1. Introduction
Alzheimer’s disease (AD) is characterized by the abnormal extracellular accumulation of the amyloid-beta (Aβ) protein and/or its assembly into paired helical filaments and plaques. The progressive accumulation of Aβ aggregates is widely believed to be fundamental to the initial development of the neurodegenerative pathology and to trigger neurotoxicity, oxidative damage, and inflammation that contribute to the progression of AD [
1]. Recently, several strategies were investigated, aiming to influence the Aβ peptide aggregation pathway in order to either favor its elimination and, therefore, the balance between production and clearance, or to slow the aggregation process [
2,
3,
4]. Elucidation of Aβ fibril structural properties enabled the design and discovery of drugs that specifically interfere with Aβ–Aβ interaction and polymerization, thereby inhibiting the formation of fibrils or dissolving preformed fibrillar aggregates of Aβ.
Amongst these different drugs, a pentapeptide (Lys-Leu-Val-Phe-Phe, named KLVFF) specifically designed to target a central region of Aβ was confirmed to inhibit the elongation of fibrillar aggregates in vitro [
5,
6], by interacting with a homologous sequence on the oligomeric form of fragment Aβ-42 [
6]. Both electrostatic forces and hydrogen bonds lead to the formation of a link between the pentapeptide and the Aβ-42 fragment, causing a change in the secondary structure to an antiparallel β-sheet formation [
7]. This conformation interferes with the amyloid plaque formation process by reducing aggregation and non-toxic minor fibril formation [
8,
9,
10], and more importantly, KLVFF can disaggregate fibrils by binding to hydrophobic regions of Aβ [
11,
12]. Unfortunately, the KLVFF peptide displays poor aqueous solubility, low stability, slight cellular uptake, and the inability to cross the blood–brain barrier (BBB). To increase KLVFF’ s stability, several modifications in the peptide structure, such as methylation [
13] or the addition of an amino acid such as proline [
14], were tested, but the poor properties still hamper in vivo applications.
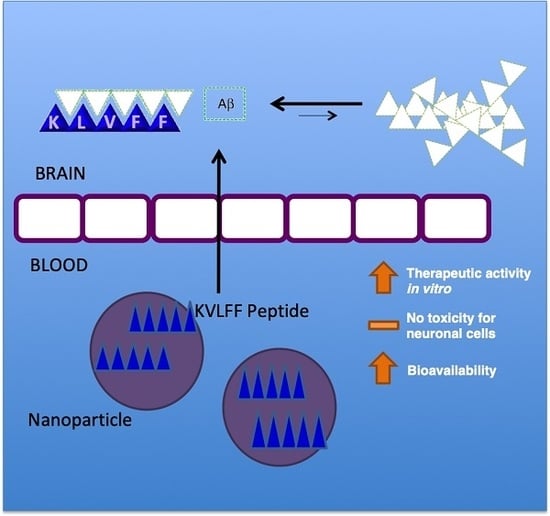
To overcome the limitations, a powerful approach to improve the pharmacokinetic profile of drugs is based on the exploitation of polymeric nanocarriers. Very recent experiments described the use of a nanocomposite featured by small-sized particles (lower than 20 nm) with KLVFF integrated on the surface [
15], with interesting readouts in terms of changes in Aβ aggregate morphology and a decrease in neuron damage in vitro and in AD mice, confirming the possible role of KLVFF to remarkably impact on the Aβ aggregation process.
The simultaneous use of both peptides and polymers was also positively applied by recent findings, describing the production and testing of self-destructive nanosweepers based on multifunctional peptide-polymers, such as chitosan and polyethylene glycol (PEG) [
16], or poly (hydroxypropyl methacrylamide) [
17], able to capture and clear Aβ for the effective treatment of AD.
All of these approaches aim to stabilize peptides, but may be limited by the nature of the polymer used in terms of impact on slowing down the rapid metabolism and systemic elimination or modulating the peptide’s release. In this view, the use of polymeric nanoparticles (NPs) can increase the water solubility, improving drug biodistribution and bioavailability. Amongst the different materials, few polymers guarantee the safety profiles; thus, given its favorable biocompatibility and biodegradability properties, poly(
d,
l-lactide-
co-glycolide) (PLGA) is one of the most successful polymers used in the development of drug delivery systems, particularly in brain targeting [
18], and is widely used for drug delivery across the BBB by means of tailored PLGA NPs. This has been proven in several in vivo studies using relevant pathological models [
19,
20,
21,
22]. Thus, application of this drug delivery technology could indeed be a promising option for the in vivo treatment of AD.
The poor solubility of KLVFF in water-based phases (buffer or surfactant solution) negatively affects the routinely proposed methods of nanoparticle preparation to load peptides (for example, double emulsion technology), by inhibiting encapsulation efficiency. Thus, in this research, we developed a novel biocompatible nanomedicine-based carrier able to control the release and stabilize KLVFF. Nanocarriers were prepared by adapting the nanoprecipitation procedure using an unusual mixture of organic solvents (Dimethyl sulfoxide (DMSO)/Acetone) to dissolve the polymer and peptide, forming stable loaded NPs. The effect of released KLVFF by NPs in inhibiting the amyloid formation or in inducing disaggregation of formed or in-formation plaques was then tested in in vitro models.
2. Materials and Methods
2.1. Materials
Poly(d,l-lactide-co-glycolide) acid (PLGA RG-503H 50:50, inherent viscosity in 0.1% (w/v) CHCl3 at 25 °C = 0.38 dL/g) was used as received from the manufacturer (Boehringer-Ingelheim, Ingelheim am Rhein, Germany). According to the experimental titration results of the carboxylic end of the polymers (4.94 mg KOH/g polymer), the molecular weight of RG-503H was calculated to be 11,000 Da. Peptide KLVFF (Lys-Leu-Val-Phe-Phe, C42H63N9O8, MW 822.01) was obtained from Mimotopes (Springvale Rd Mulgrave, Victoria, Australia). Pluronic F68 (molecular weight of 8500–9000 Da) was purchased from Sigma-Aldrich. Primary antibodies were purchased from Synaptic Systems (Homer1) and Abcam (MAP2). Fetal bovine serum (FBS) and phosphate-buffered saline (PBS) were purchased from Euroclone Celbio (Milan, Italy). Secondary Alexa Fluor conjugated antibodies were purchased from Invitrogen. Thioflavin was obtained from Sigma Aldrich. Synthetic Aβ peptide, human Aβ(1–42), was purchased from Abcam. All the solvents were of analytical grade, and all other chemicals and media were used as received from the manufacturers, and unless otherwise indicated, obtained from Sigma-Aldrich.
2.2. Preparation of KLVFF-Loaded NPs
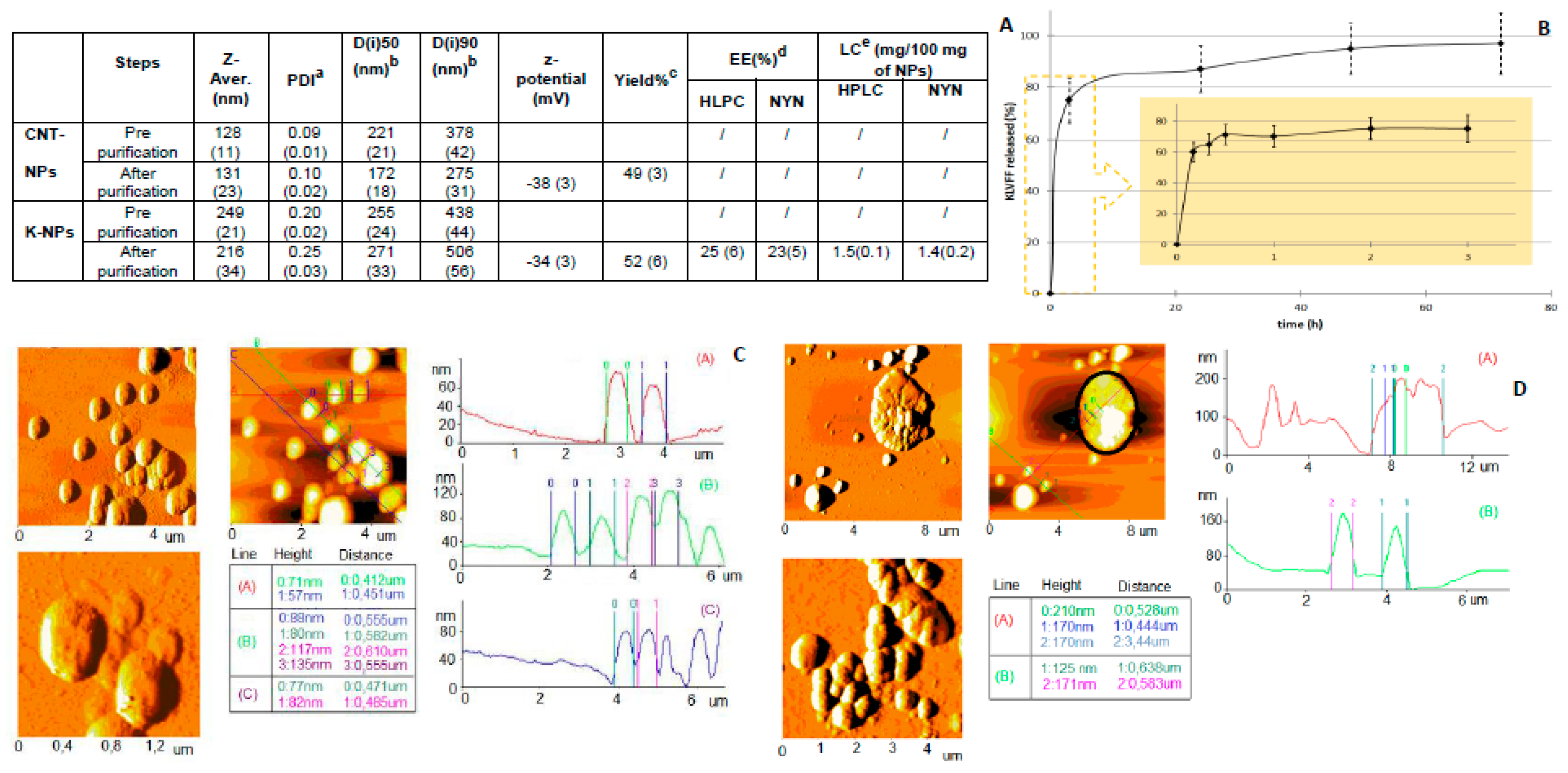
KLVFF-loaded NPs (K-NPs) were obtained with an optimized nanoprecipitation procedure. Preliminary KLVFF, PLGA, and KLVFF/PLGA-mix solubility tests were carried out in water and diffusible organic solvent (see
Supplementary Table S1). K-NPs were prepared by solubilizing a fixed amount of KLVFF (3 mg) into DMSO (0.3 mL) and adding it to a heated (50 °C) acetone solution (3.7 mL) of PLGA RG-503H (50 mg). This hot organic mixture was added dropwise into a 3% (
w/
v) Pluronic F68 aqueous solution (12.5 mL). The solution was then stirred at room temperature (r.t.) for 15 min, and the organic solvent was removed at 30 °C under reduced pressure (10 mm Hg, Buchi Rotavapor R-300 Evaporation Systems, Flawil, Switzerland). The same procedure was applied to produce control NPs (CNT-NPs), with the unique difference of using a solution of DMSO without KLVFF. NP batches were then purified by centrifugation (16,000 rpm for 10 min, 4 °C; J2-21 model, Beckman centrifuge, NewPort Pagnell, UK), washed and re-suspended in water (4 mL). Samples were stored at −20 °C before use, or freeze-dried (LyoLab 3000, Heto-Holten, Allerod, Denmark) to allow weight yield evaluation.
2.3. Characterization of NPs
The average diameter (Z-average), the size distribution (expressed as D(50) and D(90)) and the polydispersity index (PDI) of the NP samples were assessed by Photon Correlation Spectroscopy (PCS) using a Zetasizer Nano ZS (Malvern, Malvern, UK; Laser 4 mW He–Ne, 633 nm, laser attenuator automatic, transmission 100–0.0003%, detector avalanche photodiode, quantum efficiency (QE) >50% at 633 nm, at 25 °C). For each formulation, the mean diameter and PDI were calculated from three replicates of three different batches (nine measurements). The zeta potential (z-potential) (average of 10 measurements) was analyzed using the same equipment, with a combination of laser Doppler velocimetry and phase analysis light scattering (PALS). The Atomic Force Microscope (AFM) measurements were performed with an AFM (Park Instruments, Sunnyvale, CA, USA) operating at about 20 °C in air and in non-contact mode using a commercial silicon tip-cantilever (high resolution noncontact “GOLDEN” Silicon Cantilevers NSG-11, NT-MDT, tip diameter 5–10 nm; Zelenograd, Moscow, Russia) with a stiffness of about 40 mN and a resonance frequency around 150 kHz. After the purification, the NP sample was dispersed in distilled water before being applied on a freshly cleaved mica disk (1 cm × 1 cm); after 2 min, the excess water was removed using filter paper. The AFM images (a topographical image, and a second one showing the “error signal”) were obtained with a scan rate of 1 Hz. The error signal was obtained by comparing the image representing the amplitude of the vibrations of the cantilever with the amplitude of a reference point. The images obtained by this method showed small superficial variations of the samples. Images were processed using ProScan Data Acquisition software.
2.4. Evaluation of the Weight Yield
Freeze-dried samples were used to calculate weight recovery. In particular, the weight yield (WY%) was calculated as follows:
2.5. Extraction of KLVFF from NPs
Briefly, a weight amount of NPs (about 5 mg) was solubilized into 1 mL of dichloromethane (DCM) by using a bath sonicator (Bransonic® bath Ultrasonic, Optolab Instrument, Modena, Italy) for 2 min; 1 mL of 1% (w/v) Polyvinyl Alcohol (PVA) solution was then added to extract the peptide. The mixture was shaken and then centrifuged (Spectrafuge 24D) to separate the two phases. The aqueous phase was collected, and the organic phase was again subjected to two further extractions.
2.6. Quantification of KLVFF
Quantification of the peptide was obtained by using HPLC and a colorimetric assay with ninhydrin. In detail, using an HPLC instrument (JASCO Europe, Cremella, Italy) with a Model PU2089 pump and an injection valve with a 50 µL sample loop (Model 7725i, Jasco), and a UV/VIS detector (UV1575, Jasco), the freshly prepared solutions and mobile phases were filtered through a 0.45 µm hydrophilic polypropylene (PP) membrane filter (Sartorius, Goettingen, Germany). A C8 column (Aeris Widepure; 150 × 4.60 mm) equipped with a security guard was used as an analytical column. Elution was obtained using gradient steps of solvents A (0.1% trifluoroacetic acid in MilliQ water (pH 2.5)) and B (0.1% trifluoroacetic acid in acetonitrile) starting from 80:20 (A:B) to 20:80 in 13 min at a flow rate of 1.2 mL/min. After the run was complete, the column was re-equilibrated for 2 min. All analyses were carried out under isothermal conditions at 70 °C (Column Heater, model 7971, Jones Chromatography). The chromatographic peak area of the samples was recorded and analyzed using a JASCO software (ChromNav 2.0-Chromatography Data System program, Jasco). The concentration was calculated using a calibration curve (linearity was assumed in the range 2–38 µg/mL; R2 = 0.9994).
Ninhydrin assay: 100 μL of the sample (extracted from NPs) was added, in duplicates, in the wells of a 96-well plate with 75 μL of ninhydrin color reagent. The plate was then incubated at 80 °C for 30 min. Subsequently, the plate was cooled to r.t., and 100 μL of stabilizing solution (50% v/v ethanol) was added to each well. Finally, the spectrophotometric reading at 570 nm was performed through a Microplate Reader Multiscan (Spectrum Finstruments®). A standard concentration of peptide 1% w/v of PVA aqueous solution was used to generate a calibration curve. Linearity was assumed in the range of 2–35 µg/mL (R2 = 0.9979). All the data are shown as the mean of at least three measurements.
2.7. Encapsulation Efficiency and Loading Capacity
The amount of KLVFF loaded into NPs was calculated as the percentage of loading capacity (LC) and encapsulation efficiency (EE) using the following equations:
where
D is the amount of the encapsulated drug,
W is the weight of NPs (polymer + drug), and
Td is the amount of drug added for the preparation. All data are expressed as the means of at least three determinations.
2.8. In Vitro Release Studies
For each time point, an exact amount of K-NPs (about 10 mg, KLVFF content about 0.1 mg) was suspended in 1 mL FBS:PBS (50:50
v/
v) at 37 °C under magnetic stirring. After an incubation time (10 min, 1, 3, 24, 48, or 72 h), the suspension was centrifuged (13,000 rpm for 10 min) to separate K-NPs from the supernatant. Serum proteins that may be adsorbed on the NP surface were quantified using a colorimetric bicinchoninic acid (BCA) assay (Micro BCA protein assay kit composed of reagent A (alkaline tartrate–carbonate buffer), reagent B (bicinchoninic acid solution), and reagent C (copper sulfate solution)—Thermo Fisher Scientific Inc., Milan, Italy). The amount of adsorbed proteins was subtracted from the final value, representing the amount of plain NPs recovered after centrifugation. The KLVFF amount at each incubation time was determined after extraction from the NPs, as described above. The percentage of drug released was calculated using the following formula:
where KLVFF tot is the initial drug content in the nanoparticle sample (
t = 0), and the KLVFF residual is the amount of drug in the same sample after the incubation period.
2.9. Cell Culture
Primary hippocampal cultures were prepared from rat hippocampi (BrainBits, Loughborough, UK). In brief, hippocampi were washed and transferred to 1800 μL HBSS (Hank’s Balanced Salt Solution, Sigma Aldrich) and then trypsinized with 200 μL Trypsin 2.5% for 20 min at 37 °C. After washing the cells five times with HBSS, they were re-suspended in 1600 μL HBSS with 400 μL DNAse 1 (0.01%). The suspension was then filtered through a 125 μm sieve and incubated with 18 mL of DMEM (Dulbecco’s Modified Eagle Medium, Sigma Aldrich, Arklow, Ireland) containing 10% Fetal Calf Serum (FCS), 1% glutamine, and 1% penicillin-streptomycin (DMEM+++). After cell-counting using a Neubauer chamber, the cells were seeded on poly-l-lysine (PLL) (0.1 mg/mL) coated glass coverslips in a 24-well plate at a density of 30,000 cells/well. After 24 h, the medium was changed to Neurobasal medium (Life Technologies via Biosciences, Dun Laoghaire, Ireland), complemented with B27 supplement (Life Technologies), 0.5 mM l-Glutamine (Life Technologies), and 100 U/mL penicillin/streptomycin (Life Technologies) (NB+++) and maintained at 37 °C in 5% CO2.
2.10. Treatment of Hippocampal Cells
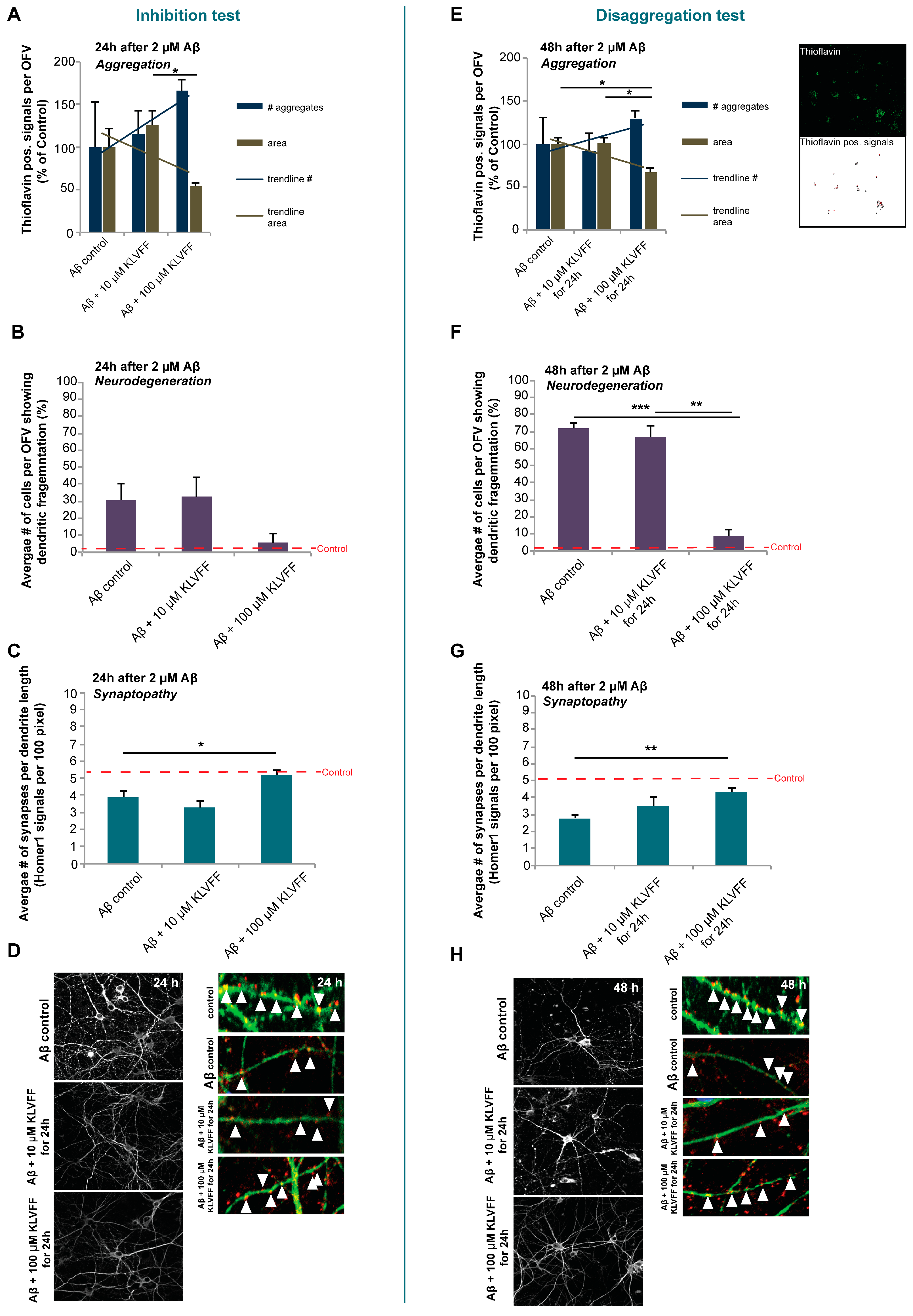
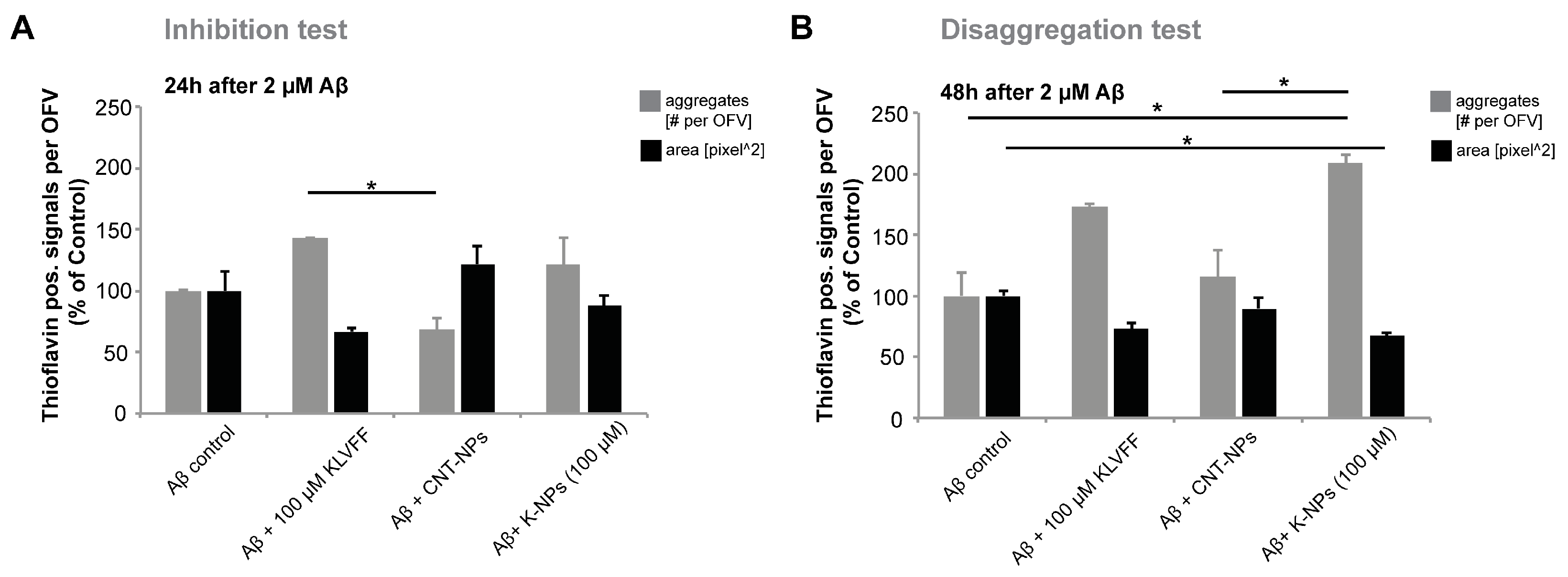
To investigate the two possible impacts of treatment with KLVFF (inhibition of aggregation, and disaggregation and inhibition of re-aggregation), we set up two different experiments: to clarify the potency of inhibition of KLVFF on plaque formation (“inhibition test”), Aβ(1–42) peptides (2 M) and KLVFF peptides (10 or 100 M) (or corresponding dose of KLVFF delivered by KLVFF-loaded NPs) were co-administered at time 0 and the experiments were performed over 24 h.
To evaluate the effect of KLVFF on the disaggregation of formed A aggregates and inhibition of re-aggregation (“disaggregation test”), Aβ(1–42) peptides (2 M) were administered to cells at time 0 and after 24 h, free KLVFF peptides (10 or 100 M) (or corresponding doses of KLVFF delivered by KLVFF-loaded NPs) were administered to cells.
To generate 200 μM (mainly monomeric) Aβ stock solution, lyophilized Aβ(1–42) was dissolved in DMSO, vortexed for 30 min at RT, and centrifuged for 1 h (15,000× g) at 4 °C. The supernatant was aliquoted, snap-frozen, and stored at −20 °C. Further dilutions of the peptides were performed in the culture medium (NB+++). For the treatments, neurons were seeded in a total volume of 500 μL per well (24-well plate). Freeze-dried NP samples were re-suspended in HBSS under sonication (Emmi-05P Sonicator, Emag, Germany) to have a starting batch for further dilutions in NB+++. All the NP stock solutions were prepared according to the calculated KLVFF content, and the volume of HBSS was adjusted accordingly. Stock solutions of CNT-NPs were prepared to match the amount of NPs per mL of K-NPs. After the incubation period, cells were analyzed using immunocytochemistry and cell health assays.
2.11. Immunocytochemistry (ICC)
For ICC, the cells were fixed with 4% paraformaldehyde (PFA)/4% sucrose/PBS at 4 °C for 20 min. After washing three times for 5 min with 1× PBS containing 0.2% Triton X-100 at r.t., blocking was performed using a blocking solution (BS) containing 10% FBS in 1x PBS for 1 h at r.t. Then, samples were incubated with the primary antibodies (Homer1, MAP2) in BS at r.t. for 2 h or at 4 °C overnight, followed by three 5 min washing-steps with 1× PBS and incubation with the secondary antibody coupled with Alexa488, Alexa568, or Alexa647 in BS for 1 h. Cell nuclei were counterstained with DAPI (4′,6-diamidino-2-phenylindole). Finally, coverslips were mounted using Vecta Mount (Vector Laboratories, Burlingame, CA, USA). Aβ aggregates were visualized using Thioflavin T.
2.12. Cell Health Assay
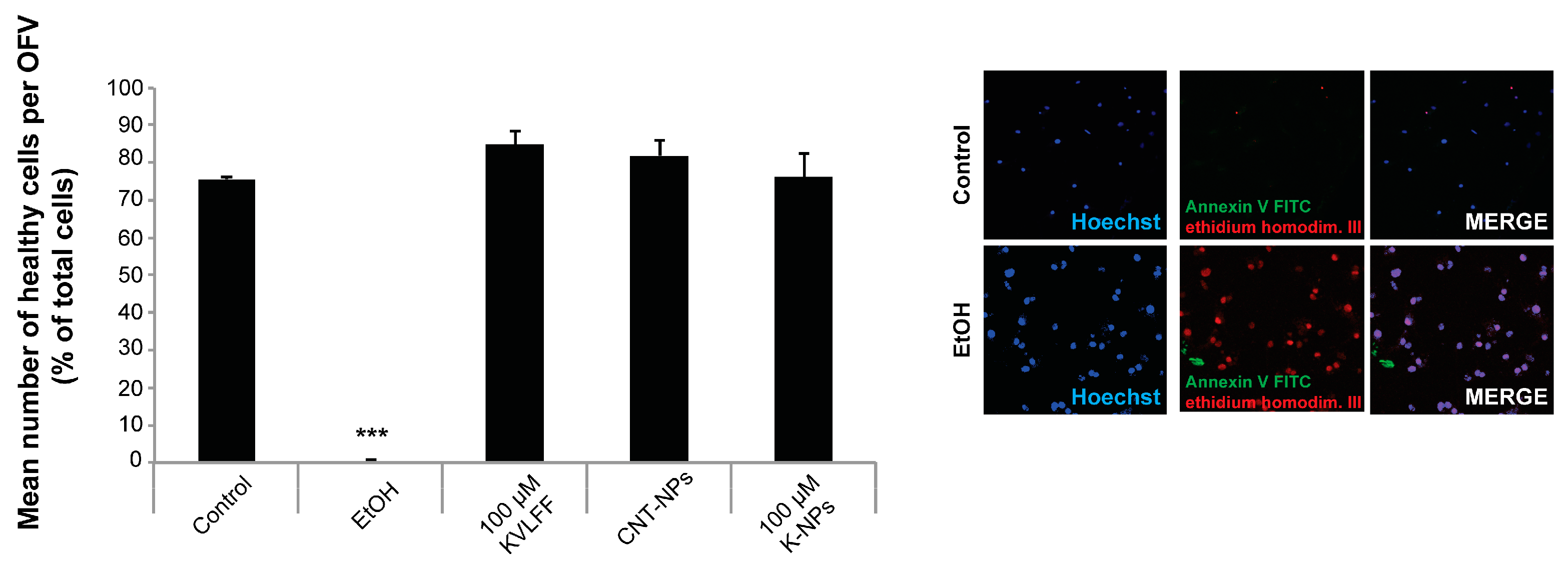
The cell health of the neuronal cultures after treatment was measured using the healthy/apoptotic/necrotic cell detection kit (Promokine) according to the manufacturer’s protocol. In brief, cells were treated with 1× binding buffer and then incubated for 15 min at r.t. protected from light with a staining solution containing 100 μL of 1× binding buffer, 5 μL of FITC-Annexin V, 5 μL of Ethidium Homodimer III, and 5 μL of Hoechst 33342. Subsequently, cells were washed twice with the 1× binding buffer. Finally, the cells were fixed with (PFA)/PBS and 1.25 mM CaCl2 at r.t. for 15 min and coverslips mounted using Vecta Mount. As a positive control, apoptosis was induced with 500 μL of 70% ethanol. Fluorescence images were obtained with an inverted confocal LSM710 microscope (Zeiss) and ZEN software v.2.5. Quantification was performed using ImageJ 1.49j.
Alternatively, cells were seeded at a density of 5,000 cells per well on PLL-coated E-Plate 16 plate (ACEA Biosciences, San Diego, CA, USA) and treated with 2, 5, or 10 µM Aβ(1–42) peptide after 24 h of growth. Controls were treated with vehicle (DMSO) only. Impedance was measured every 5 min in the following 36 h of treatment using the xCELLigence RTCA Systems. A decrease in impedance is associated with a decreased proliferation and/or the detachment of cells, and therefore a sign impaired cell health. Data are shown as slope, calculated according to the RTCA DPlus Software Manual V1.0.
2.13. Quantitative Real-Time PCR (qRT-PCR)
Isolation of total RNA was performed using the RNeasy Mini kit (Qiagen, Manchester, UK) according to the manufacturer’s instructions. First-strand synthesis and real-time qRT-PCR amplification (Roche LightCycler 480 II) were carried out in a one-step, single-tube format using the Rotor-Gene SYBR Green RT-PCR kit (Qiagen), and using validated primer pairs from Qiagen (Quantitect primer assay). The hydroxymethylbilane synthase (HMBS) gene was used as an internal standard. All reactions were run at least in technical triplicates, and mean ct values were transformed into virtual mRNA levels according to the formula: virtual mRNA level = 10 × ((ct(target) – ct(standard))/slope of the standard curve).
2.14. Statistics
Statistical analysis was performed with SPSS version 20. All data are shown as mean ± SEM (standard error of the mean), if not indicated otherwise. For comparisons, one-way analysis of variance (ANOVA) was performed, followed by post hoc tests for within-group comparisons (Tukey test). Statistically significant differences are indicated in the figures by * p ≤ 0.05, ** p ≤ 0.01, and *** p ≤ 0.001.
4. Discussion
The brain-targeted delivery of active molecules with poor aqueous solubility, low stability, and an inability to cross the BBB is currently a major challenge in the development of novel treatment strategies for neurodegenerative disorders. In particular, recent research on AD has revealed several compounds with high potential to act as therapeutics; however, due to their chemical nature and the thereby existing impairments mentioned above, these potential therapeutic strategies cannot be easily translated to clinical practice. Among the proposed compounds, the KLVFF peptide showed promising results for the treatment of AD. Here, we report the successful development of KLVFF loaded polymeric NPs that have been previously demonstrated to cross the BBB through the addition of a g7 ligand [
28].
Recently, we demonstrated the possibility of delivering an active compound across the BBB using g7-NPs injected in a mouse model for AD [
29]. This shows a promising approach to assure the protection of the cargo in blood and a controlled release inside the brain using NPs. The KVLFF peptide is another potent candidate for AD treatment. KLVFF has been specifically designed to target a central region of Aβ and was shown to inhibit the elongation to fibrillar aggregates in vitro [
5,
6]. Here, we confirm the effects of KVLFF on Aβ aggregation at a concentration of 100 μM and in the presence of 2 μM Aβ. We observed stronger effects on existing larger aggregates (disaggregation) as on the prevention of aggregation (inhibition). However, the application of a broader range of concentrations and variations in exposure times in the future may identify conditions in which the efficacy of the peptide is further improved. The presence of KVLFF seems to have complex effects on the dynamics of aggregation and disaggregation of Aβ, and might result in a shift towards more but smaller aggregates with limited potential for growth rather than preventing aggregation altogether. More importantly, 100 μM KVLFF delivered by NPs had similar effects compared to the free peptide. Future studies will need to evaluate whether maintaining Aβ in a state of smaller aggregates or disaggregating plaques into smaller aggregates through the chronic application of the peptide is an effective treatment approach, as not soluble oligomers may be more toxic than larger Aβ aggregates, although in our study, a beneficial effect was observed on the neuropathology in vitro.
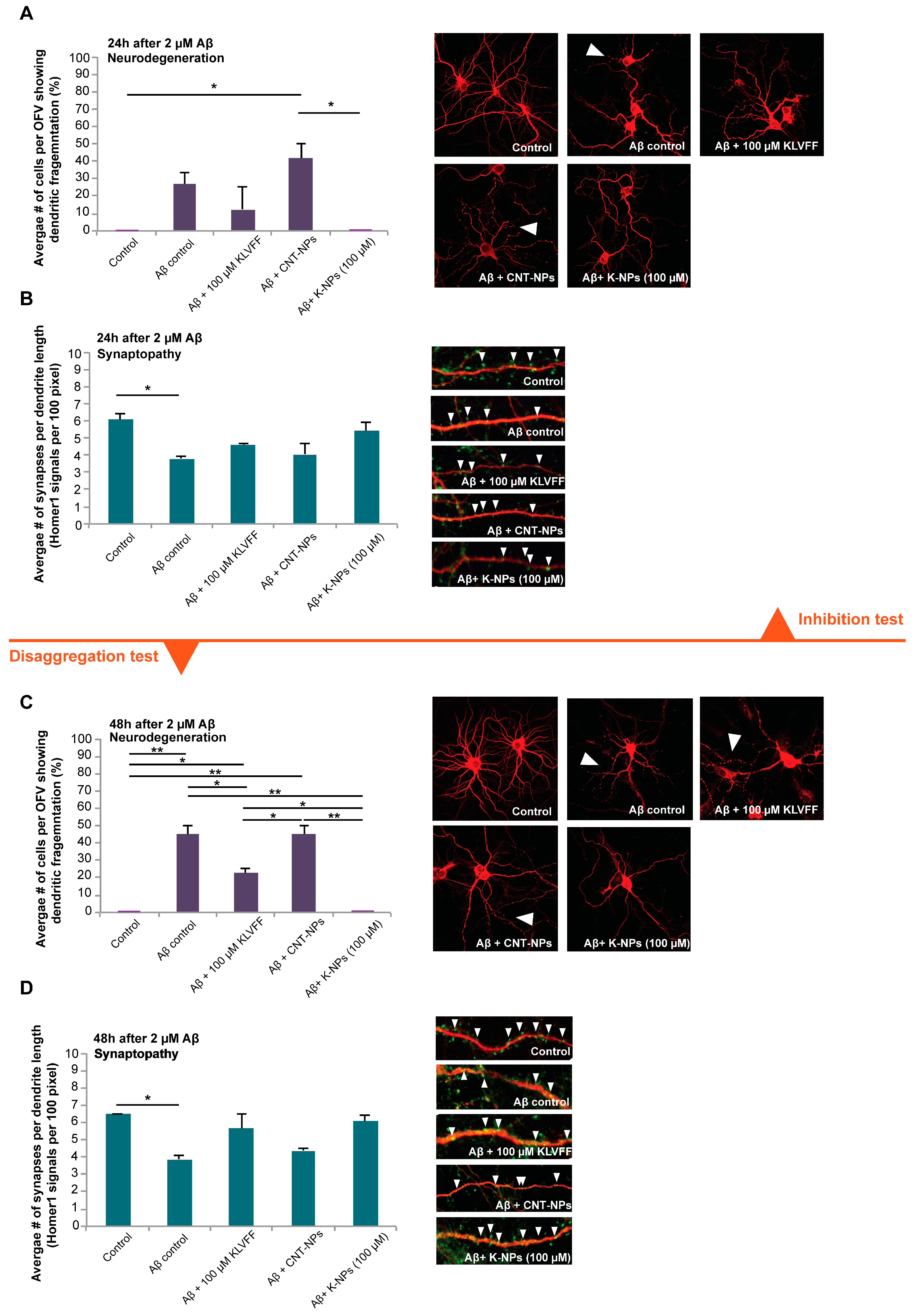
While a previous study could show the effects of KVLFF nanocomposites on the viability of PC12 cells after exposure to Aβ [
15], we performed a more thorough analysis using primary neurons. In this study, we could observe that free KVLFF and, even more potent, NP delivered KVLFF significantly reduces dendritic fragmentation, a sign of neurodegeneration. Neurodegeneration is a hallmark of AD, and Aβ-induced toxicity was shown to be a major contributor through various mechanisms, such as oxidative stress, triggering inflammatory responses, and the generation of trace metal dyshomeostasis. In addition, the presence of Aβ was shown to have direct effects on synapses [
30,
31] and zinc-depletion [
32]. Here, we could also observe a significant reduction in synapse density after the application of Aβ. This reduction was absent after treatment with free KLVFF peptide and K-NPs. Given that neurodegeneration and synapse loss may be the best correlate to the observed cognitive decline in AD, K-NPs being able to deliver KLVFF into the brain may be a promising approach. In particular, due to the results obtained, if confirmed in AD animal models, the efficacy of the KLVFF peptide could be exploited in the treatment of late-stage AD and not only in the starting steps of the disease, opening up treatment to a more relevant number of patients to be managed.

 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




