 | 42, 40 |
| (H2Mf)(HpOXA)2 | 136 134 c | 119, 118 b 117 | 123 120 c | 157 154 | 164 163 | 159 | 160 | 42, 41 |
| Organic Salt | X–O∙∙∙C | XO∙∙∙C/Å | X–O∙∙∙C/° | O∙∙∙C∙∙∙O/° | Me2NCNC/° |
|---|---|---|---|---|---|
| H2Mf(HpOXA)2∙4W(this work) | CO∙∙∙C54 | 3.142(3) | 133.4(2) | 148.8(2) | |
| CO∙∙∙C54 | 3.109(3) | 131.8(2) | 163.6(3) | ||
| H2Mf-squarate (1) a | CO∙∙∙C | 3.212 | 148.0 | −144.9 | |
| H2O∙∙∙C | 3.156 | 167.5 | 136.3 | ||
| H2Mf-squarate (2) a | CO∙∙∙C | 2.988 | 87.9 | 143.3 | |
| CO∙∙∙C | 3.023 | 87.7 | |||
| H2Mf-oxalate b | CO∙∙∙C | 3.108 | 88.9 | 143.2 | |
| H2O∙∙∙C | 3.152 | 158.3 | |||
| H2Mf-sulfonatocalix[4,5]arenes c | SO∙∙∙C | 2.938 | 132.2 | 41.4 | |
| SO∙∙∙C | 3.187 | 148.0 | 163.0 | ||
| H2Mf-acesulfame d | SO∙∙∙C | 3.130 | 128.1 | 141.6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chong-Canto, S.; García-Báez, E.V.; Martínez-Martínez, F.J.; Ramos-Organillo, A.A.; Padilla-Martínez, I.I. Mechanochemical Synthesis and Structure of the Tetrahydrate and Mesoporous Anhydrous Metforminium(2+)-N,N′-1,4-Phenylenedioxalamic Acid (1:2) Salt: The Role of Hydrogen Bonding and n→π * Charge Assisted Interactions. Pharmaceutics 2020, 12, 998. https://doi.org/10.3390/pharmaceutics12100998
Chong-Canto S, García-Báez EV, Martínez-Martínez FJ, Ramos-Organillo AA, Padilla-Martínez II. Mechanochemical Synthesis and Structure of the Tetrahydrate and Mesoporous Anhydrous Metforminium(2+)-N,N′-1,4-Phenylenedioxalamic Acid (1:2) Salt: The Role of Hydrogen Bonding and n→π * Charge Assisted Interactions. Pharmaceutics. 2020; 12(10):998. https://doi.org/10.3390/pharmaceutics12100998
Chicago/Turabian StyleChong-Canto, Sayuri, Efrén V. García-Báez, Francisco J. Martínez-Martínez, Angel A. Ramos-Organillo, and Itzia I. Padilla-Martínez. 2020. "Mechanochemical Synthesis and Structure of the Tetrahydrate and Mesoporous Anhydrous Metforminium(2+)-N,N′-1,4-Phenylenedioxalamic Acid (1:2) Salt: The Role of Hydrogen Bonding and n→π * Charge Assisted Interactions" Pharmaceutics 12, no. 10: 998. https://doi.org/10.3390/pharmaceutics12100998