Direct Quantification of Drug Loading Content in Polymeric Nanoparticles by Infrared Spectroscopy



Abstract
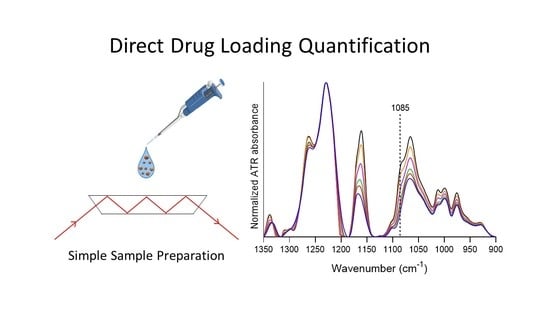
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Preparation of Stock Nanoparticle Dispersion and Drug Solution
2.3. Calibration Samples for NAR in SFN
2.4. NAR-Loaded SFN Samples
2.5. Spectra Acquisition and Data Pretreatment
3. Results and Discussion
3.1. Specificity of ATR-FTIR Spectroscopy for Identifying Drugs in SFN
3.2. Identification of the Best Marker Band for NAR in SFNNAR Samples
3.3. Calibration Curve
3.4. Parameters for Method Validation
3.4.1. Range and Linearity of the Calibration Curve
3.4.2. Robustness of the Method
3.4.3. Limit of Detection, Limit of Quantification and Reproducibility (Precision) of the Method
3.4.4. Accuracy of the Method
3.5. Drug Quantification in NAR-Loaded SFN
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bobo, D.; Robinson, K.J.; Islam, J.; Thurecht, K.J.; Corrie, S.R. Nanoparticle-Based Medicines: A Review of FDA-Approved Materials and Clinical Trials to Date. Pharm. Res. 2016, 33, 2373–2387. [Google Scholar] [CrossRef] [PubMed]
- Kalepu, S.; Nekkanti, V. Insoluble drug delivery strategies: Review of recent advances and business prospects. Acta Pharm. Sin. B 2015, 5, 442–453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karimi, M.; Ghasemi, A.; Sahandi Zangabad, P.; Rahighi, R.; Moosavi Basri, S.M.; Mirshekari, H.; Amiri, M.; Shafaei Pishabad, Z.; Aslani, A.; Bozorgomid, M.; et al. Smart micro/nanoparticles in stimulus-responsive drug/gene delivery systems. Chem. Soc. Rev. 2016, 45, 1457–1501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montalbán, M.G.; Carissimi, G.; Lozano-Pérez, A.A.; Cenis, J.L.; Coburn, J.M.; Kaplan, D.L.; Víllora, G. Biopolymeric Nanoparticle Synthesis in Ionic Liquids. In Recent Advances in Ionic Liquids; IntechOpen: London, UK, 2018; pp. 3–26. [Google Scholar]
- Matsumura, Y.; Maeda, H. A New Concept for Macromolecular Therapeutics in Cancer Chemotherapy: Mechanism of Tumoritropic Accumulation of Proteins and the Antitumor Agent Smancs. Cancer Res. 1986, 46, 6387–6392. [Google Scholar] [PubMed]
- Fang, J.; Islam, R.; Islam, W.; Yin, H.; Subr, V.; Etrych, T.; Ulbrich, K.; Maeda, H. Augmentation of EPR Effect and Efficacy of Anticancer Nanomedicine by Carbon Monoxide Generating Agents. Pharmaceutics 2019, 11, 343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elzoghby, A.O.; Samy, W.M.; Elgindy, N.A. Albumin-based nanoparticles as potential controlled release drug delivery systems. J. Control. Release 2012, 157, 168–182. [Google Scholar] [CrossRef] [PubMed]
- Barenholz, Y. Doxil®-The first FDA-approved nano-drug: Lessons learned. J. Control. Release 2012, 160, 117–134. [Google Scholar] [CrossRef]
- Lim, S.B.; Banerjee, A.; Önyüksel, H. Improvement of drug safety by the use of lipid-based nanocarriers. J. Control. Release 2012, 163, 34–45. [Google Scholar] [CrossRef]
- Ding, B.; Wahid, M.A.; Wang, Z.; Xie, C.; Thakkar, A.; Prabhu, S.; Wang, J. Triptolide and celastrol loaded silk fibroin nanoparticles show synergistic effect against human pancreatic cancer cells. Nanoscale 2017, 9, 11739–11753. [Google Scholar] [CrossRef]
- Zhao, Z.; Li, Y.; Xie, M. Bin Silk fibroin-based nanoparticles for drug delivery. Int. J. Mol. Sci. 2015, 16, 4880–4903. [Google Scholar] [CrossRef] [Green Version]
- Mottaghitalab, F.; Farokhi, M.; Shokrgozar, M.A.; Atyabi, F.; Hosseinkhani, H. Silk fibroin nanoparticle as a novel drug delivery system. J. Control. Release 2015, 206, 161–176. [Google Scholar] [CrossRef] [PubMed]
- Montalbán, M.; Coburn, J.; Lozano-Pérez, A.; Cenis, J.; Víllora, G.; Kaplan, D. Production of Curcumin-Loaded Silk Fibroin Nanoparticles for Cancer Therapy. Nanomaterials 2018, 8, 126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raveendran, P.; Fu, J.; Wallen, S.L. Completely “Green” Synthesis and Stabilization of Metal Nanoparticles. J. Am. Chem. Soc. 2003, 125, 13940–13941. [Google Scholar] [CrossRef]
- Lozano-Pérez, A.A.; Rivero, H.C.; del Carmen Párez Hernández, M.; Pagán, A.; Montalbán, M.G.; Víllora, G.; Cénis, J.L. Silk fibroin nanoparticles: Efficient vehicles for the natural antioxidant quercetin. Int. J. Pharm. 2017, 518, 11–19. [Google Scholar]
- Fuster, M.G.; Carissimi, G.; Montalbán, M.G.; Víllora, G. Improving Anticancer Therapy with Naringenin-Loaded Silk Fibroin Nanoparticles. Nanomaterials 2020, 10, 718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathur, A.B.; Gupta, V. Silk fibroin-derived nanoparticles for biomedical applications. Nanomedicine 2010, 5, 807–820. [Google Scholar] [CrossRef]
- Tian, Y.; Jiang, X.; Chen, X.; Shao, Z.; Yang, W. Doxorubicin-loaded magnetic silk fibroin nanoparticles for targeted therapy of multidrug-resistant cancer. Adv. Mater. 2014, 26, 7393–7398. [Google Scholar] [CrossRef]
- Amgoth, C.; Dharmapuri, G. Synthesis and Characterization of Polymeric Nanoparticles and Capsules as Payload for Anticancer Drugs and Nanomedicines. Mater. Today Proc. 2016, 3, 3833–3837. [Google Scholar] [CrossRef]
- Bian, X.; Wu, P.; Sha, H.; Qian, H.; Wang, Q.; Cheng, L.; Yang, Y.; Yang, M.; Liu, B. Anti-EGFR-iRGD recombinant protein conjugated silk fibroin nanoparticles for enhanced tumor targeting and antitumor efficiency. Onco. Targets. Ther. 2016, 9, 3153–3162. [Google Scholar]
- Kumari, A.; Yadav, S.K.; Pakade, Y.B.; Singh, B.; Yadav, S.C. Development of biodegradable nanoparticles for delivery of quercetin. Colloids Surfaces B Biointerfaces 2010, 80, 184–192. [Google Scholar] [CrossRef]
- Trucillo, P.; Campardelli, R.; Reverchon, E. Supercritical CO2 assisted liposomes formation: Optimization of the lipidic layer for an efficient hydrophilic drug loading. J. CO2 Util. 2017, 18, 181–188. [Google Scholar] [CrossRef]
- Seib, F.P.; Jones, G.T.; Rnjak-Kovacina, J.; Lin, Y.; Kaplan, D.L.; Philipp Seib, F.; Jones, G.T.; Rnjak-Kovacina, J.; Lin, Y.; Kaplan, D.L. pH-Dependent anticancer drug release from silk nanoparticles. Adv. Healthc. Mater. 2014, 2, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subia, B.; Kundu, S.C. Drug loading and release on tumor cells using silk fibroin–albumin nanoparticles as carriers. Nanotechnology 2013, 24, 035103. [Google Scholar] [CrossRef] [PubMed]
- Wongpinyochit, T.; Uhlmann, P.; Urquhart, A.J.; Seib, F.P. PEGylated Silk Nanoparticles for Anticancer Drug Delivery. Biomacromolecules 2015, 16, 3712–3722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suktham, K.; Koobkokkruad, T.; Wutikhun, T.; Surassmo, S. Efficiency of resveratrol-loaded sericin nanoparticles: Promising bionanocarriers for drug delivery. Int. J. Pharm. 2018, 537, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Pham, D.T.; Saelim, N.; Tiyaboonchai, W. Paclitaxel loaded EDC-crosslinked fibroin nanoparticles: A potential approach for colon cancer treatment. Drug Deliv. Transl. Res. 2020, 10, 413–424. [Google Scholar] [CrossRef]
- Wu, J.; Wang, J.; Zhang, J.; Zheng, Z.; Kaplan, D.L.; Li, G.; Wang, X. Oral Delivery of Curcumin Using Silk Nano- and Microparticles. ACS Biomater. Sci. Eng. 2018, 4, 3885–3894. [Google Scholar] [CrossRef]
- Perteghella, S.; Sottani, C.; Coccè, V.; Negri, S.; Cavicchini, L.; Alessandri, G.; Cottica, D.; Torre, M.L.; Grignani, E.; Pessina, A. Pessina Paclitaxel-Loaded Silk Fibroin Nanoparticles: Method Validation by UHPLC-MS/MS to Assess an Exogenous Approach to Load Cytotoxic Drugs. Pharmaceutics 2019, 11, 285. [Google Scholar] [CrossRef] [Green Version]
- Wallace, S.J.; Li, J.; Nation, R.L.; Boyd, B.J. Drug release from nanomedicines: Selection of appropriate encapsulation and release methodology. Drug Deliv. Transl. Res. 2012, 2, 284–292. [Google Scholar] [CrossRef] [Green Version]
- Lv, Y.; He, H.; Qi, J.; Lu, Y.; Zhao, W.; Dong, X.; Wu, W. Visual validation of the measurement of entrapment efficiency of drug nanocarriers. Int. J. Pharm. 2018, 547, 395–403. [Google Scholar] [CrossRef]
- Khanmohammadi, M.; Mobedi, E.; Garmarudi, A.B.; Mobedi, H.; Kargosha, K. Simultaneous determination of levodopa and carbidopa in levodopa-carbidopa tablets by ATR-FTIR spectrometry. Pharm. Dev. Technol. 2007, 12, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Koleva, B.B.; Kolev, T.M.; Tsalev, D.L.; Spiteller, M. Determination of phenacetin and salophen analgetics in solid binary mixtures with caffeine by infrared linear dichroic and Raman spectroscopy. J. Pharm. Biomed. Anal. 2008, 46, 267–273. [Google Scholar] [CrossRef] [PubMed]
- de la Asunción-Nadal, V.; Armenta, S.; Garrigues, S.; de la Guardia, M. Identification and determination of synthetic cannabinoids in herbal products by dry film attenuated total reflectance-infrared spectroscopy. Talanta 2017, 167, 344–351. [Google Scholar] [CrossRef] [PubMed]
- Wartewig, S.; Neubert, R.H.H. Pharmaceutical applications of Mid-IR and Raman spectroscopy. Adv. Drug Deliv. Rev. 2005, 57, 1144–1170. [Google Scholar] [CrossRef]
- Kalinkova, G.N. Infrared spectroscopy in pharmacy. Vib. Spectrosc. 1999, 19, 307–320. [Google Scholar] [CrossRef]
- Bunaciu, A.A.; Aboul-Enein, H.Y.; Fleschin, S. Application of fourier transform infrared spectrophotometry in pharmaceutical drugs analysis. Appl. Spectrosc. Rev. 2010, 45, 206–219. [Google Scholar] [CrossRef]
- Pandey, R.P.; Gurung, R.B.; Sohng, J.K. Dietary sources, bioavailability and biological activities of naringenin and its derivatives. In Apigenin and Naringenin Natural Sources, Pharmacology and Role in Cancer Prevention; Nova Science Publishers: New York, NY, USA, 2015; pp. 151–172. [Google Scholar]
- Kawaii, S.; Tomono, Y.; Katase, E.; Ogawa, K.; Yano, M. Quantitation of flavonoids constituents in Citrus fruits. J. Agric. Food Chem. 1999, 47, 3565–3571. [Google Scholar] [CrossRef]
- Treml, J.; Šmejkal, K. Flavonoids as Potent Scavengers of Hydroxyl Radicals. Compr. Rev. Food Sci. Food Saf. 2016, 15, 720–738. [Google Scholar] [CrossRef]
- Bodet, C.; La, V.D.; Epifano, F.; Grenier, D. Naringenin has anti-inflammatory properties in macrophage and ex vivo human whole-blood models. J. Periodontal Res. 2008, 43, 400–407. [Google Scholar] [CrossRef]
- Liao, A.C.H.; Kuo, C.C.; Huang, Y.C.; Yeh, C.W.; Hseu, Y.C.; Liu, J.Y.; Hsu, L.S. Naringenin inhibits migration of bladder cancer cells through downregulation of AKT and MMP-2. Mol. Med. Rep. 2014, 10, 1531–1536. [Google Scholar] [CrossRef] [Green Version]
- Bao, L.; Liu, F.; Guo, H.; Li, Y.; Tan, B.; Zhang, W.; Peng, Y. hui Naringenin inhibits proliferation, migration, and invasion as well as induces apoptosis of gastric cancer SGC7901 cell line by downregulation of AKT pathway. Tumor Biol. 2016, 37, 11365–11374. [Google Scholar] [CrossRef] [PubMed]
- Gumushan Aktas, H.; Akgun, T. Naringenin inhibits prostate cancer metastasis by blocking voltage-gated sodium channels. Biomed. Pharmacother. 2018, 106, 770–775. [Google Scholar] [CrossRef] [PubMed]
- Rajamani, S.; Radhakrishnan, A.; Sengodan, T.; Thangavelu, S. Augmented anticancer activity of naringenin-loaded TPGS polymeric nanosuspension for drug resistive MCF-7 human breast cancer cells. Drug Dev. Ind. Pharm. 2018, 44, 1752–1761. [Google Scholar] [CrossRef] [PubMed]
- Lim, W.; Park, S.; Bazer, F.W.; Song, G. Naringenin-Induced Apoptotic Cell Death in Prostate Cancer Cells Is Mediated via the PI3K/AKT and MAPK Signaling Pathways. J. Cell. Biochem. 2017, 118, 1118–1131. [Google Scholar] [CrossRef]
- Carissimi, G.; Baronio, C.M.; Montalbán, M.G.; Víllora, G.; Barth, A. On the Secondary Structure of Silk Fibroin Nanoparticles Obtained Using Ionic Liquids: An Infrared Spectroscopy Study. Polymers 2020, 12, 1294. [Google Scholar] [CrossRef]
- Boulet-Audet, M.; Buffeteau, T.; Boudreault, S.; Daugey, N.; Pézolet, M. Quantitative determination of band distortions in diamond attenuated total reflectance infrared spectra. J. Phys. Chem. B 2010, 114, 8255–8261. [Google Scholar] [CrossRef]
- Goormaghtigh, E.; Raussens, V.; Ruysschaert, J.M. Attenuated total reflection infrared spectroscopy of proteins and lipids in biological membranes. Biochim. Biophys. Acta-Rev. Biomembr. 1999, 1422, 105–185. [Google Scholar] [CrossRef]
- Hu, X.; Kaplan, D.; Cebe, P. Determining beta-sheet crystallinity in fibrous proteins by thermal analysis and infrared spectroscopy. Macromolecules 2006, 39, 6161–6170. [Google Scholar] [CrossRef]
- Kumar, S.; Singh, S.K. Fabrication and characterization of fibroin solution and nanoparticle from silk fibers of Bombyx mori. Part. Sci. Technol. 2017, 35, 304–313. [Google Scholar] [CrossRef]
- Carissimi, G.; Lozano-Pérez, A.A.; Montalbán, M.G.; Aznar-Cervantes, S.D.; Cenis, J.L.; Víllora, G. Revealing the Influence of the Degumming Process in the Properties of Silk Fibroin Nanoparticles. Polymers (Basel). 2019, 11, 2045. [Google Scholar] [CrossRef] [Green Version]
- Baranović, G.; Šegota, S. Infrared spectroscopy of flavones and flavonols. Reexamination of the hydroxyl and carbonyl vibrations in relation to the interactions of flavonoids with membrane lipids. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2018, 192, 473–486. [Google Scholar] [CrossRef] [PubMed]
- Machado, N.F.L.; Batista de Carvalho, L.A.E.; Otero, J.C.; Marques, M.P.M. A conformational study of hydroxyflavones by vibrational spectroscopy coupled to DFT calculations. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2013, 109, 116–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zar, J.H. Biostatistical analysis, 2nd ed.; Prentice-Hall: Englewood-Cliffs, NJ, USA, 1984. [Google Scholar]
- IUPAC. Compendium of Chemical Terminology; Nič, M., Jirát, J., Košata, B., Jenkins, A., McNaught, A., Eds.; IUPAC: Research Triangle Park, NC, USA, 2009. [Google Scholar]
- Thompson, M.; Ellison, S.L.R.; Wood, R. Harmonized guidelines for single-laboratory validation of methods of analysis (IUPAC Technical Report). Pure Appl. Chem. 2002, 74, 835–855. [Google Scholar] [CrossRef]
- Pandey, S.; Pandey, P.; Tiwari, G.; Tiwari, R.; Rai, A.K. FTIR spectroscopy: A tool for quantitative analysis of ciprofloxacin in tablets. Indian J. Pharm. Sci. 2012, 74, 86–90. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Fitting Parameters | UMU | SU | All Data Points |
|---|---|---|---|
| Mean ± σ | |||
| Best-fit values | |||
| Slope * | 0.030 ± 0.002 | 0.031 ± 0.001 | 0.029 |
| Y-intercept | 0.145 ± 0.009 | 0.146 ± 0.008 | 0.152 |
| Goodness of Fit | |||
| R2 ** | 0.978 ± 0.007 | 0.982 ± 0.019 | 0.973 |
| Quantity | Value |
|---|---|
| Average of absorbance at 1085 cm−1 (Ā) | 0.151 |
| Standard deviation (σ) | 0.003 |
| Reproducibility (%) | 2.0 |
| LOD of absorbance (ALOD) | 0.161 |
| LOQ of absorbance (ALOQ) | 0.181 |
| DLCLOD (%) | 0.3 |
| DLCLOQ (%) | 1.0 |
| Sample | Recovery (%) |
|---|---|
| 1 | 103% |
| 2 | 104% |
| 3 | 102% |
| 4 | 106% |
| Average | 104% |
| Standard deviation | 2% |
| Sample | DLC (%) | EE (%) |
|---|---|---|
| Mean ± σ (RSD) | ||
| SFNNAR-0.3 | 0.87 ± 0.07 (8.7%) | 41.0 ± 3.0 (7.3%) |
| SFNNAR-1.2 | 3.13 ± 0.08 (2.6%) | 38.0 ± 1.0 (2.6%) |
| SFNNAR-4.5 | 4.06 ± 0.06 (1.6%) | 13.2 ± 0.2 (1.5%) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carissimi, G.; Montalbán, M.G.; Víllora, G.; Barth, A. Direct Quantification of Drug Loading Content in Polymeric Nanoparticles by Infrared Spectroscopy. Pharmaceutics 2020, 12, 912. https://doi.org/10.3390/pharmaceutics12100912
Carissimi G, Montalbán MG, Víllora G, Barth A. Direct Quantification of Drug Loading Content in Polymeric Nanoparticles by Infrared Spectroscopy. Pharmaceutics. 2020; 12(10):912. https://doi.org/10.3390/pharmaceutics12100912
Chicago/Turabian StyleCarissimi, Guzmán, Mercedes G. Montalbán, Gloria Víllora, and Andreas Barth. 2020. "Direct Quantification of Drug Loading Content in Polymeric Nanoparticles by Infrared Spectroscopy" Pharmaceutics 12, no. 10: 912. https://doi.org/10.3390/pharmaceutics12100912