Praziquantel–Clays as Accelerated Release Systems to Enhance the Low Solubility of the Drug

 ,
,
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
2.2.1. Preparation of Praziquantel–Clay Minerals Interaction Products
2.2.2. X-ray Diffraction
2.2.3. Thermal Analysis
2.2.4. Fourier Transform Infrared (FTIR) Spectroscopy
2.2.5. In Vitro Release Studies
2.2.6. Solubility Studies
2.2.7. Cell Culture
2.2.8. Cytotoxicity Studies
2.2.9. Cell Cycle Studies
3. Results
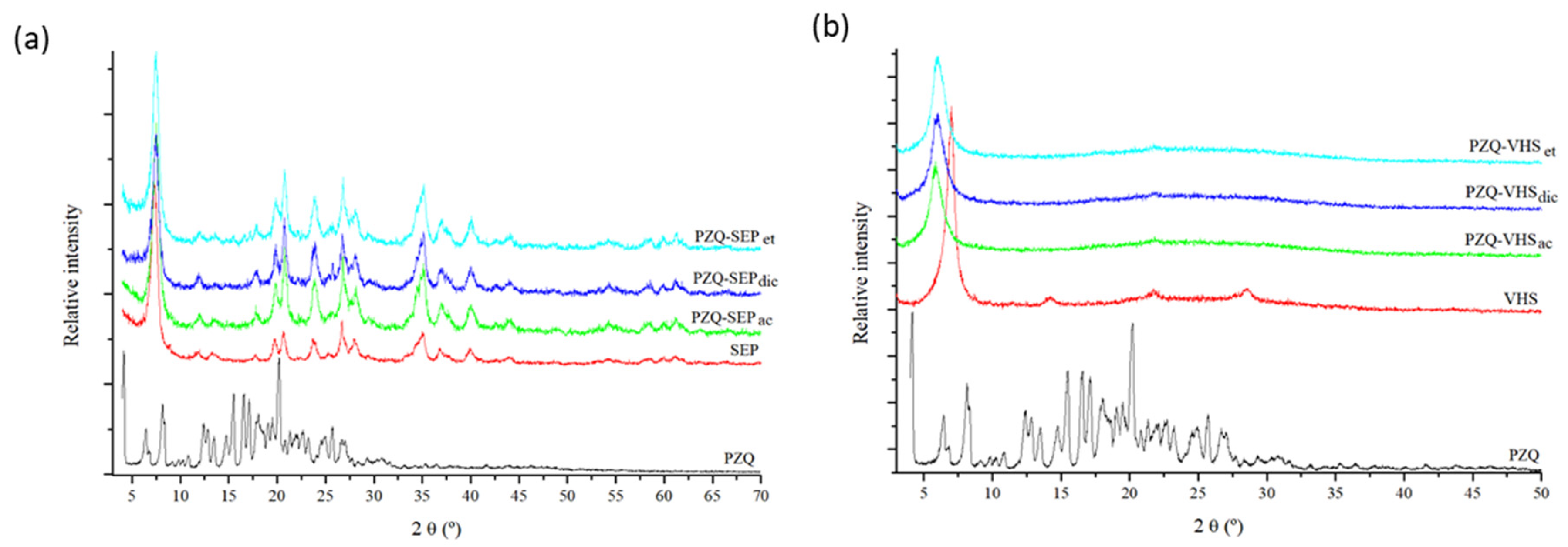
3.1. X-ray Diffraction
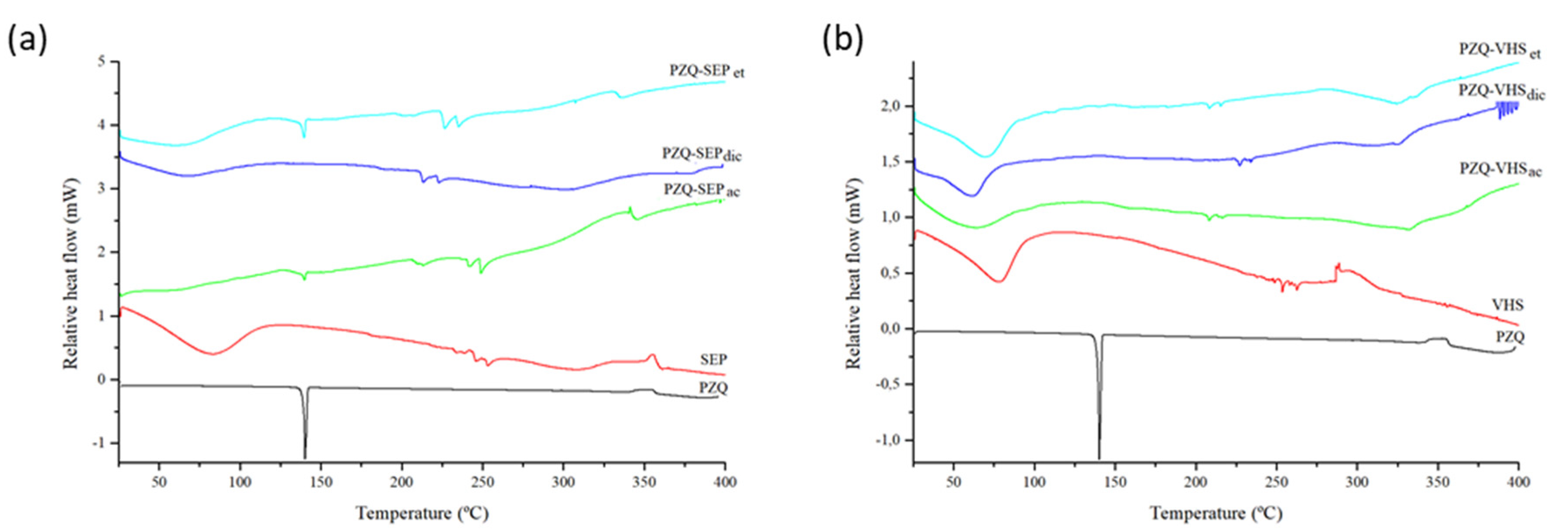
3.2. Thermal Analysis
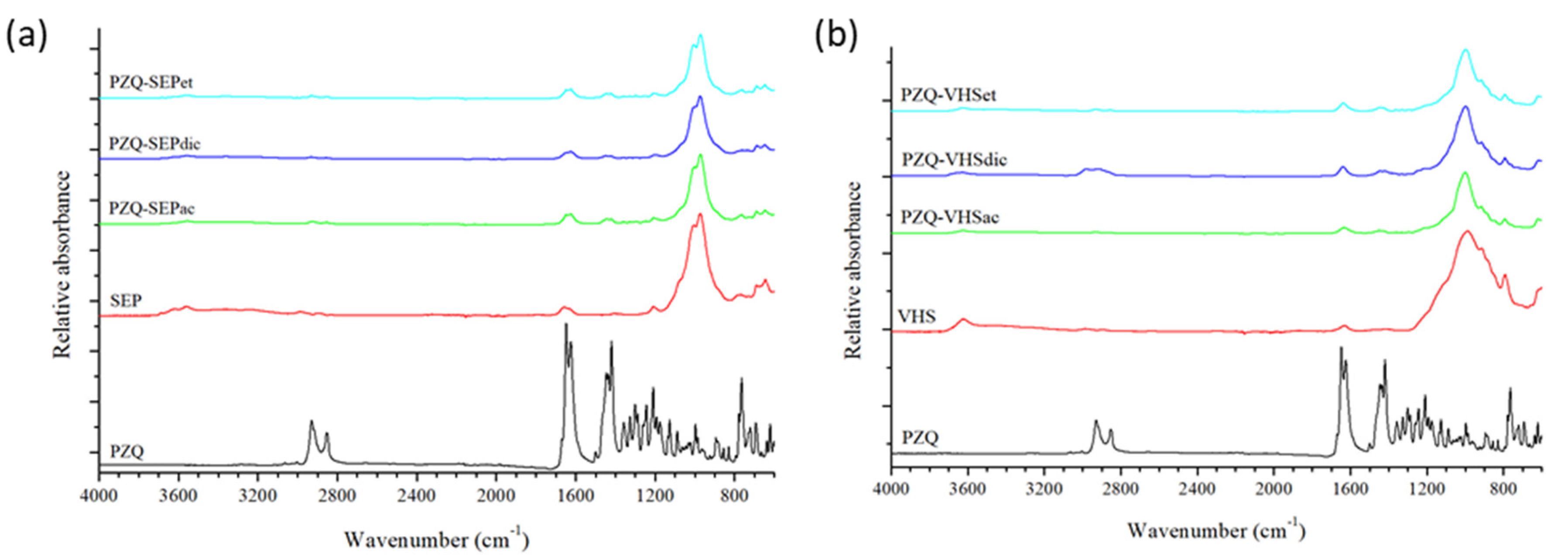
3.3. Fourier Transform Infrared Spectroscopy
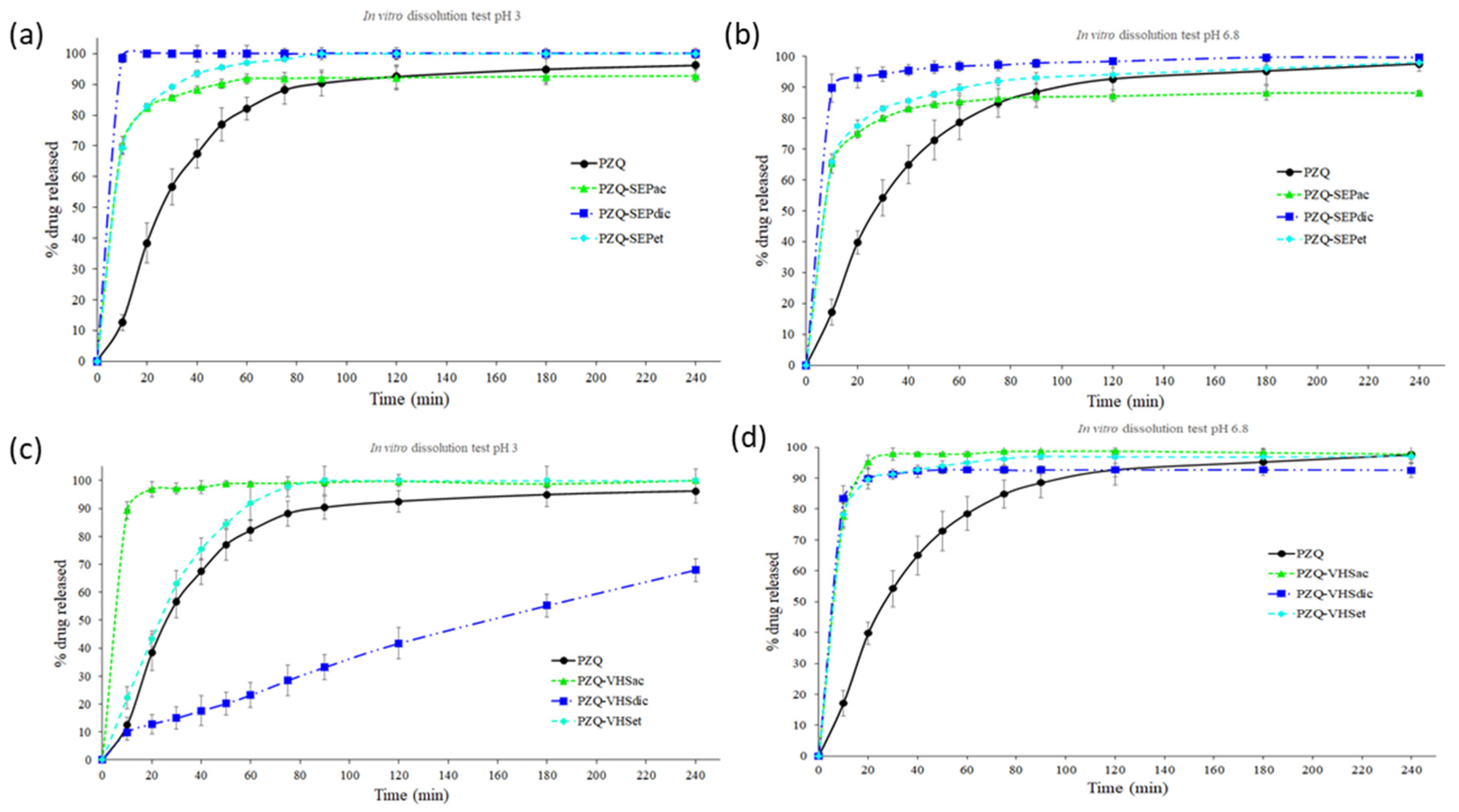
3.4. In Vitro Release Studies
3.5. Solubility Studies
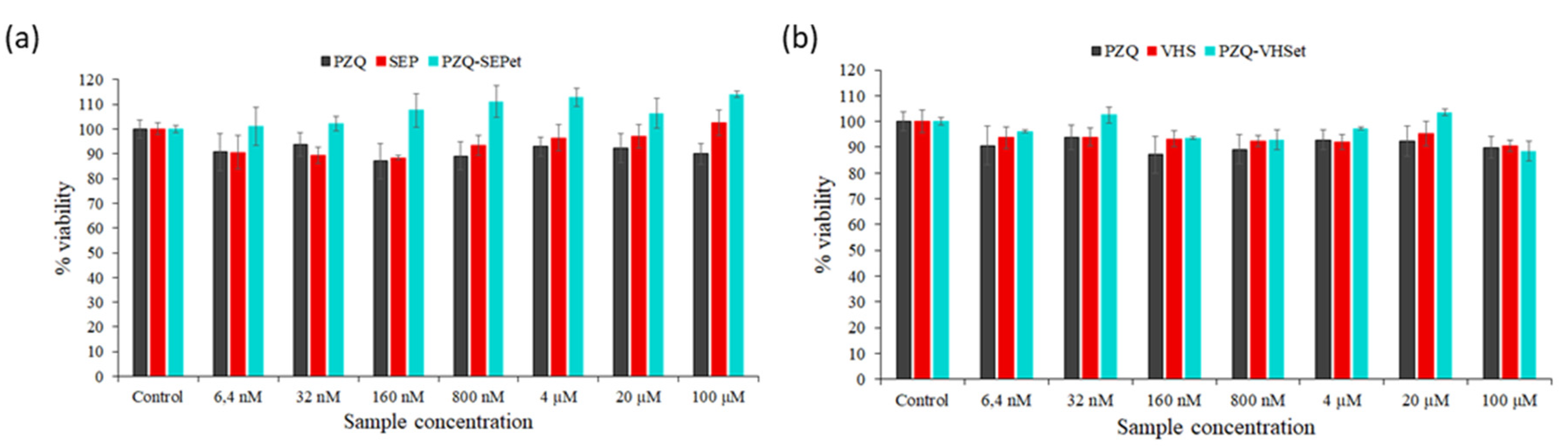
3.6. Cytotoxicity Studies
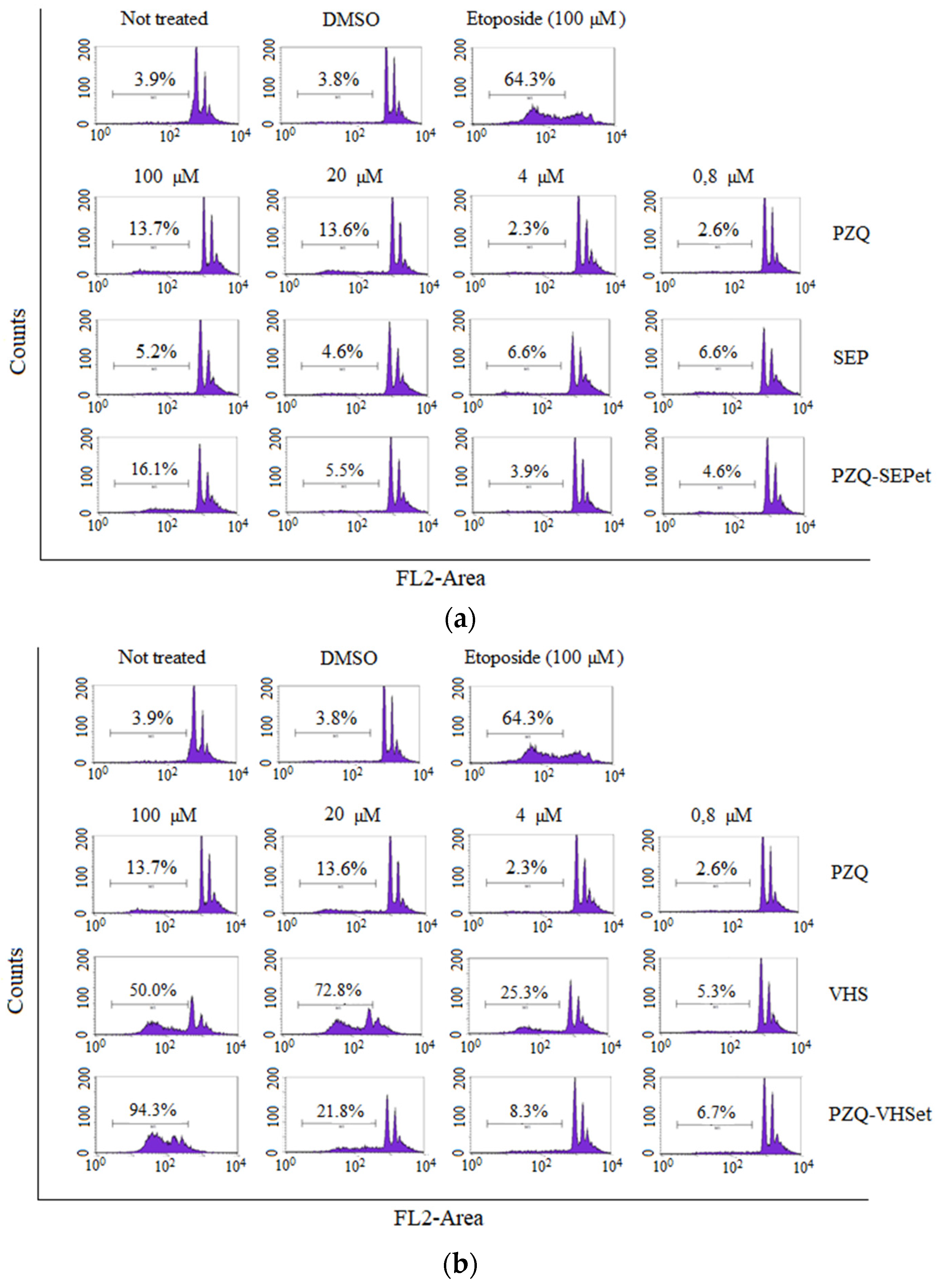
3.7. Cell Cycle Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- World Health Organization (WTO). Available online: https://www.who.int/neglected_diseases/en/ (accessed on 1 June 2020).
- World Health Organization (WTO). Available online: https://www.who.int/news-room/fact-sheets/detail/schistosomiasis (accessed on 1 June 2020).
- Chitsulo, L.; Engels, D.; Montresor, A.; Savioli, L. The global status of schistosomiasis and its control. Acta Trop. 2000, 77, 41–51. [Google Scholar] [CrossRef] [Green Version]
- Steinmann, P.; Keiser, J.; Bos, R.; Tanner, M.; Utzinger, J. Schistosomiasis and water resources development: Systematic review, meta-analysis, and estimates of people at risk. Lancet Infect. Dis. 2006, 6, 411–425. [Google Scholar] [CrossRef]
- Wang, W.; Wang, L.; Liang, Y.-S. Susceptibility or resistance of praziquantel in human schistosomiasis: A review. Parasitol. Res. 2012, 111, 1871–1877. [Google Scholar] [CrossRef]
- Andrews, P. Praziquantel: Mechanisms of anti-schistosomal activity. Pharmacol. Ther. 1985, 29, 129–156. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, X.; Wang, J.K.; Ching, C.B. Structural characterization and enantioseparation of the chiral compound praziquantel. J. Pharm. Sci. 2004, 93, 3039–3046. [Google Scholar] [CrossRef] [PubMed]
- Borrego-Sánchez, A.; Viseras, C.; Aguzzi, C.; Sainz-Díaz, C.I. Molecular and crystal structure of praziquantel. Spectroscopic properties and crystal polymorphism. Eur. J. Pharm. Sci. 2016, 92, 266–275. [Google Scholar] [CrossRef]
- U.S. Food and Drug Administration (FDA). Available online: https://www.fda.gov/AboutFDA/CentersOffices/OfficeofMedicalProductsandTobacco/CDER/ucm128219.htm (accessed on 1 June 2020).
- Amidon, G.L.; Lennernäs, H.; Shah, V.P.; Crison, J.R. A theoretical basis for a biopharmaceutic drug classification: The correlation of in vitro drug product dissolution and in vivo bioavailability. Pharm. Res. 1995, 12, 413–420. [Google Scholar] [CrossRef] [Green Version]
- González-Esquivel, D.; Rivera, J.; Castro, N.; Yepez-Mulia, L.; Helgi, J.C. In vitro characterization of some biopharmaceutical properties of praziquantel. Int. J. Pharm. 2005, 295, 93–99. [Google Scholar] [CrossRef]
- Bergaya, F.; Lagaly, G. General introduction: Clays, clay minerals, and clay science. In Developments in Clay Science; Handbook of Clay Science, Elsevier: Amsterdam, The Netherlands, 2006; Volume 1, pp. 1–18. [Google Scholar] [CrossRef]
- Guggenheim, S.; Adams, J.M.; Bain, D.C.; Bergaya, F.; Brigatti, M.F.; Drits, V.A.; Formoso, M.L.L.; Galán, E.; Kogure, T.; Stanjek, H. Summary of recommendations of nomenclature committees relevant to clay mineralogy: Report of the Association Internationale Pour l’Etude des Argiles (AIPEA) nomenclature committee for 2006. Clays Clay Miner. 2006, 54, 761–772. [Google Scholar] [CrossRef]
- López-Galindo, A.; Viseras, C. Pharmaceutical and Cosmetic Applications of Clays. Interface Sci. Technol. 2004, 1, 267–289. [Google Scholar] [CrossRef]
- Aguzzi, C.; Viseras, C.; Cerezo, P.; Rossi, S.; Ferrari, F.; López-Galindo, A.; Caramella, C. Influence of dispersion conditions of two pharmaceutical grade clays on their interaction with some tetracyclines. Appl. Clay Sci. 2005, 30, 79–86. [Google Scholar] [CrossRef]
- Aguzzi, C.; Cerezo, P.; Viseras, C.; Caramella, C. Use of clays as drug delivery systems: Possibilities and limitations. Appl. Clay Sci. 2007, 36, 1–3. [Google Scholar] [CrossRef]
- Viseras, C.; Cerezo, P.; Sánchez, R.; Salcedo, I.; Aguzzi, C. Current challenges in clay minerals for drug delivery. Appl. Clay Sci. 2010, 48, 291–295. [Google Scholar] [CrossRef]
- Borrego-Sánchez, A.; Carazo, E.; Albertini, B.; Passerini, N.; Perissutti, B.; Cerezo, P.; Viseras, C.; Hernández-Laguna, A.; Aguzzi, C.; Sainz-Díaz, C.I. Conformational polymorphic changes in the crystal structure of the chiral antiparasitic drug praziquantel and interactions with calcium carbonate. Eur. J. Pharm. Biopharm. 2018, 132, 180–191. [Google Scholar] [PubMed]
- Borrego-Sánchez, A.; Sánchez-Espejo, R.; Albertini, B.; Passerini, N.; Cerezo, P.; Viseras, C.; Sainz-Díaz, C.I. Ground calcium carbonate as a low cost and biosafety excipient for solubility and dissolution improvement of praziquantel. Pharmaceutics 2019, 11, 533. [Google Scholar]
- Trofimov, A.D.; Ivanova, A.A.; Zyuzin, M.V.; Timin, A.S. Porous inorganic carriers based on silica, calcium carbonate and calcium phosphate for controlled/modulated drug delivery: Fresh outlook and future perspectives. Pharmaceutics 2018, 10, 167. [Google Scholar]
- Rodrigues, S.G.; Chaves, I.S.; Melo, N.F.S.; de Jesus, M.B.; Fraceto, L.F.; Fernandes, S.A.; de Paula, E.; de Freitas, M.P.; Pinto, L.M.A. Computational analysis and physico-chemical characterization of an inclusion compound between praziquantel and methyl-β-cyclodextrin for use as an alternative in the treatment of schistosomiasis. J. Incl. Phenom. Macrocycl. Chem. 2010, 70, 19–28. [Google Scholar] [CrossRef]
- Maragos, S.; Archontaki, H.; Macheras, P.; Valsami, G. Effect of cyclodextrin complexation on the aqueous solubility and solubility/dose ratio of praziquantel. AAPS Pharm. Sci. Tech. 2009, 10, 1444. [Google Scholar]
- Becket, G.; Schep, L.J.; Tan, M.Y. Improvement of the in vitro dissolution of praziquantel by complexation with α-, β-, and γ-cyclodextrins. Int. J. Pharm. 1999, 179, 65–71. [Google Scholar]
- De la Torre, P.; Torrado, S.; Torrado, S. Preparation, dissolution and characterization of Praziquantel solid dispersions. Chem. Pharm. Bull. 1999, 47, 1629–1633. [Google Scholar] [CrossRef] [Green Version]
- Costa, E.D.; Priotti, J.; Orlandi, S.; Leonardi, D.; Lamas, M.C.; Nunes, T.G.; Diogo, H.P.; Salomon, C.J.; Ferreira, M.J. Unexpected solvent impact in the crystallinity of praziquantel/poly(vinylpyrrolidone) formulations. A solubility, DSC and solid-state NMR study. Int. J. Pharm. 2016, 511, 983–993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Passerini, N.; Albertini, B.; Perissutti, B.; Rodriguez, L. Evaluation of melt granulation and ultrasonic spray congealing as techniques to enhance the dissolution of praziquantel. Int. J. Pharm. 2006, 318, 92–102. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wang, T.; Ding, W.; Dong, C.; Wang, X.; Chen, J.; Li, Y. Dissolution and oral bioavailability enhancement of praziquantel by solid dispersions. Drug Deliv. Transl. Res. 2018, 8, 580–590. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Lei, L.; Guo, S. In vitro and in vivo evaluation of praziquantel loaded implants based on PEG/PCL blends. Int. J. Pharm. 2010, 387, 129–138. [Google Scholar] [CrossRef]
- Chaud, M.V.; Lima, A.C.; Vila, M.M.D.C.; Paganelli, M.O.; Paula, F.C.; Pedreiro, L.N.; Gremião, M.P.D. Development and evaluation of praziquantel solid dispersions in sodium starch glycolate. Trop. J. Pharm. Res. 2013, 12, 163–168. [Google Scholar] [CrossRef] [Green Version]
- Mourao, S.C.; Costa, P.I.; Salgado, H.R.N.; Gremiao, M.P.D. Improvement of antischistosomal activity of praziquantel by incorporation into phosphatidylcholine-containing liposomes. Int. J. Pharm. 2005, 295, 157–162. [Google Scholar] [CrossRef]
- Timóteo, T.R.R.; Melo, C.G.; Danda, L.J.A.; Silva, L.C.P.B.B.; Fontes, D.A.F.; Silva, P.C.D.; Aguilera, C.S.B.; Siqueira, L.P.; Rolim, L.A.; Neto, P.J.R. Layered double hydroxides of CaAl: A promising drug delivery system for increased dissolution rate and thermal stability of praziquantel. Appl. Clay Sci. 2019, 180, 105197. [Google Scholar] [CrossRef]
- El-Feky, G.S.; Mohamed, W.S.; Nasr, H.E.; El-Lakkany, N.M.; Seif el-Din, S.H.; Botros, S.S. Praziquantel in a clay nanoformulation shows more bioavailability and higher efficacy against murine schistosoma mansoni infection. antimicrob. Agents Chemother. 2015, 59, 3501–3508. [Google Scholar]
- Borrego-Sánchez, A.; Carazo, E.; Aguzzi, C.; Viseras, C.; Sainz-Díaz, C.I. Biopharmaceutical improvement of praziquantel by interaction with montmorillonite and sepiolite. Appl. Clay Sci. 2018, 160, 173–179. [Google Scholar] [CrossRef]
- Sainz-Díaz, C.I.; Borrego-Sánchez, A.; Viseras, C.; Aguzzi, C. Method for Preparing a Nano-Structured Material of Praziquantel and a Silicate, Material Obtained and Use as an Antiparasitic. Patent WO/2020/012044, 15 January 2019. [Google Scholar]
- Lauroba, J.; Doménech, J. La liberación como factor limitativo de la absorción gastrointestinal. In Biofarmacia y Farmacocinética; Lauroba, J., Doménech, J., Plá, J.M., Eds.; Síntesis: Madrid, Spain, 1998; Volume 2, pp. 241–274. [Google Scholar]
- Costa, P.; Souza Lobo, J.M. Modeling and comparison of dissolution profiles. Eur. J. Pharm. Sci. 2001, 13, 123–133. [Google Scholar] [CrossRef]
- Siepman, J.; Göpferich, A. Mathematical modeling of bioerodible, polymeric drug delivery systems. Adv. Drug Deliv. Rev. 2001, 48, 229–247. [Google Scholar] [CrossRef]
- Cheikh, D.; García-Villén, F.; Majdoub, H.; Viseras, C.; Zayania, M.B. Chitosan/beidellite nanocomposite as diclofenac carrier. Int. J. Biol. Macromol. 2019, 126, 44–53. [Google Scholar] [CrossRef] [PubMed]
- Akaike, H. A new look at the statistical model identification. IEEE Trans. Automat. Contr. 1974, 19, 716–723. [Google Scholar]
- Gong, J.P.; Traganos, F.; Darzynkiewicz, Z. A selective procedure for DNA extraction from apoptotic cells applicable for gel electrophoresis and flow cytometry. Anal. Biochem. 1994, 218, 314–319. [Google Scholar] [CrossRef] [PubMed]
- El-Subbagh, H.I.; Al-Badr, A.A. Praziquantel. In Analytical Profiles of Drug Substances and Excipients; Florey, K., Ed.; Academic Press Inc.: New York, NY, USA, 1998; Volume 25, pp. 463–500. [Google Scholar]
- Meteleva, E.S.; Chistyachenko, Y.S.; Suntsova, L.P.; Khvostov, M.V.; Polyakov, N.E.; Selyutina, O.Y.; Tolstikova, T.G.; Frolova, T.S.; Mordvinov, V.A.; Dushkin, A.V.; et al. Disodium salt of glycyrrhizic acid–A novel supramolecular delivery system for anthelmintic drug praziquantel. J. Drug Deliv. Sci. Technol. 2019, 50, 66–77. [Google Scholar] [CrossRef]
- Borrego-Sánchez, A.; Hernández-Laguna, A.; Sainz-Díaz, C.I. Molecular modeling and infrared and Raman spectroscopy of the crystal structure of the chiral antiparasitic drug Praziquantel. J. Mol. Model. 2017, 23, 106. [Google Scholar] [CrossRef]
- Borrego-Sánchez, A.; Viseras, C.; Sainz-Díaz, C.I. Molecular interactions of praziquantel drug with nanosurfaces of sepiolite and montmorillonite. Appl. Clay Sci. 2020, 197, 105774. [Google Scholar]
- Viseras, C.; López-Galindo, A. Pharmaceutical applications of some Spanish clays (sepiolite, palygorskite, bentonite): Some preformulation studies. Appl. Clay Sci. 1999, 14, 69–82. [Google Scholar] [CrossRef]
- Frost, R.L.; Locos, O.B.; Ruan, H.; Kloprogge, J.T. Near-infrared and mid-infrared spectroscopic study of sepiolites and palygorskites. Vib. Spectrosc. 2001, 27, 1–13. [Google Scholar] [CrossRef]
- Ortega-Castro, J.; Hernández-Haro, N.; Muñoz-Santiburcio, D.; Hernández-Laguna, A.; Sainz-Díaz, C.I. Crystal structure and hydroxyl group vibrational frequencies of phyllosilicates by DFT methods. Theochem. J. Mol. Struct. 2009, 912, 82–87. [Google Scholar] [CrossRef]
- Langenbucher, F. Letters to the editor: Linearization of dissolution rate curves by the Weibull distribution. J. Pharm. Pharmacol. 1972, 24, 979–981. [Google Scholar] [CrossRef] [PubMed]
- Weibul, W. Wide applicability. J. Appl. Mech. 1951, 103, 293–297. [Google Scholar]
- Hixson, A.W.; Crowell, J.H. Dependence of reaction velocity surface and agitation. Ind. Eng. Chem. 1931, 23, 923–931. [Google Scholar] [CrossRef]
- Neibergall, P.J.; Milosovich, G.; Goyan, J.E. Dissolution rate studies. II. Dissolution of particles under conditions of rapid agitation. J. Pharm. Sci. 1963, 52, 236–241. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HCl 0.001 M Medium, pH = 3 | SIF Medium, pH = 6.8 | |||
|---|---|---|---|---|
| Solubility (mg/mL) | Increase (%) | Solubility (mg/mL) | Increase (%) | |
| PZQ | 0.50 | 0.45 | ||
| PZQ–SEPet | 0.71 | 42 | 0.61 | 36 |
| PZQ–VHSet | 0.74 | 48 | 0.65 | 44 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Borrego-Sánchez, A.; Sánchez-Espejo, R.; García-Villén, F.; Viseras, C.; Sainz-Díaz, C.I. Praziquantel–Clays as Accelerated Release Systems to Enhance the Low Solubility of the Drug. Pharmaceutics 2020, 12, 914. https://doi.org/10.3390/pharmaceutics12100914
Borrego-Sánchez A, Sánchez-Espejo R, García-Villén F, Viseras C, Sainz-Díaz CI. Praziquantel–Clays as Accelerated Release Systems to Enhance the Low Solubility of the Drug. Pharmaceutics. 2020; 12(10):914. https://doi.org/10.3390/pharmaceutics12100914
Chicago/Turabian StyleBorrego-Sánchez, Ana, Rita Sánchez-Espejo, Fátima García-Villén, César Viseras, and C. Ignacio Sainz-Díaz. 2020. "Praziquantel–Clays as Accelerated Release Systems to Enhance the Low Solubility of the Drug" Pharmaceutics 12, no. 10: 914. https://doi.org/10.3390/pharmaceutics12100914