Comparison of In Vitro and In Vivo Results Using the GastroDuo and the Salivary Tracer Technique: Immediate Release Dosage Forms under Fasting Conditions

 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Dosage Forms Investigated
2.2.1. Immediate Release Tablets
2.2.2. Capsules
2.3. In Vitro Investigations
2.3.1. Compendial Dissolution
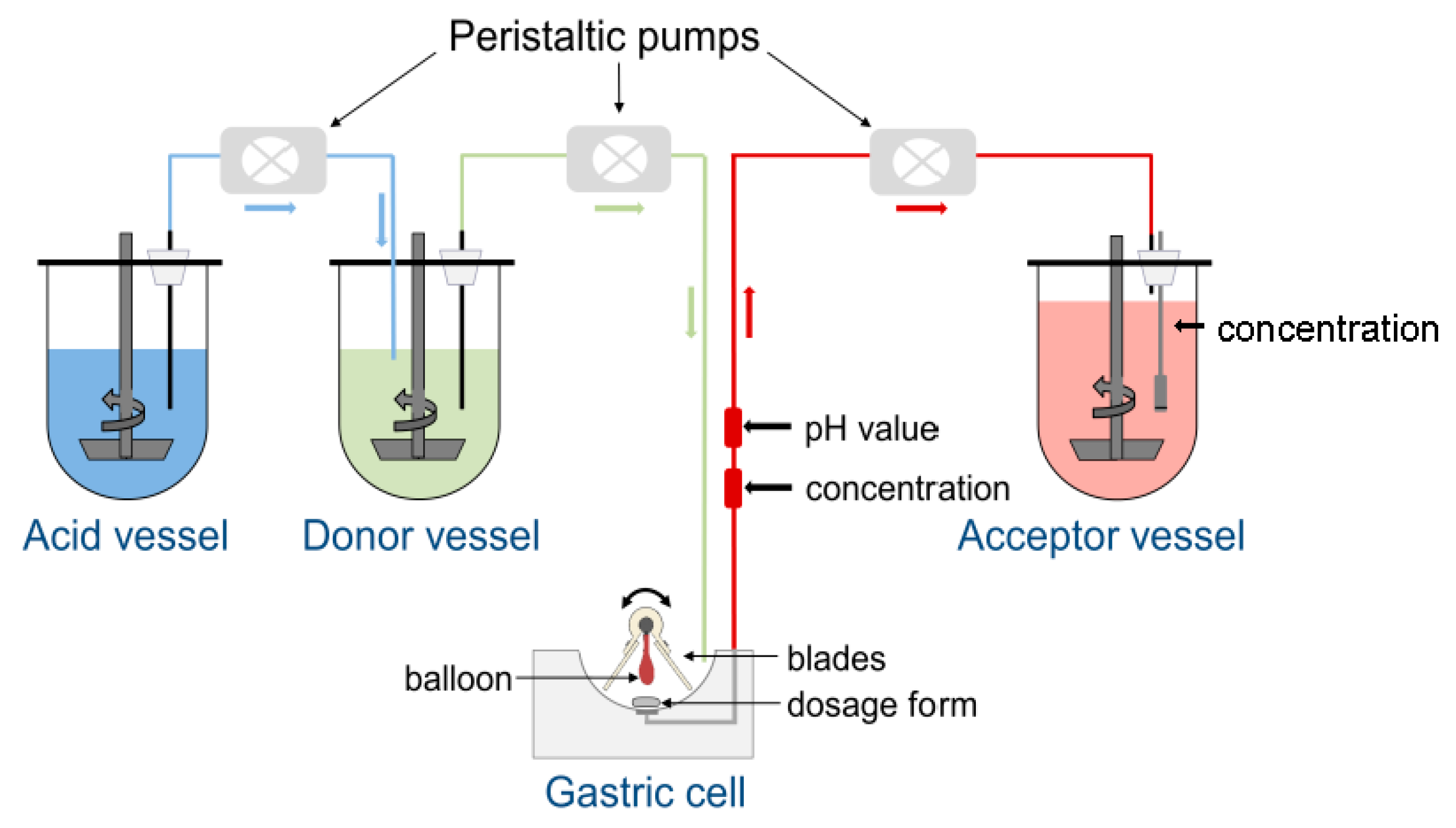
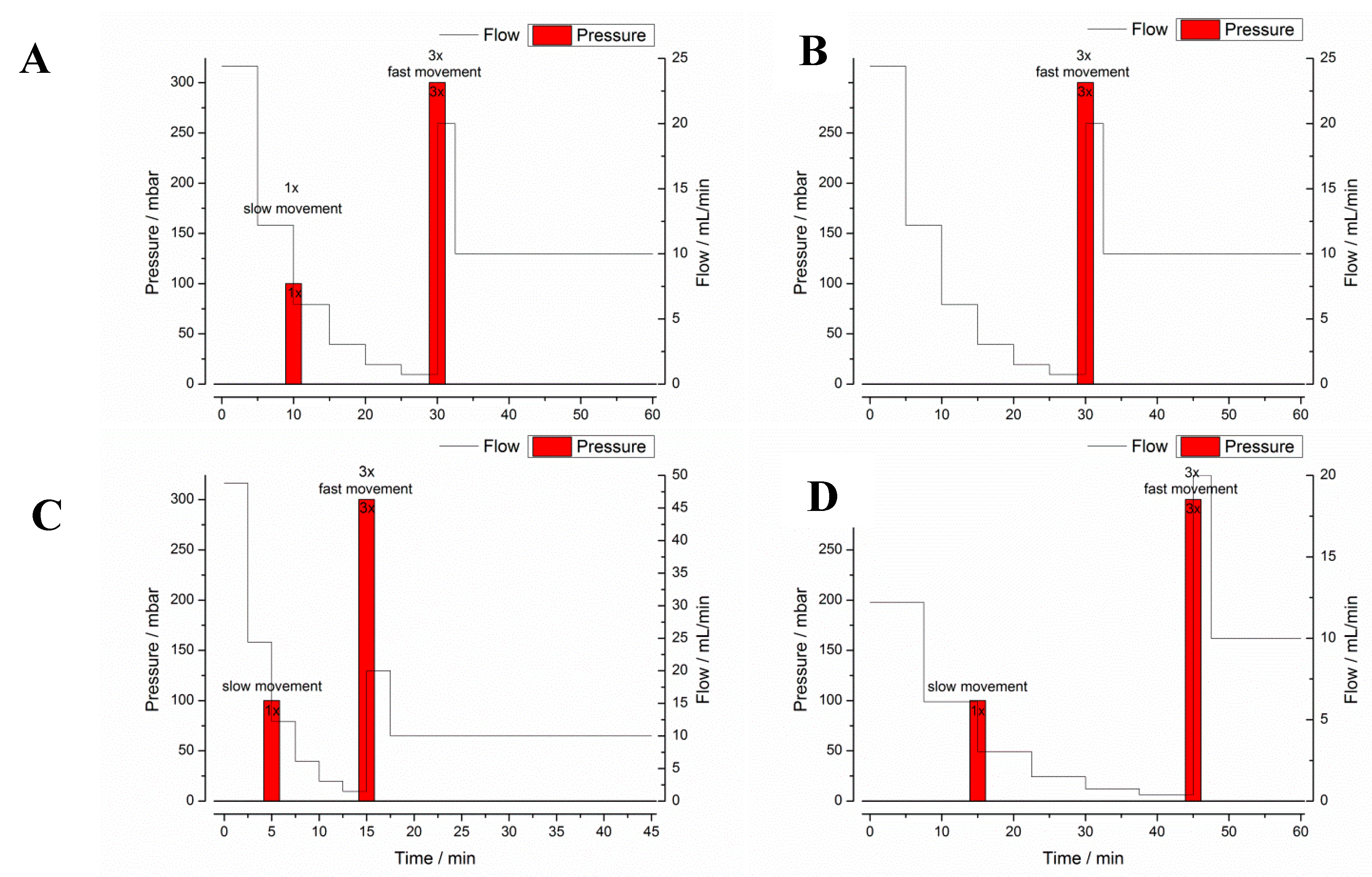
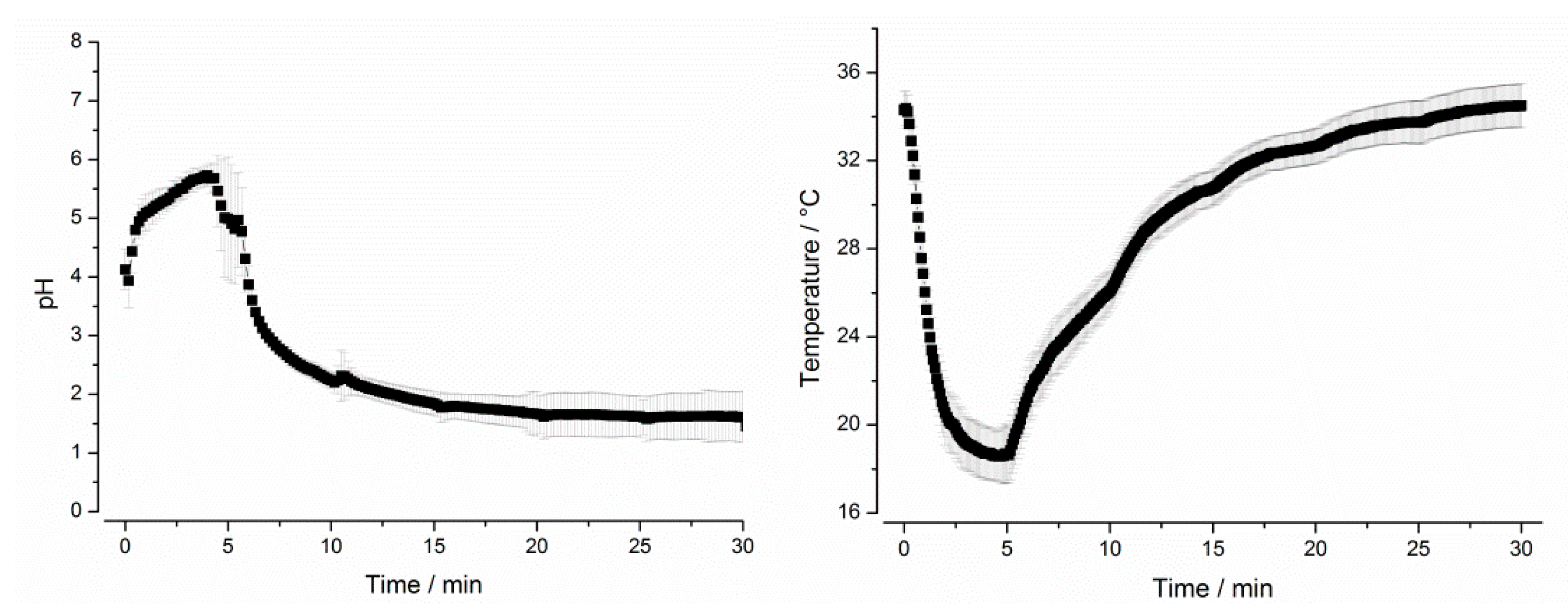
2.3.2. GastroDuo
2.4. Determination of In Vitro Disintegration Parameters
2.5. In Vivo Study with the Salivary Tracer Method
2.5.1. Study Design
2.5.2. Preparation and Analysis of Saliva Samples
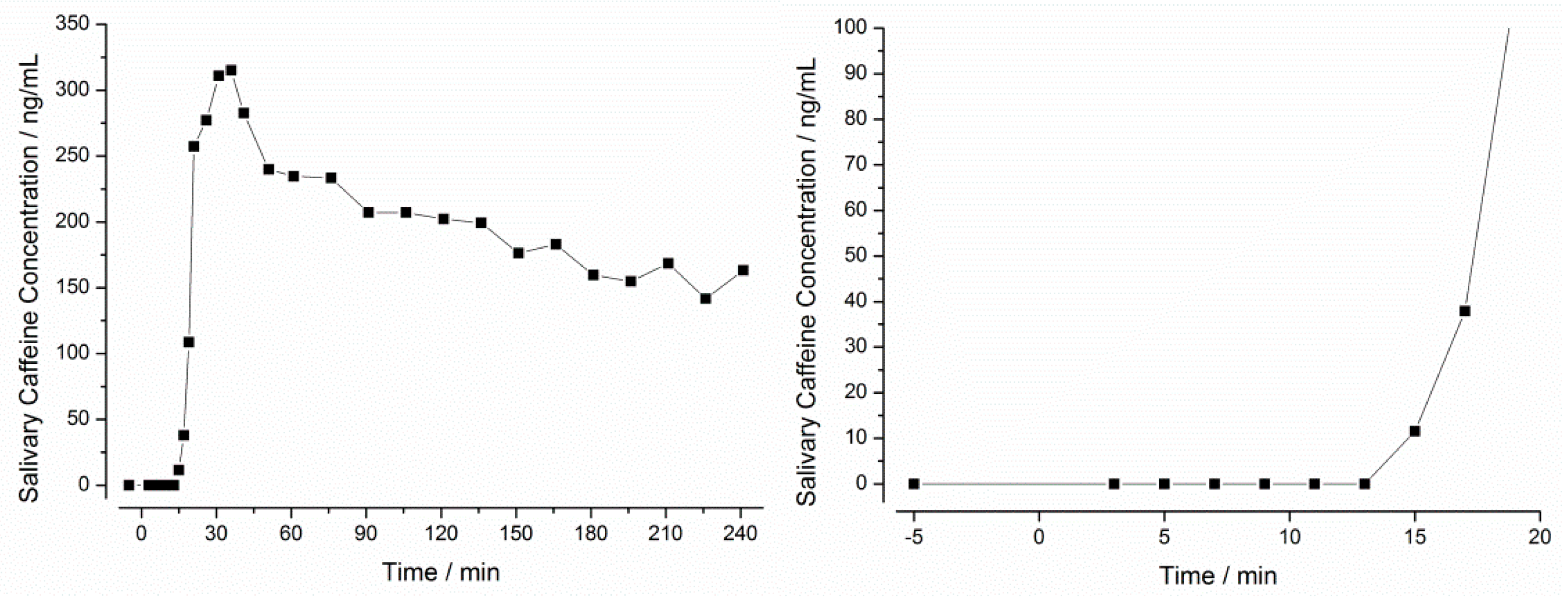
2.5.3. Determination of the In Vivo Disintegration Parameters
2.5.4. Statistical Comparison
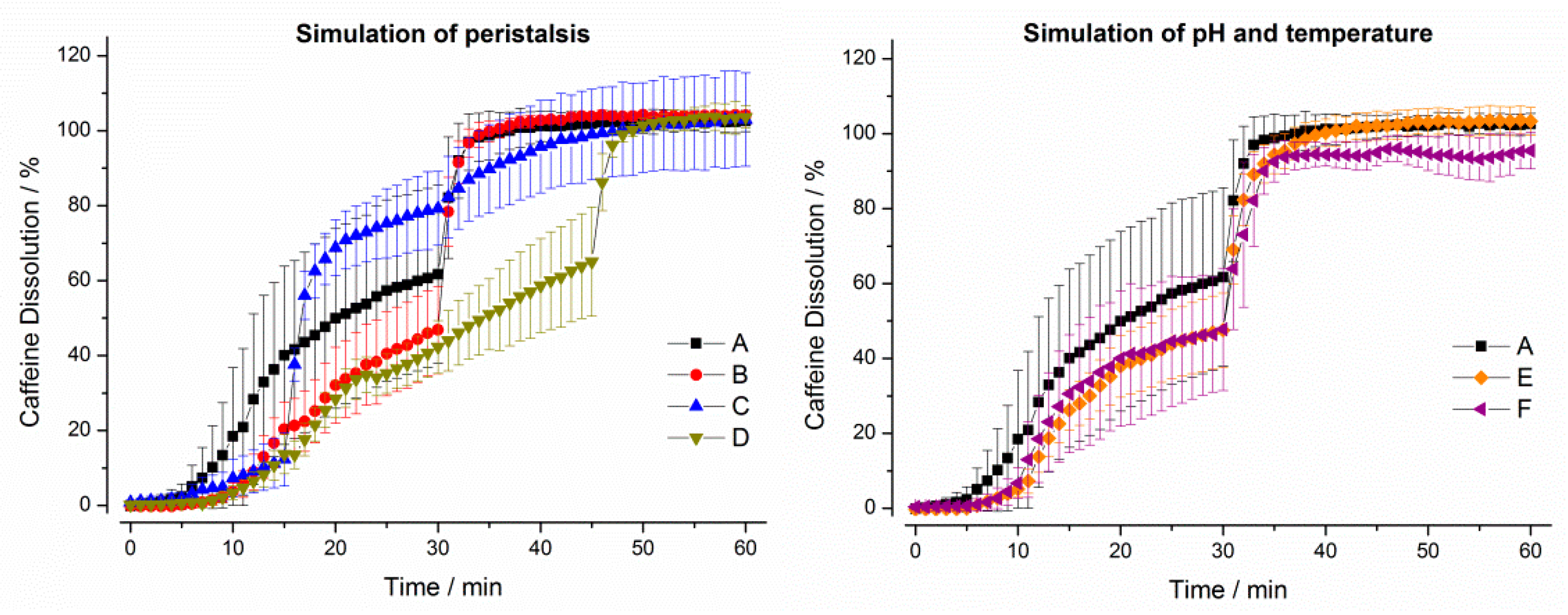
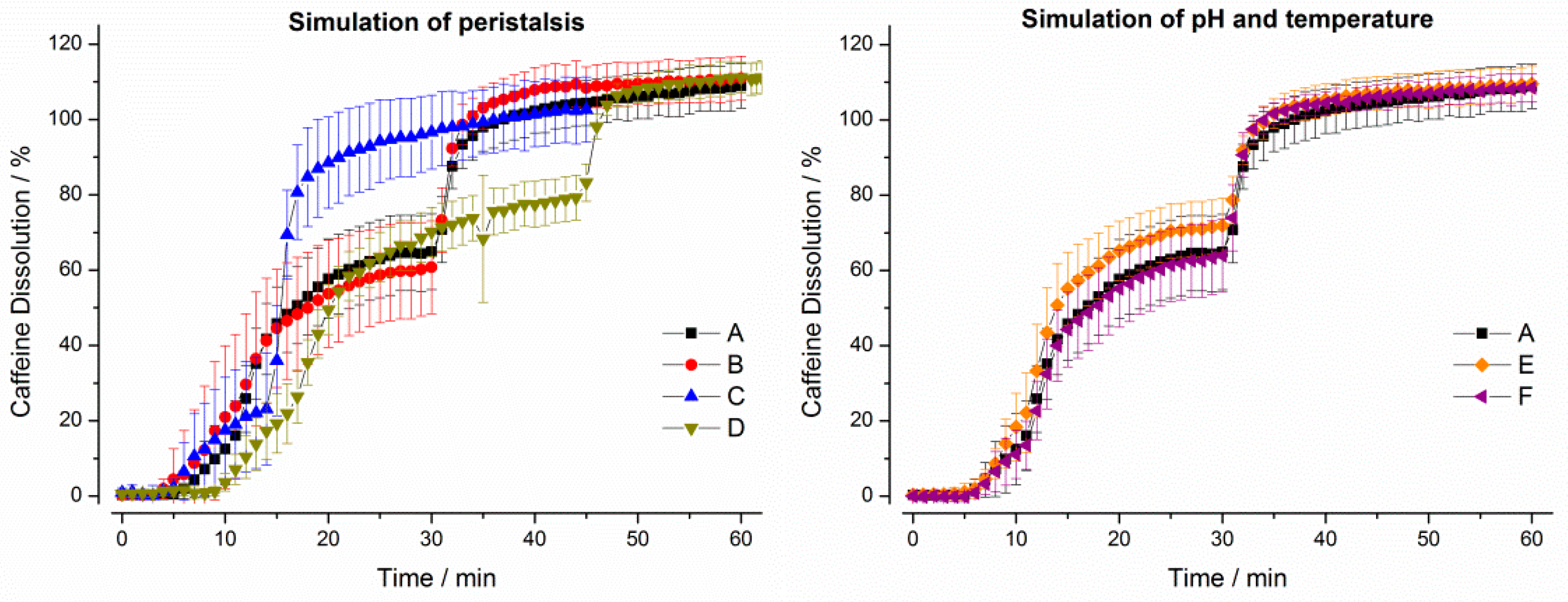
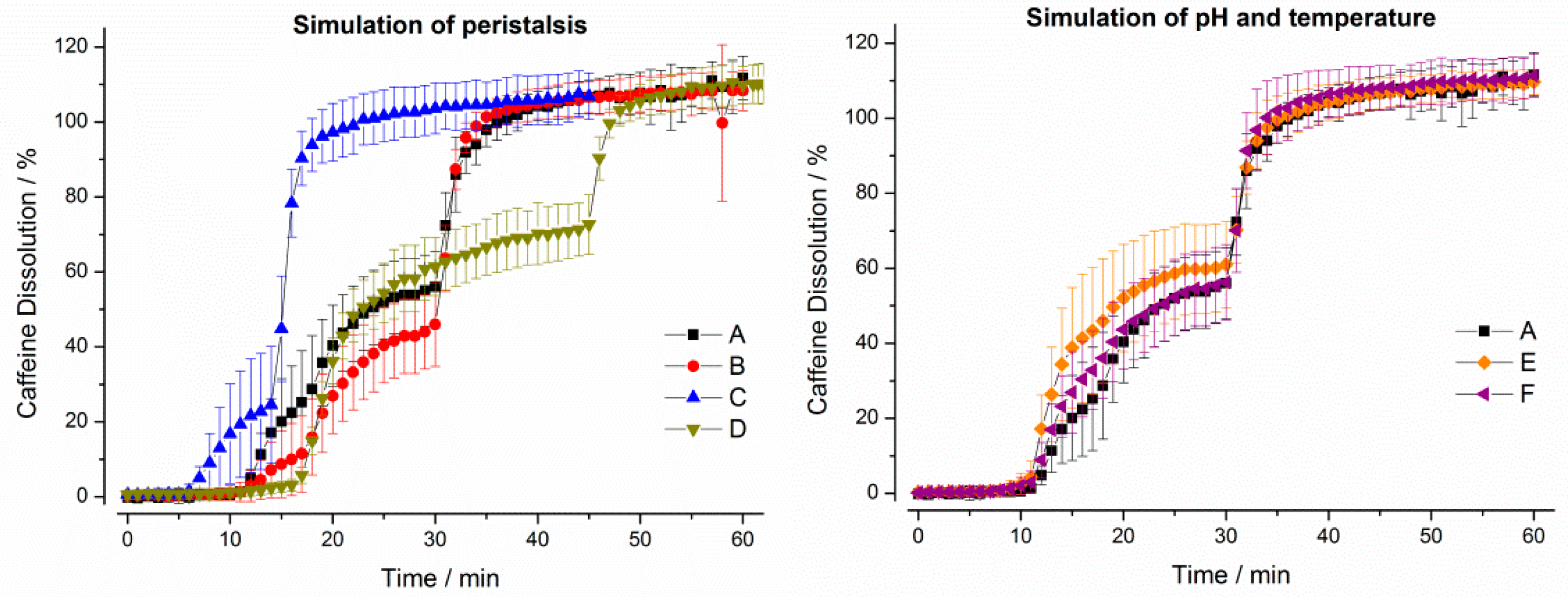
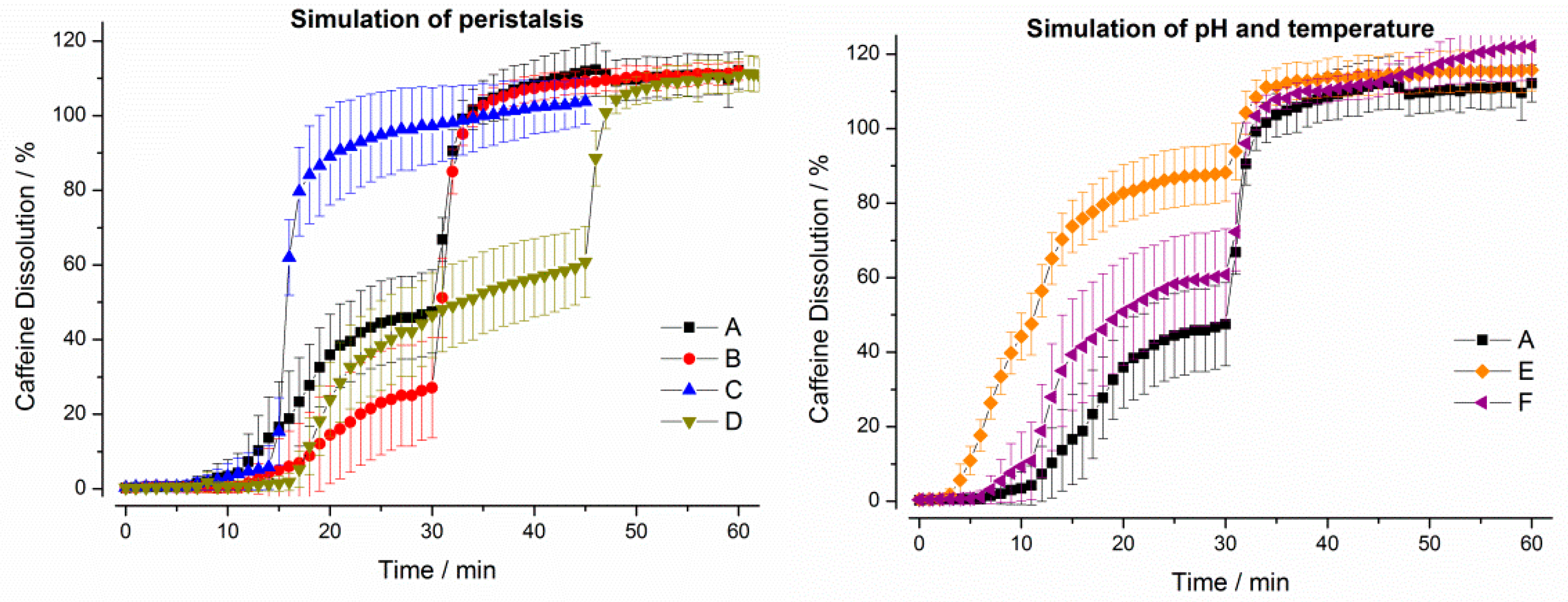
3. Results
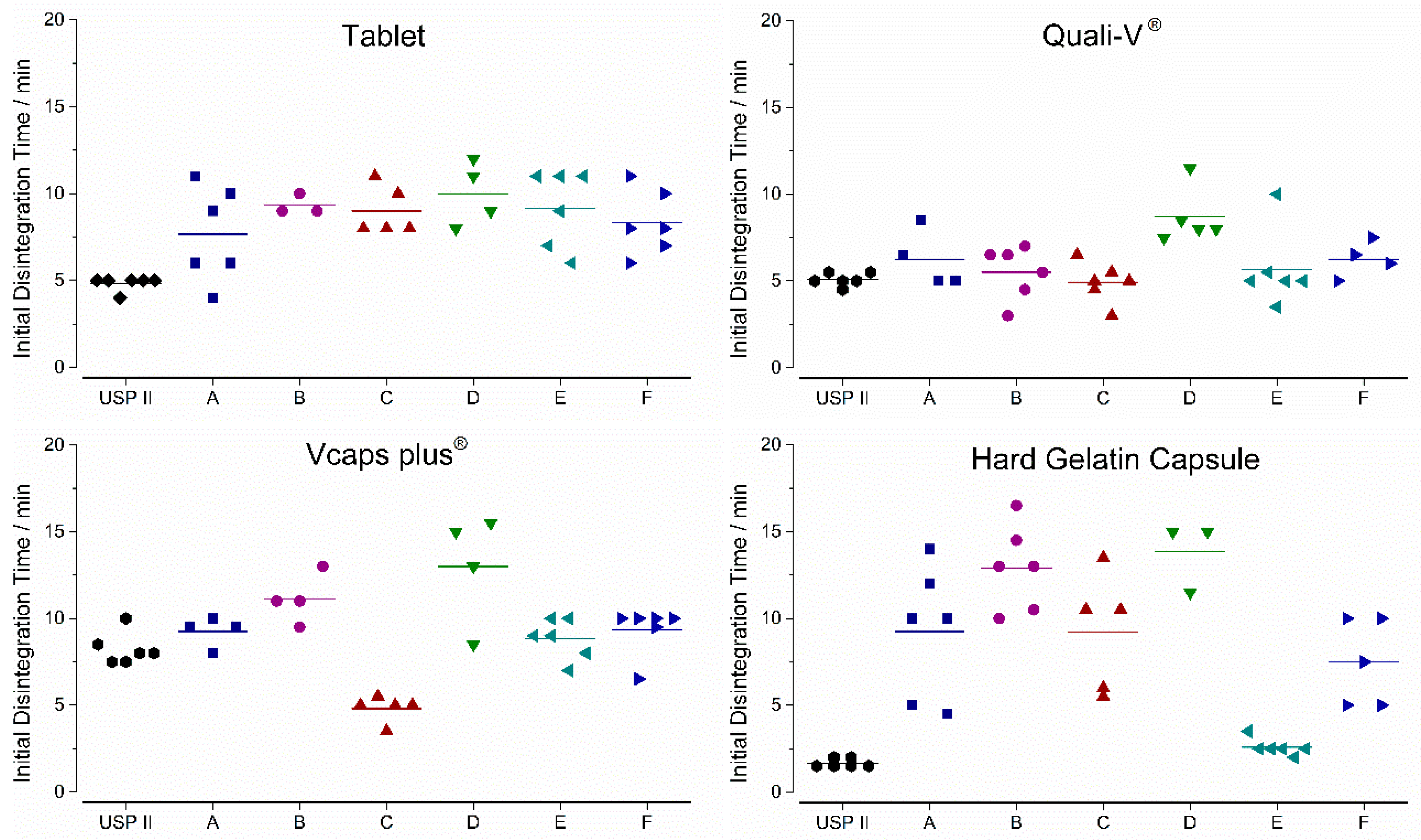
3.1. In Vitro Experiments
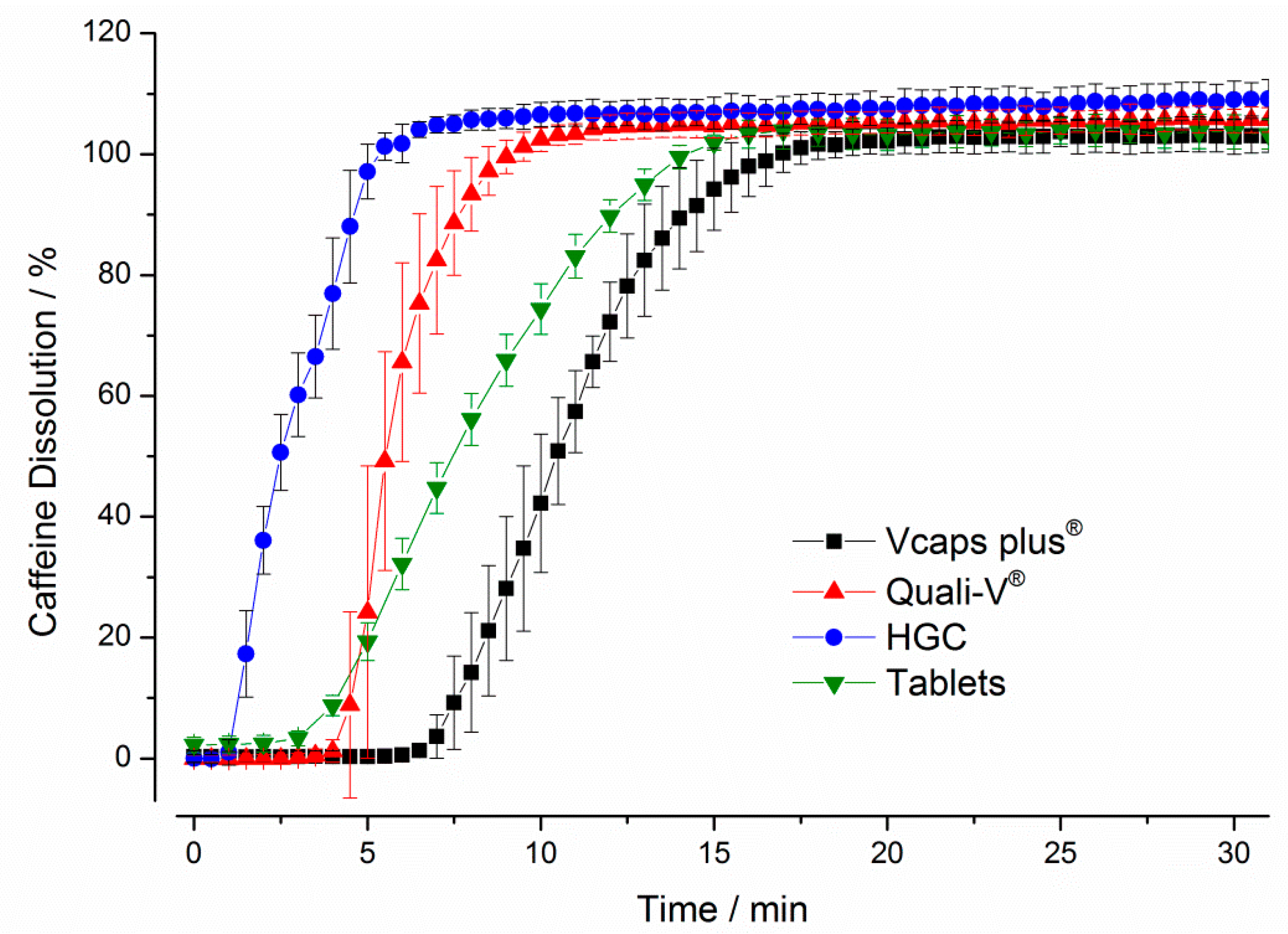
3.1.1. Compendial Dissolution Testing
3.1.2. GastroDuo Experiments
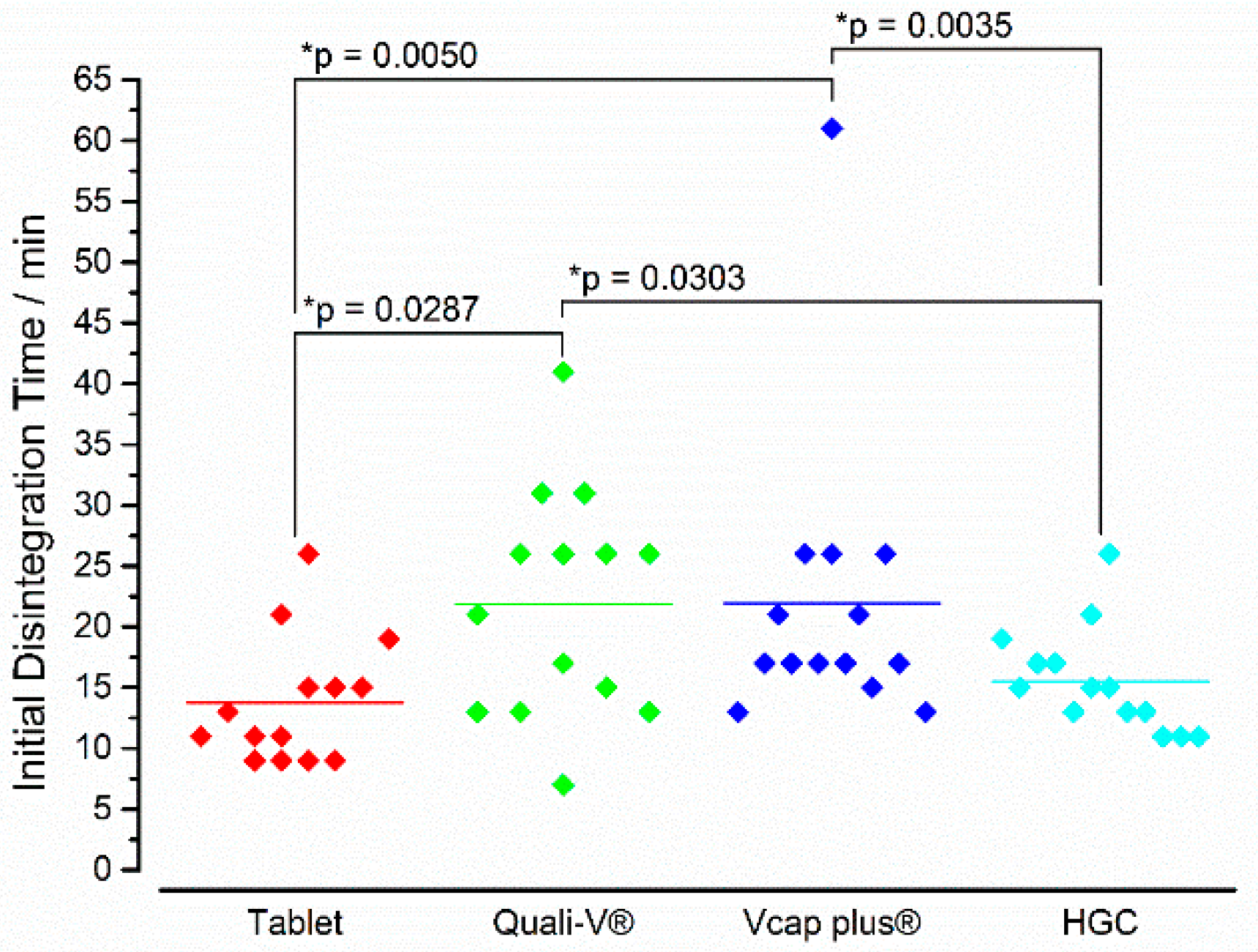
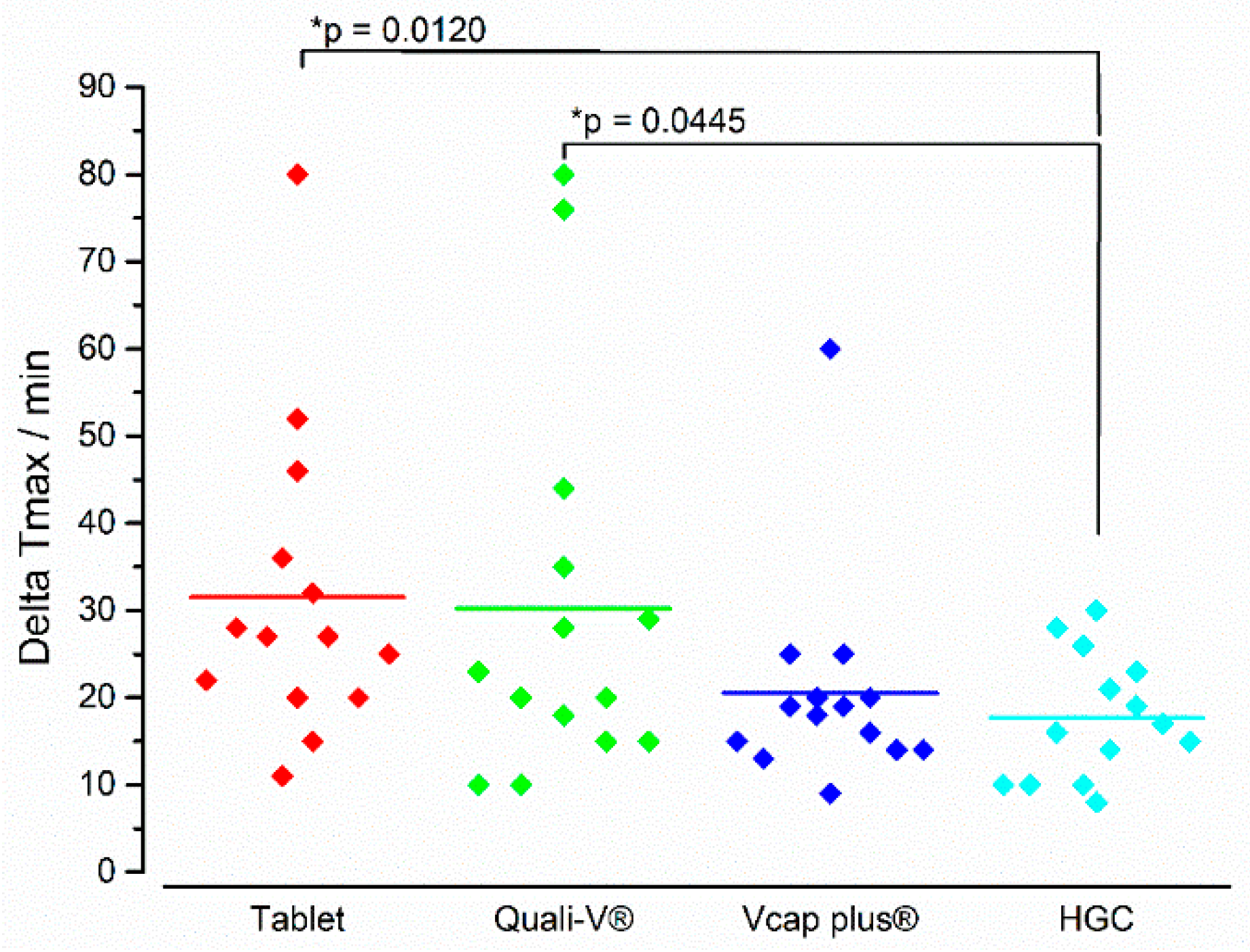
3.2. In Vivo Experiments
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Food and Drug Administration. Guidance for Industry Bioavailability and Bioequivalence Studies for Orally Administered Drug Products—General Guidance for Industry Bioavailability and Bioequivalence; Food and Drug Administration: Washington, DC, USA, 2002; pp. 1–24.
- EMA. Guideline on the Investigation of Bioequivalence; European Medicines Agency: London, UK, 2010; Volume 1, pp. 1–27. [Google Scholar]
- Schneider, F.; Grimm, M.; Koziolek, M.; Modeß, C.; Dokter, A.; Roustom, T.; Siegmund, W.; Weitschies, W. Resolving the physiological conditions in bioavailability and bioequivalence studies: Comparison of fasted and fed state. Eur. J. Pharm. Biopharm. 2016, 108, 214–219. [Google Scholar] [CrossRef] [PubMed]
- Koziolek, M.; Grimm, M.; Becker, D.; Iordanov, V.; Zou, H.; Shimizu, J.; Wanke, C.; Garbacz, G.; Weitschies, W. Investigation of pH and temperature profiles in the GI tract of fasted human subjects using the intellicap® system. J. Pharm. Sci. 2015, 104, 2855–2863. [Google Scholar] [CrossRef] [PubMed]
- Mudie, D.M.; Murray, K.; Hoad, C.L.; Pritchard, S.E.; Garnett, M.C.; Amidon, G.L.; Gowland, P.A.; Spiller, R.C.; Amidon, G.E.; Marciani, L. Quantification of gastrointestinal liquid volumes and distribution following a 240 mL dose of water in the fasted state. Mol. Pharm. 2014, 11, 3039–3047. [Google Scholar] [CrossRef] [PubMed]
- Grimm, M.; Koziolek, M.; Kühn, J.P.; Weitschies, W. Interindividual and intraindividual variability of fasted state gastric fluid volume and gastric emptying of water. Eur. J. Pharm. Biopharm. 2018, 127, 309–317. [Google Scholar] [CrossRef]
- Butler, J.; Hens, B.; Vertzoni, M.; Brouwers, J.; Berben, P.; Dressman, J.; Andreas, C.J.; Schaefer, K.J.; Mann, J.; Jamei, M.; et al. In vitro models for the prediction of in vivo performance of oral dosage forms: Recent progress from partnership through the IMI OrBiTo collaboration. Eur. J. Pharm. Biopharm. 2018, 136, 70–83. [Google Scholar] [CrossRef]
- Vardakou, M.; Mercuri, A.; Naylor, T.A.; Rizzo, D.; Butler, J.M.; Connolly, P.C.; Wickham, M.S.J.; Faulks, R.M. Predicting the human in vivo performance of different oral capsule shell types using a novel in vitro dynamic gastric model. Int. J. Pharm. 2011, 419, 192–199. [Google Scholar] [CrossRef]
- Minekus, M.; Marteau, P.; Havenaar, R.; Huis in ’t Veld, J.H.J. A multicompartmental dynamic computer-controlled model simulating the stomach and small intestine. Altern. Lab. Anim. 1995, 23, 197–209. [Google Scholar]
- Kourentas, A.; Vertzoni, M.; Khadra, I.; Symillides, M.; Clark, H.; Halbert, G.; Butler, J.; Reppas, C. Evaluation of the impact of excipients and an albendazole salt on albendazole concentrations in upper small intestine using an in vitro biorelevant gastrointestinal transfer (BioGIT) system. J. Pharm. Sci. 2016, 105, 2896–2903. [Google Scholar] [CrossRef] [Green Version]
- Kostewicz, E.S.; Abrahamsson, B.; Brewster, M.; Brouwers, J.; Butler, J.; Carlert, S.; Dickinson, P.A.; Dressman, J.; Holm, R.; Mann, J.; et al. In vitro models for the prediction of in vivo performance of oral dosage forms. Eur. J. Pharm. Sci. 2014, 57, 342–366. [Google Scholar] [CrossRef]
- Sager, M.; Schneider, F.; Jedamzik, P.; Wiedmann, M.; Schremmer, E.; Koziolek, M.; Weitschies, W. Effect of coadministered water on the in vivo performance of oral formulations containing N-acetylcysteine: An in vitro approach using the dynamic open flow-through test apparatus. Mol. Pharm. 2017, 14, 4272–4280. [Google Scholar] [CrossRef]
- Schick, P.; Sager, M.; Wegner, F.; Wiedmann, M.; Schapperer, E.; Weitschies, W.; Koziolek, M. Application of the GastroDuo as an in vitro dissolution tool to simulate the gastric emptying of the postprandial stomach. Mol. Pharm. 2019, 16, 4651–4660. [Google Scholar] [CrossRef] [PubMed]
- Sager, M.; Jedamzik, P.; Merdivan, S.; Grimm, M.; Schneider, F.; Kromrey, M.L.; Hasan, M.; Oswald, S.; Kühn, J.; Weitschies, W.; et al. Low dose caffeine as a salivary tracer for the determination of gastric water emptying in fed and fasted state: A MRI validation study. Eur. J. Pharm. Biopharm. 2018, 127, 443–452. [Google Scholar] [CrossRef] [PubMed]
- Garbacz, G.; Cadé, D.; Benameur, H.; Weitschies, W. Bio-relevant dissolution testing of hard capsules prepared from different shell materials using the dynamic open flow through test apparatus. Eur. J. Pharm. Sci. 2014, 57, 264–272. [Google Scholar] [CrossRef] [PubMed]
- Koziolek, M.; Görke, K.; Neumann, M.; Garbacz, G.; Weitschies, W. Development of a bio-relevant dissolution test device simulating mechanical aspects present in the fed stomach. Eur. J. Pharm. Sci. 2014, 57, 250–256. [Google Scholar] [CrossRef] [PubMed]
- Garbacz, G.; Klein, S.; Weitschies, W. A biorelevant dissolution stress test device—Background and experiences. Expert Opin. Drug Deliv. 2010, 7, 1251–1261. [Google Scholar] [CrossRef]
- Sager, M.; Grimm, M.; Jedamzik, P.; Merdivan, S.; Kromrey, M.L.; Hasan, M.; Koziolek, M.; Tzvetkov, M.V.; Weitschies, W. Combined application of MRI and the salivary tracer technique to determine the in vivo disintegration time of immediate release formulation administered to healthy, fasted subjects. Mol. Pharm. 2019, 16, 1782–1786. [Google Scholar] [CrossRef]
- Grimm, M.; Scholz, E.; Koziolek, M.; Kühn, J.P.; Weitschies, W. Gastric water emptying under fed state clinical trial conditions is as fast as under fasted conditions. Mol. Pharm. 2017, 14, 4262–4271. [Google Scholar] [CrossRef]
- Weitschies, W.; Wedemeyer, R.S.; Kosch, O.; Fach, K.; Nagel, S.; Söderlind, E.; Trahms, L.; Abrahamsson, B.; Mönnikes, H. Impact of the intragastric location of extended release tablets on food interactions. J. Control. Release 2005, 108, 375–385. [Google Scholar] [CrossRef]
- van den Abeele, J.; Brouwers, J.; Tack, J.; Augustijns, P. Exploring the link between gastric motility and intragastric drug distribution in man. Eur. J. Pharm. Biopharm. 2017, 112, 75–84. [Google Scholar] [CrossRef]
- Digenis, G.A.; Sandefer, E.P.; Page, R.C.; Doll, W.J.; Gold, T.B.; Darwazeh, N.B. Bioequivalence study of stressed and nonstressed hard gelatin capsules using amoxicillin as a drug marker and gamma scintigraphy to confirm time and GI location of in vivo capsule rupture. Pharm. Res. 2000, 17, 572–582. [Google Scholar] [CrossRef]
- Brown, J.; Madit, N.; Cole, E.T.; Wilding, I.R.; Cade, D. Effect of cross-linking on the in-vivo performance of hard gelatin capsules. Pharm. Res. 1998, 15, 1026–1030. [Google Scholar] [CrossRef] [PubMed]
- Tuleu, C.; Khela, M.K.; Evans, D.F.; Jones, B.E.; Nagata, S.; Basit, A.W. A scintigraphic investigation of the disintegration behaviour of capsules in fasting subjects: A comparison of hypromellose capsules containing carrageenan as a gelling agent and standard gelatin capsules. Eur. J. Pharm. Sci. 2007, 30, 251–255. [Google Scholar] [CrossRef] [PubMed]
- Jones, B.E.; Basit, A.W.; Tuleu, C. The disintegration behaviour of capsules in fed subjects: A comparison of hypromellose (carrageenan) capsules and standard gelatin capsules. Int. J. Pharm. 2012, 424, 40–43. [Google Scholar] [CrossRef] [PubMed]
- Cole, E.T.; Scott, R.A.; Cade, D.; Connor, A.L.; Wilding, I.R. In vitro and in vivo pharmacoscintigraphic evaluation of ibuprofen hypromellose and gelatin capsules. Pharm. Res. 2004, 21, 793–798. [Google Scholar] [CrossRef]
- Bonferoni, M.C.; Rossi, S.; Ferrari, F.; Bertoni, M.; Bolhuis, G.K.; Caramella, C. On the employment of λ carrageenan in a matrix system. III. Optimization of a λ carrageenan-HPMC hydrophilic matrix. J. Control. Release 1998, 51, 231–239. [Google Scholar] [CrossRef]
- Ku, M.S.; Lu, Q.; Li, W.; Chen, Y. Performance qualification of a new hypromellose capsule: Part II. Disintegration and dissolution comparison between two types of hypromellose capsules. Int. J. Pharm. 2011, 416, 16–24. [Google Scholar] [CrossRef]
- Honkanen, O.; Marvola, J.; Kanerva, H.; Lindevall, K.; Lipponen, M.; Kekki, T.; Ahonen, A.; Marvola, M. Gamma scintigraphic evaluation of the fate of hydroxypropyl methylcellulose capsules in the human gastrointestinal tract. Eur. J. Pharm. Sci. 2004, 21, 671–678. [Google Scholar] [CrossRef]
- Honkanen, O.; Pia, L.; Janne, M.; Sari, E.; Raimo, T.; Martti, M. Bioavailability and in vitro oesophageal sticking tendency of hydroxypropyl methylcellulose capsule formulations and corresponding gelatine capsule formulations. Eur. J. Pharm. Sci. 2002, 15, 479–488. [Google Scholar] [CrossRef]
- Osmanoglou, E.; van der Voort, I.R.; Fach, K.; Kosch, O.; Bach, D.; Hartmann, V.; Strenzke, A.; Weitschies, W.; Wiedenmann, B.; Mönnikes, H.; et al. Oesophageal transport of solid dosage forms depends on body position, swallowing volume and pharyngeal propulsion velocity. Neurogastroenterol. Motil. 2004, 16, 547–556. [Google Scholar] [CrossRef]
- Grahnen, A.; Hammarlund, M.; Lundqvist, T. Implications of intraindividual variability in bioavailability studies of furosemide. Eur. J. Clin. Pharmacol. 1984, 27, 595–602. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dosage Form | Ingredients |
|---|---|
| Immediate release tablets with 5% HPMC coating | Caffeine, croscarmellose, lactose, magnesium stearate, silica dioxide, hydroxypropyl methylcellulose |
| Hard gelatin capsule (size 0) | Caffeine, croscarmellose, lactose, gelatin |
| Quali-V® capsules (size 0) | Caffeine, croscarmellose, lactose, hydroxypropyl methylcellulose, carrageenan |
| Vcaps® plus capsules (size 0) | Caffeine, croscarmellose, lactose, hydroxypropyl methylcellulose |
| Age | 22–31 years |
|---|---|
| Body Mass Index | 20–25 |
| Female/Male | 6/8 |
| Ethnics | Caucasian |
| Pre-existing conditions | none |
| Number of volunteers | 14 |
| Regular coffee consumers | 12 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sager, M.; Schick, P.; Mischek, M.; Schulze, C.; Hasan, M.; Kromrey, M.-L.; Benameur, H.; Wendler, M.; Tzvetkov, M.V.; Weitschies, W.; et al. Comparison of In Vitro and In Vivo Results Using the GastroDuo and the Salivary Tracer Technique: Immediate Release Dosage Forms under Fasting Conditions. Pharmaceutics 2019, 11, 659. https://doi.org/10.3390/pharmaceutics11120659
Sager M, Schick P, Mischek M, Schulze C, Hasan M, Kromrey M-L, Benameur H, Wendler M, Tzvetkov MV, Weitschies W, et al. Comparison of In Vitro and In Vivo Results Using the GastroDuo and the Salivary Tracer Technique: Immediate Release Dosage Forms under Fasting Conditions. Pharmaceutics. 2019; 11(12):659. https://doi.org/10.3390/pharmaceutics11120659
Chicago/Turabian StyleSager, Maximilian, Philipp Schick, Magdalena Mischek, Christian Schulze, Mahmoud Hasan, Marie-Luise Kromrey, Hassan Benameur, Martin Wendler, Mladen Vassilev Tzvetkov, Werner Weitschies, and et al. 2019. "Comparison of In Vitro and In Vivo Results Using the GastroDuo and the Salivary Tracer Technique: Immediate Release Dosage Forms under Fasting Conditions" Pharmaceutics 11, no. 12: 659. https://doi.org/10.3390/pharmaceutics11120659