Cisplatin Loaded Multiwalled Carbon Nanotubes Induce Resistance in Triple Negative Breast Cancer Cells


 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Carbon Nanotubes Functionalization
2.2. Quantification of Platinum Ions
2.3. FTIR Spectroscopy
2.4. Scanning Electronic Microscopy
2.5. Cell Culture
2.6. Cell Treatment
2.7. Cell Viability
2.8. The Level of Lactate Dehydrogenase (LDH) Released in Culture Medium
2.9. Cell Morphology Examination and Fluorescence Labelling of Actin Cytoskeleton
2.10. Reactive Oxygen Species (ROS) Production
2.11. Preparation of Cellular Lysate
2.12. Reduced Glutathione (GSH) Content
2.13. Glutathione S-Transferase (GST) Activity
2.14. Nuclear Factor E2-Related Factor 2 (Nrf2) Protein Expression
2.15. Statistical Analysis
3. Results and Discussion
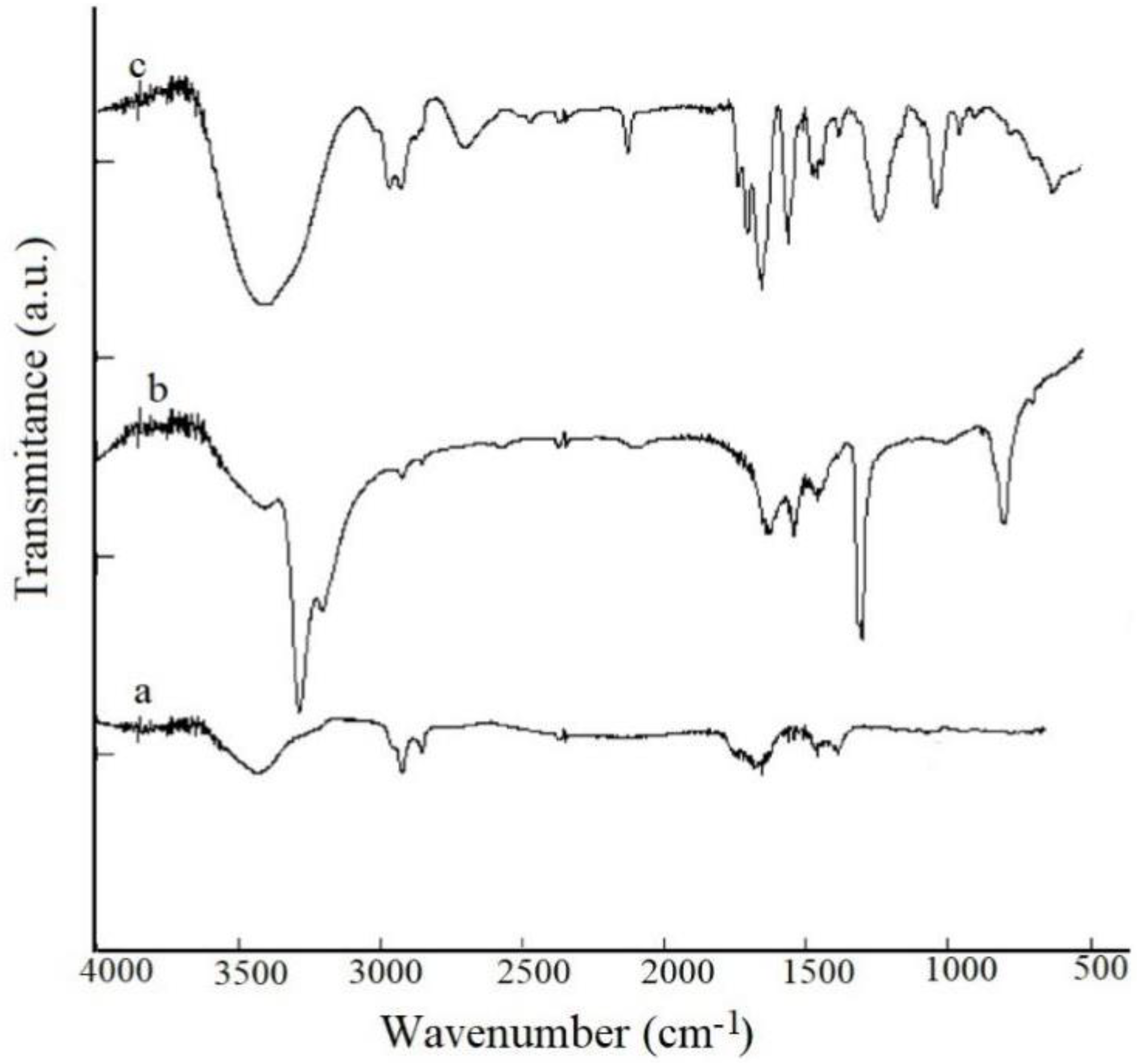
3.1. FTIR Measurements
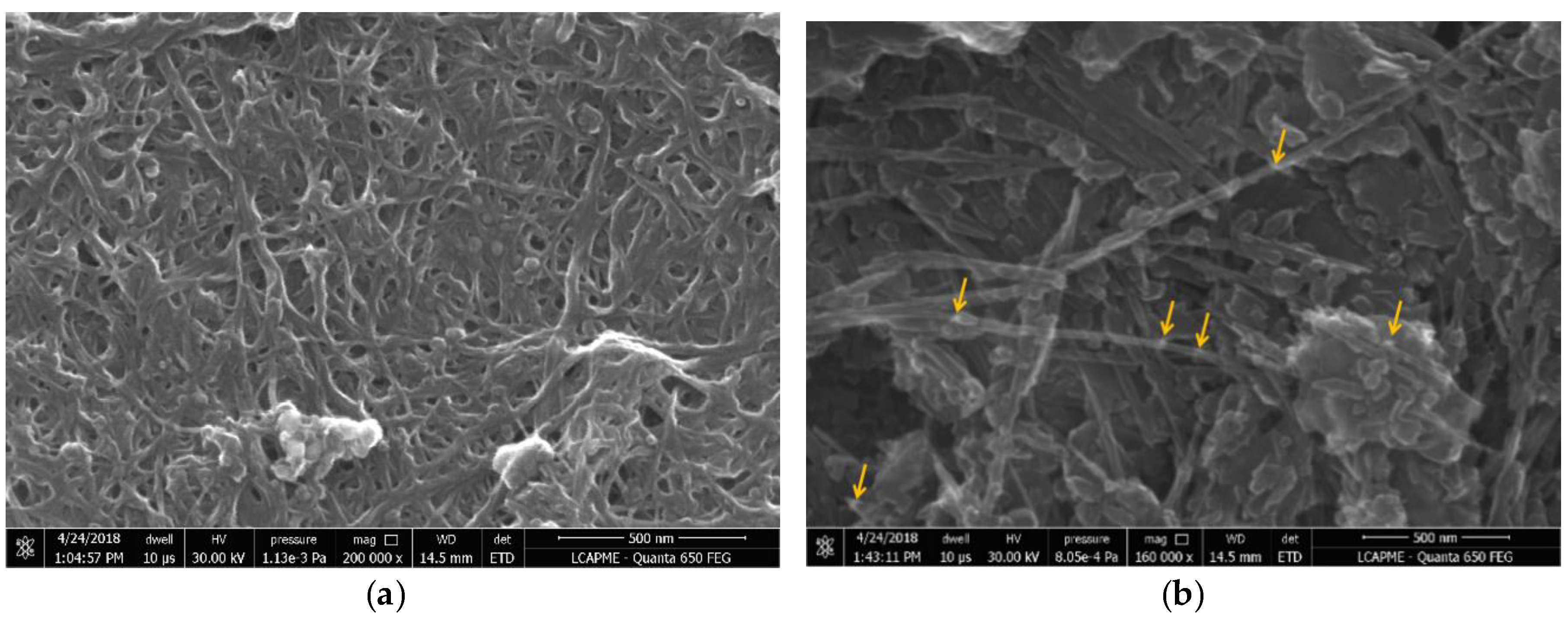
3.2. SEM Characterization
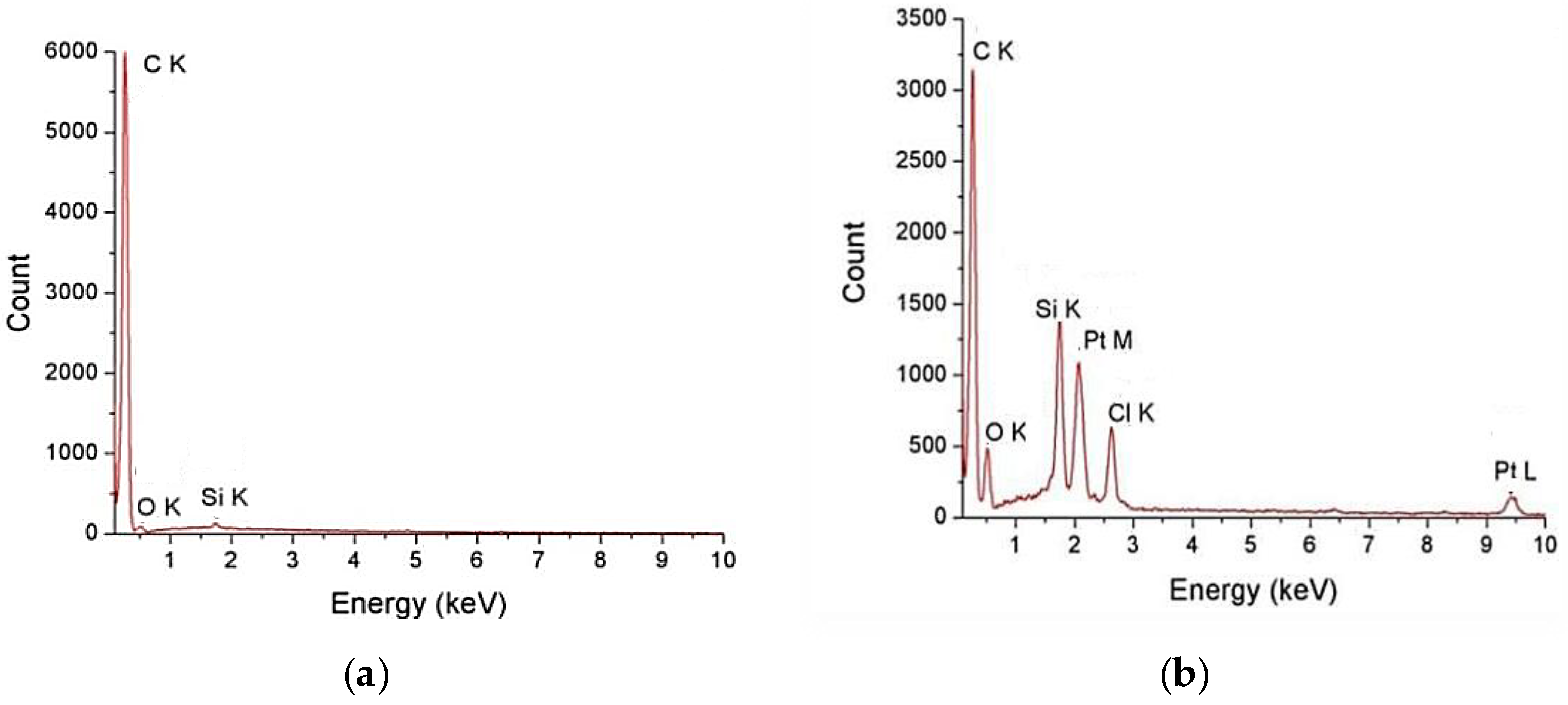
3.3. ICP-MS Analysis
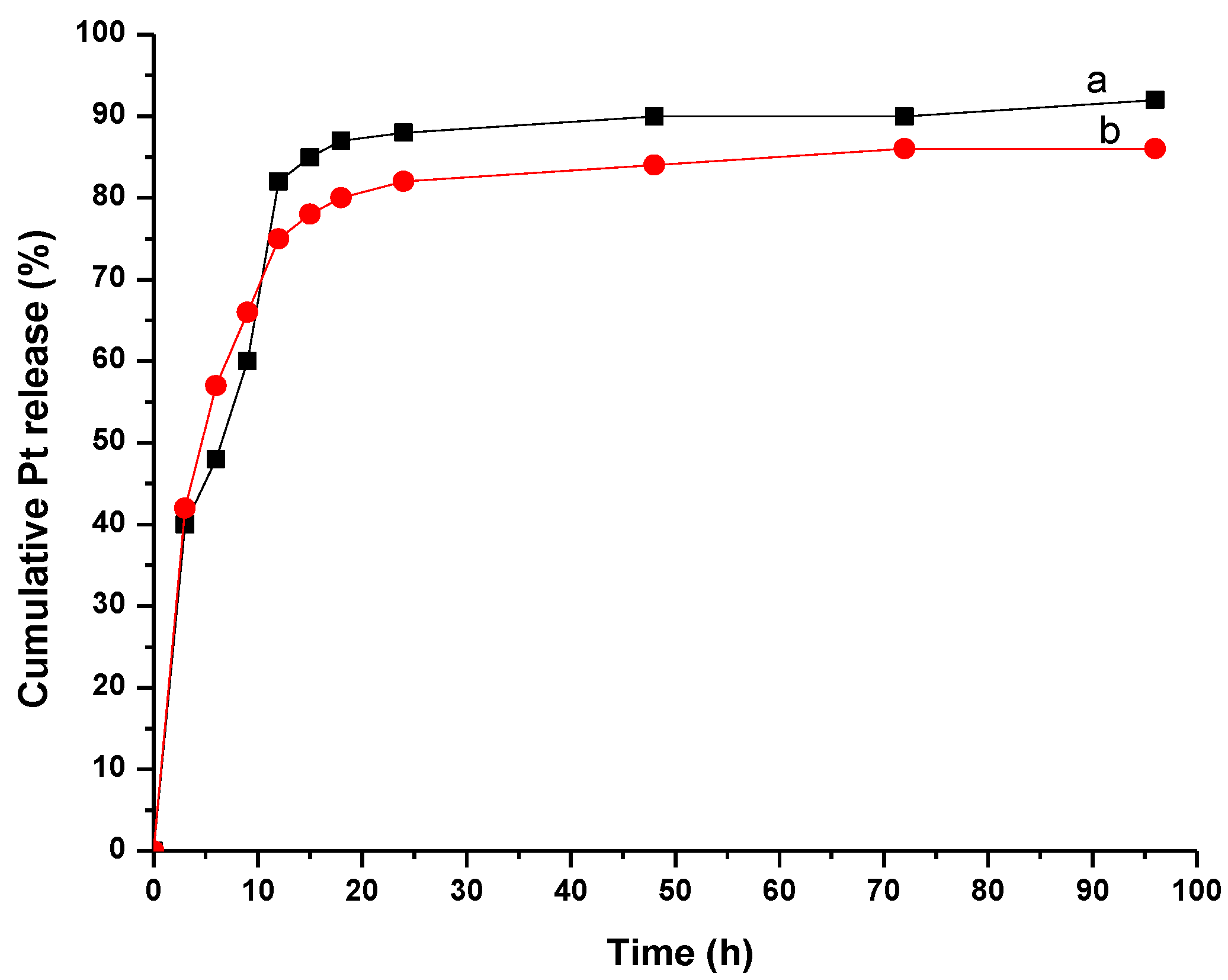
3.4. In Vitro Release of CDDP from MWCNT-COOH-CDDP Complex
3.5. In Vitro Release of CDDP from MWCNT-COOH-CDDP in Neutral and Weakly Acidic Conditions
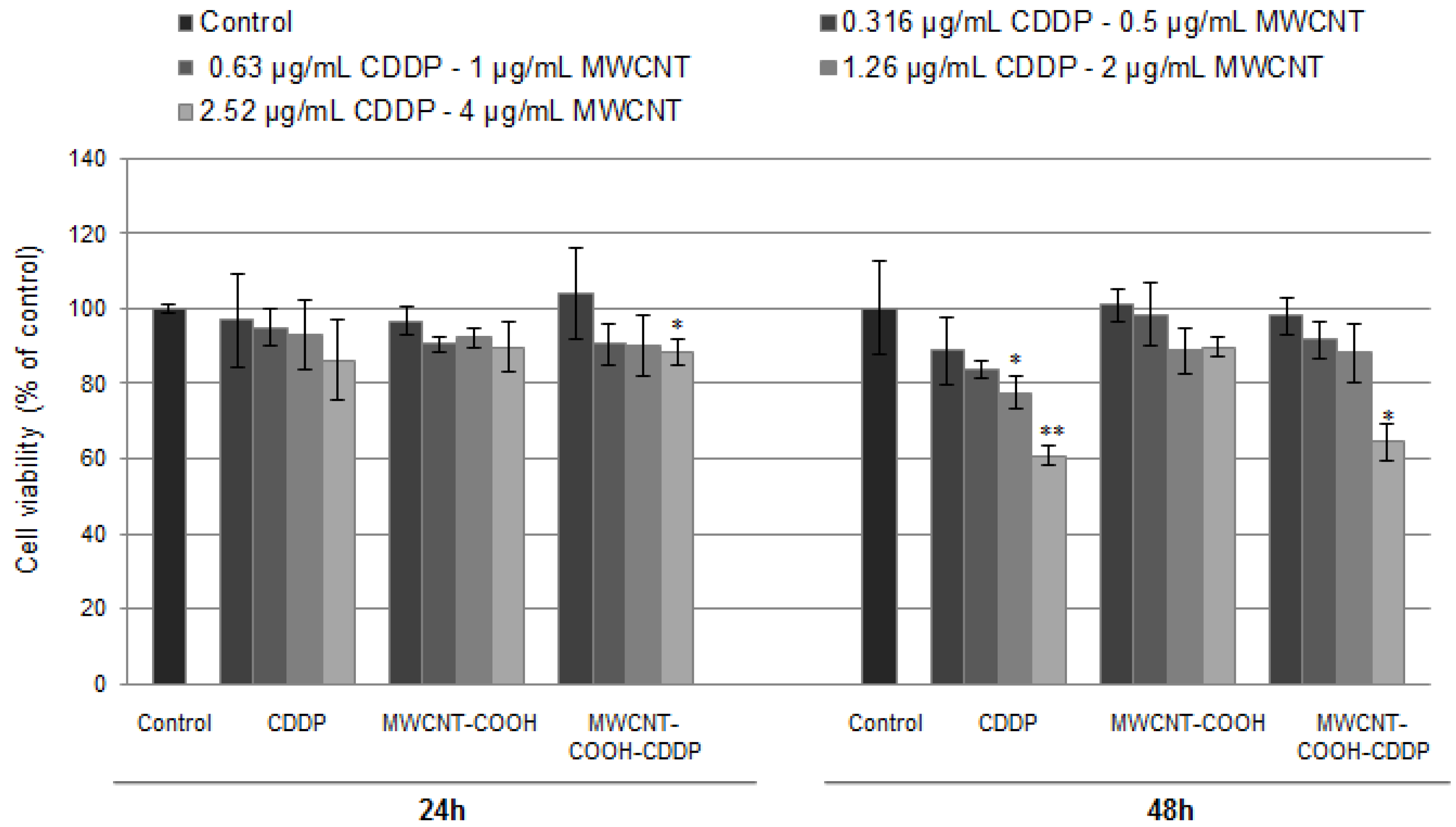
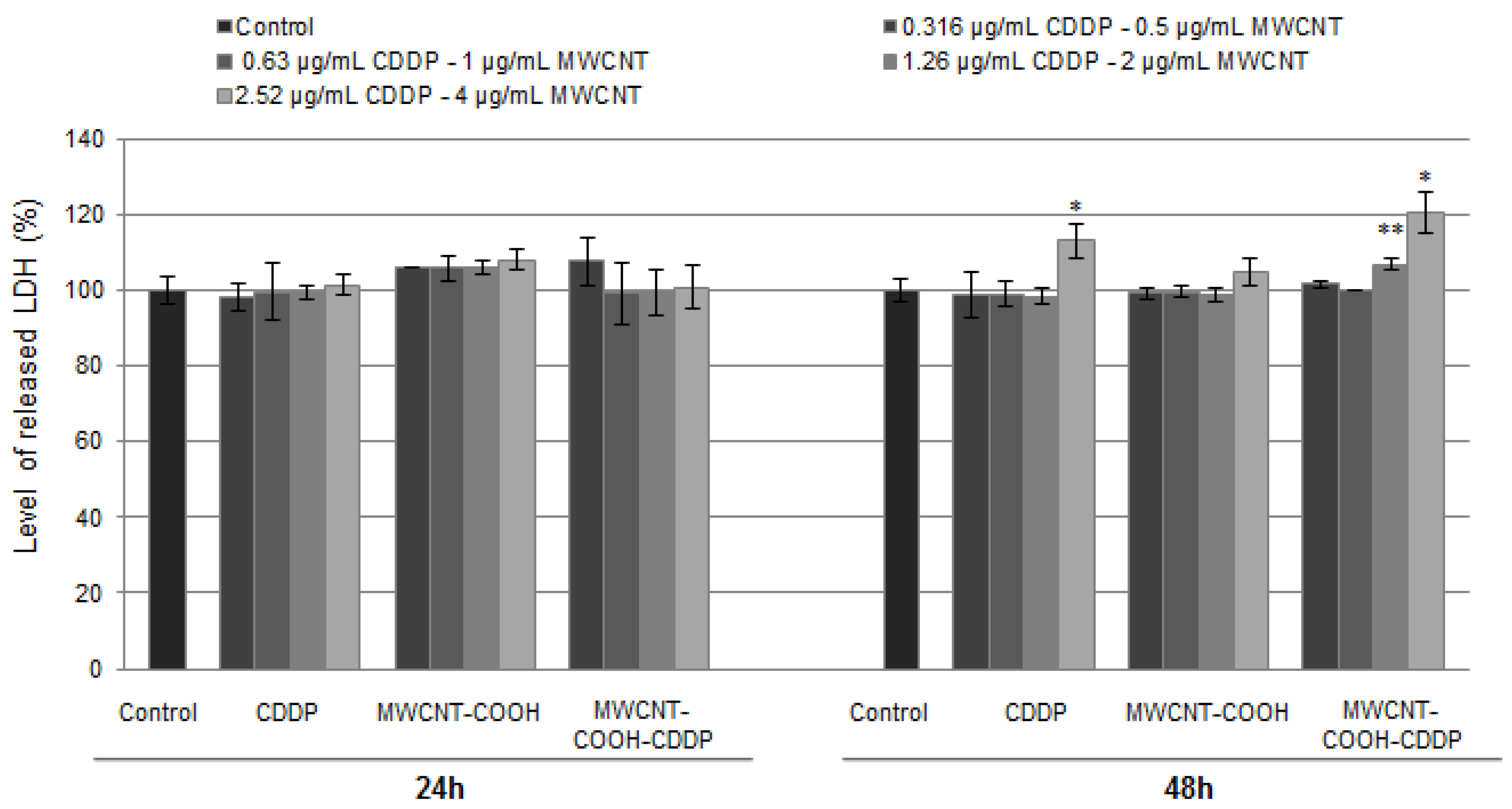
3.6. Cytotoxicity of Drug Loaded MWCNT
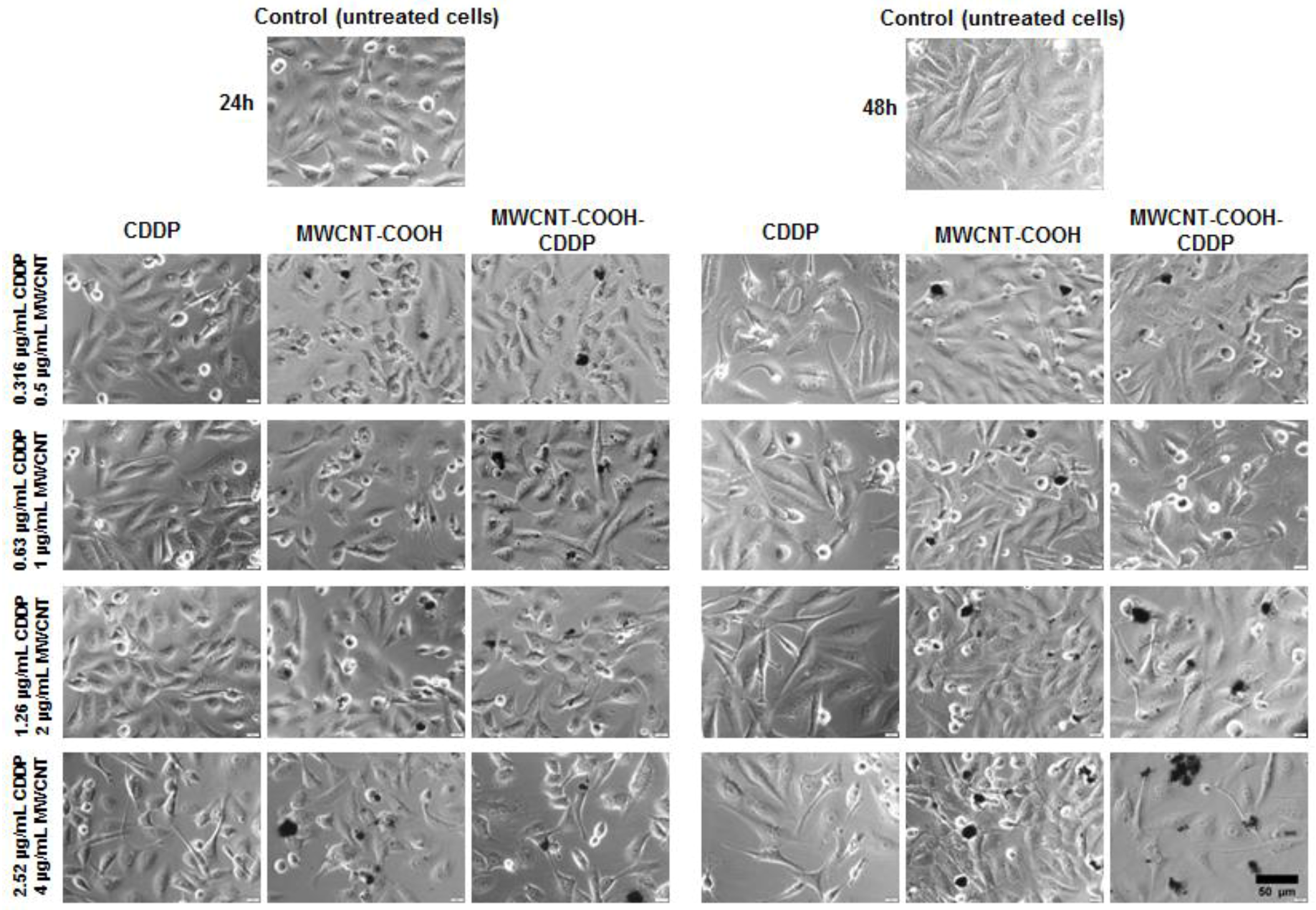
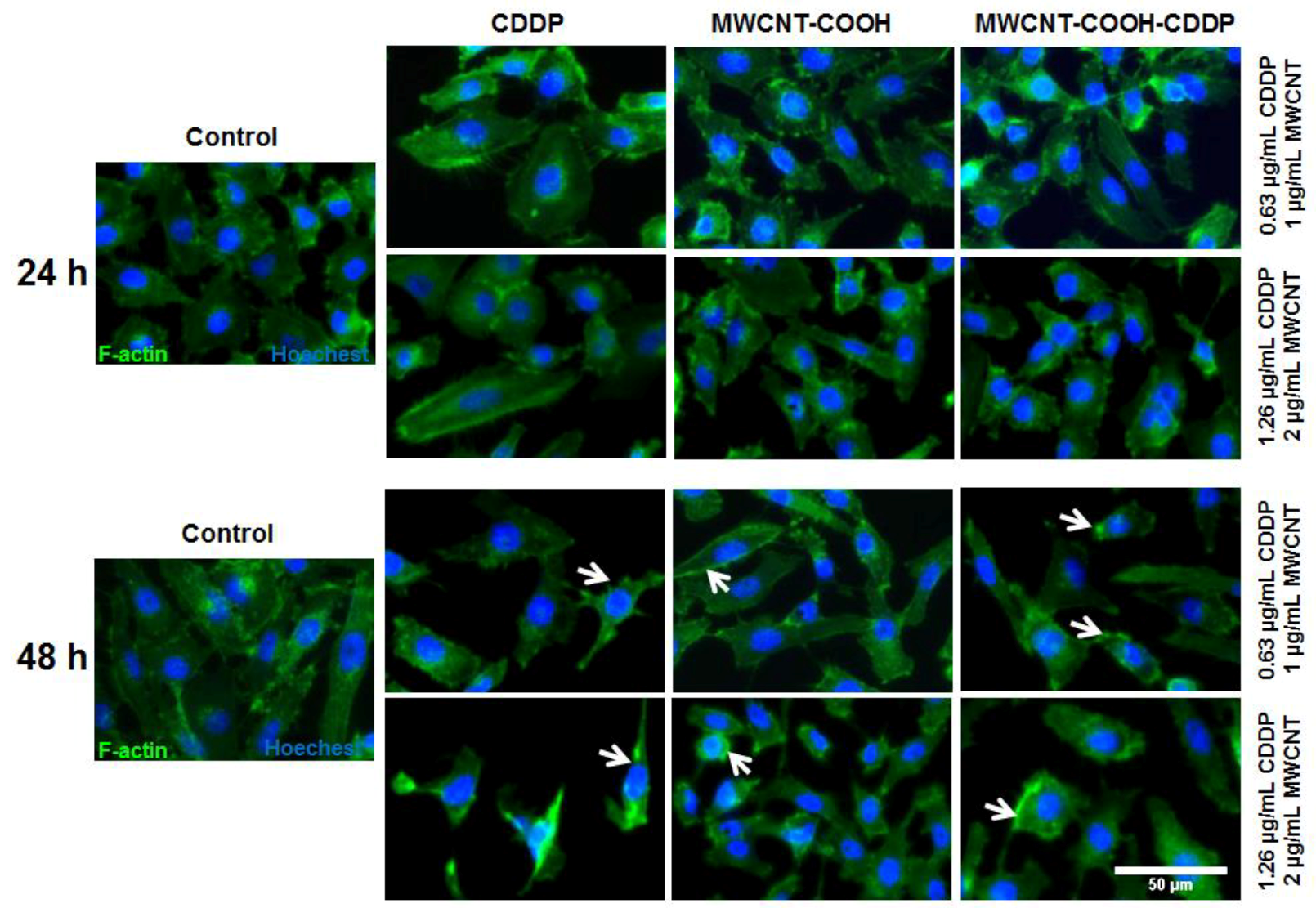
3.7. Cell Morphology and Actin Cytoskeleton Integrity
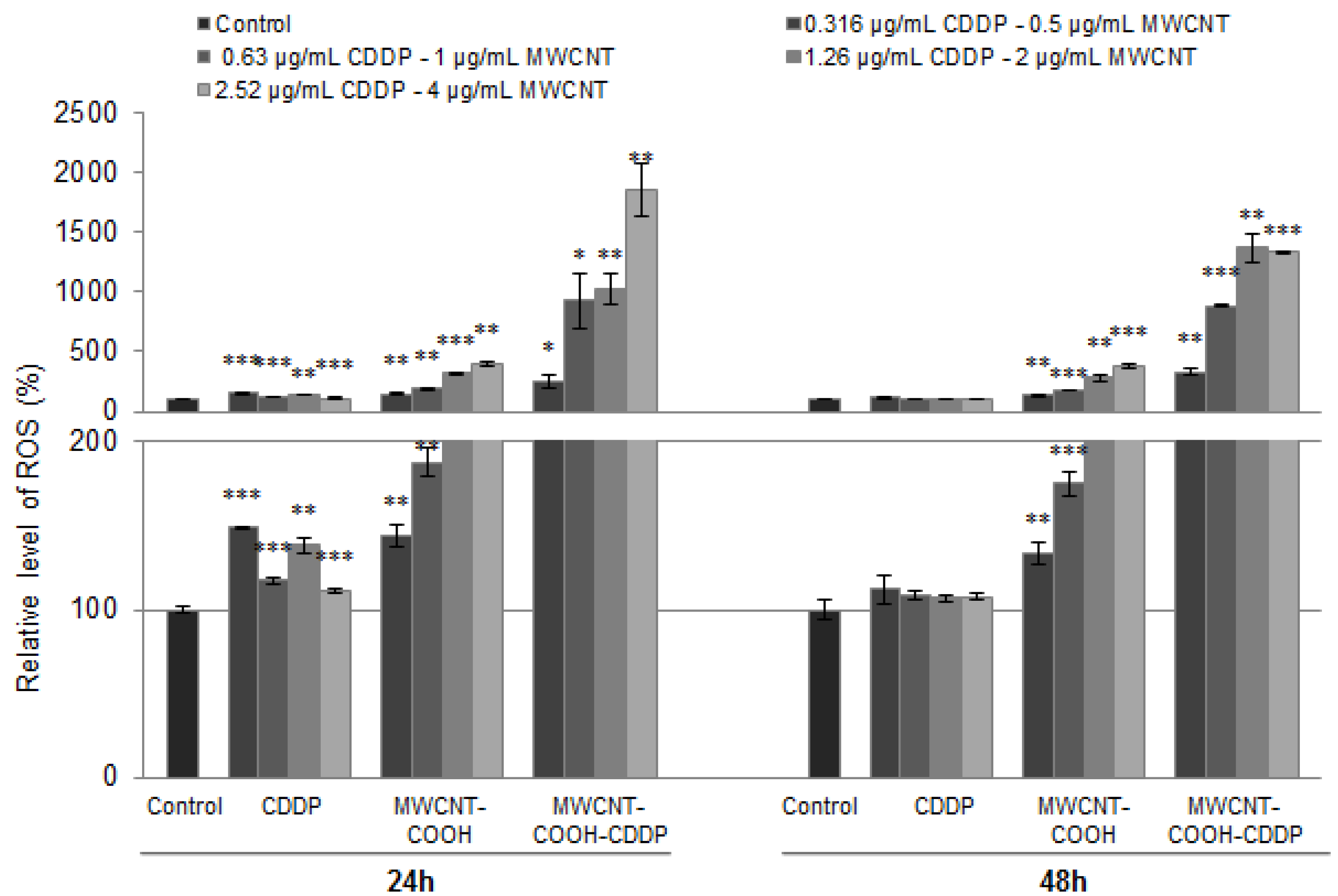
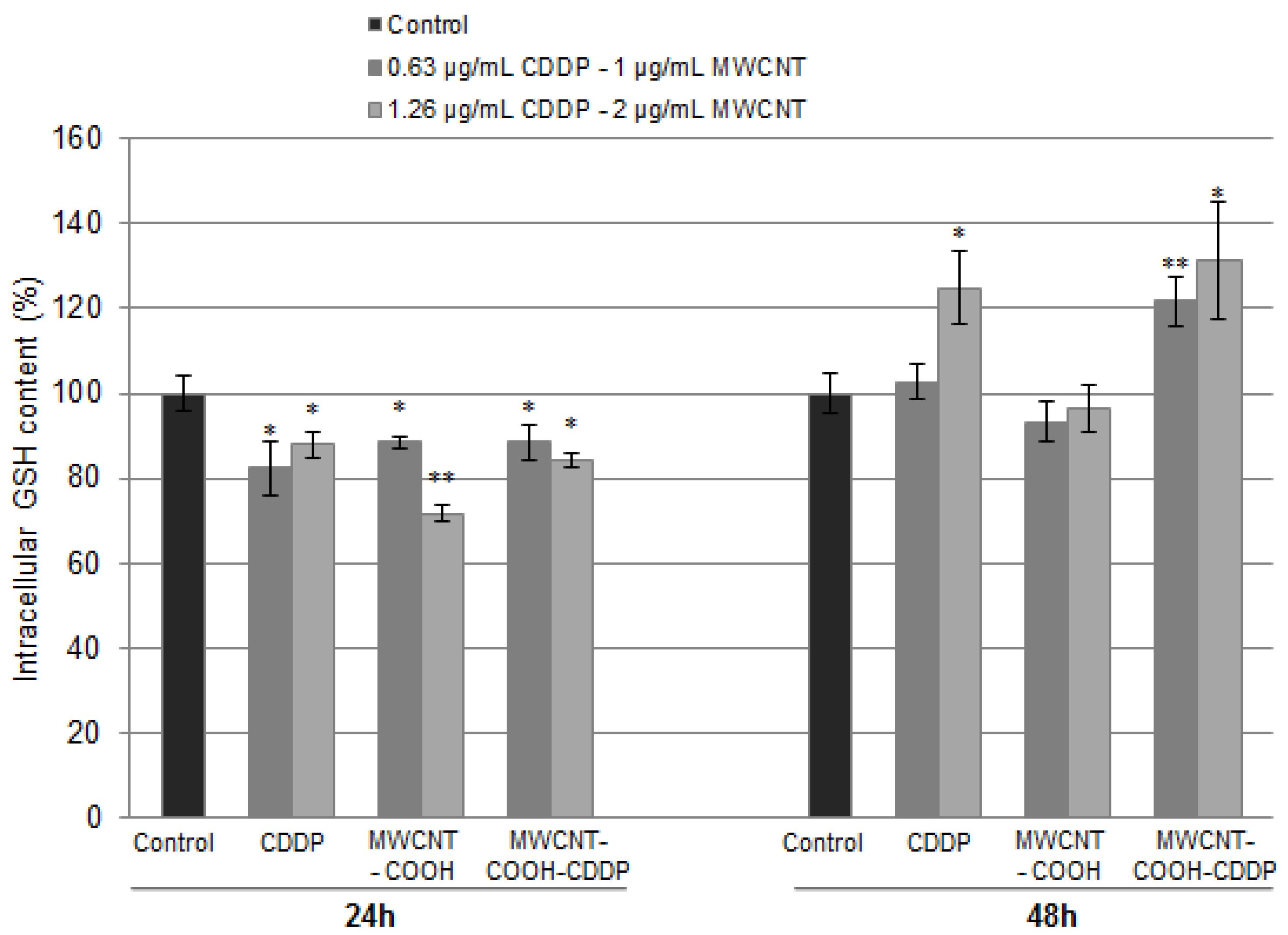
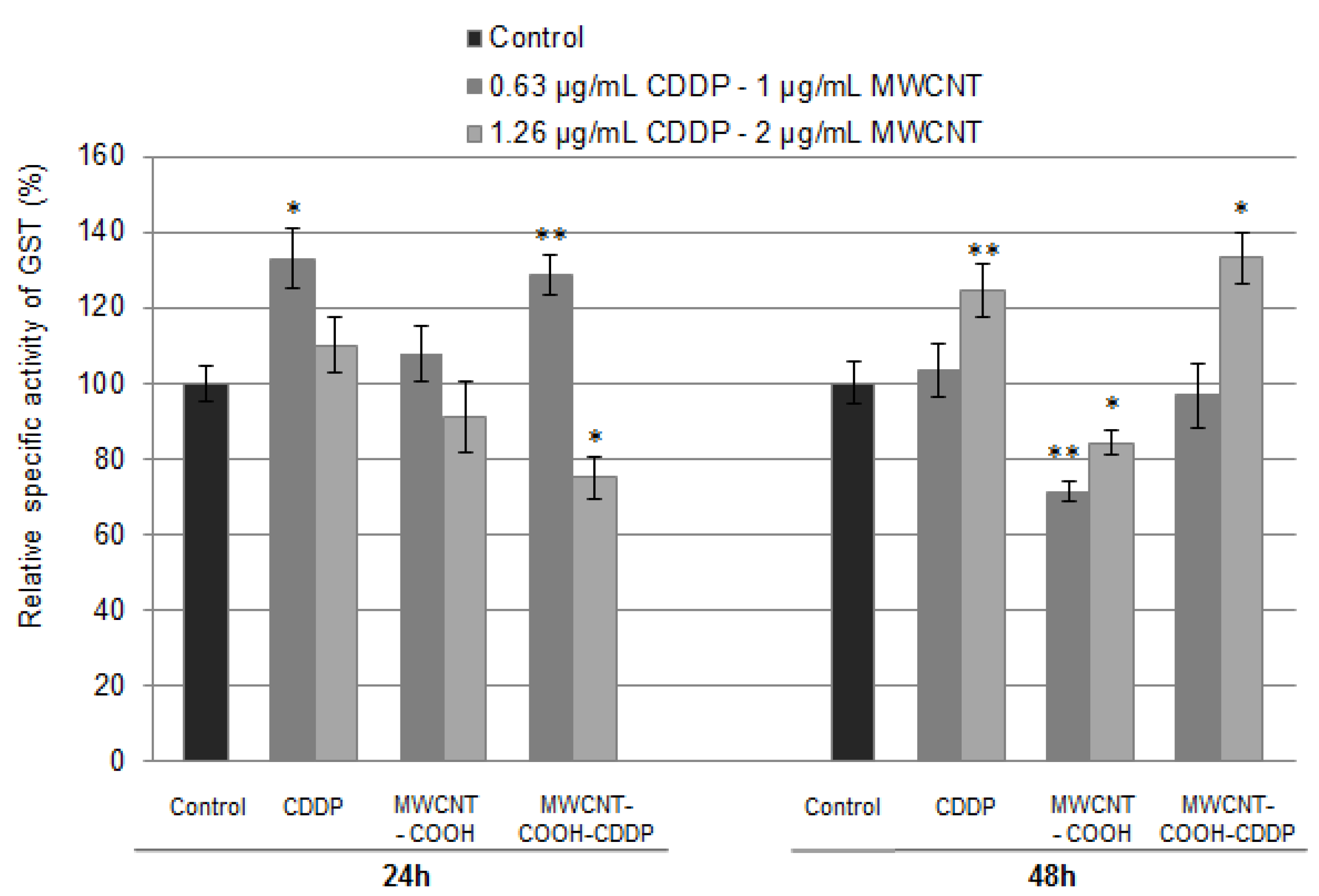
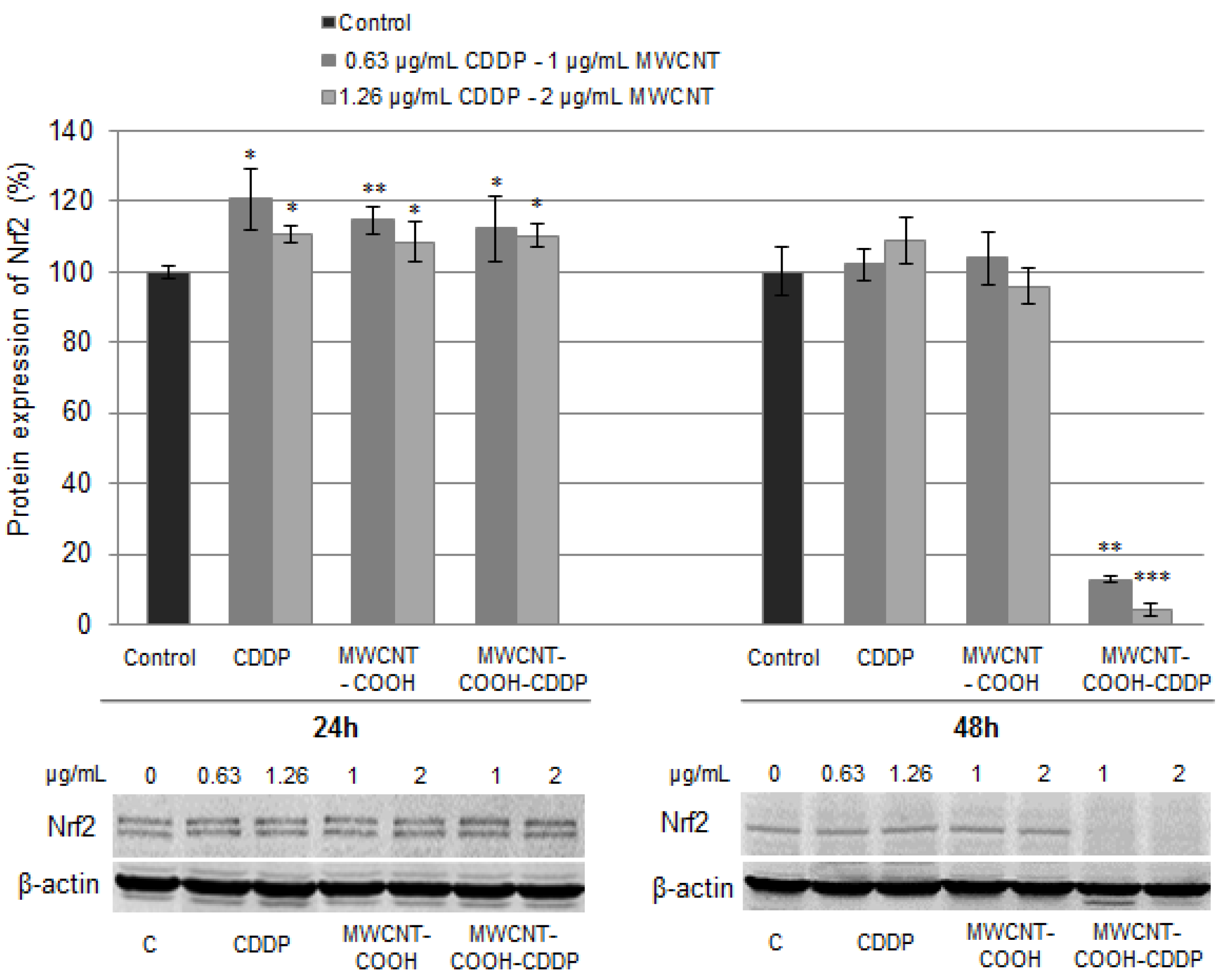
3.8. Oxidative Stress
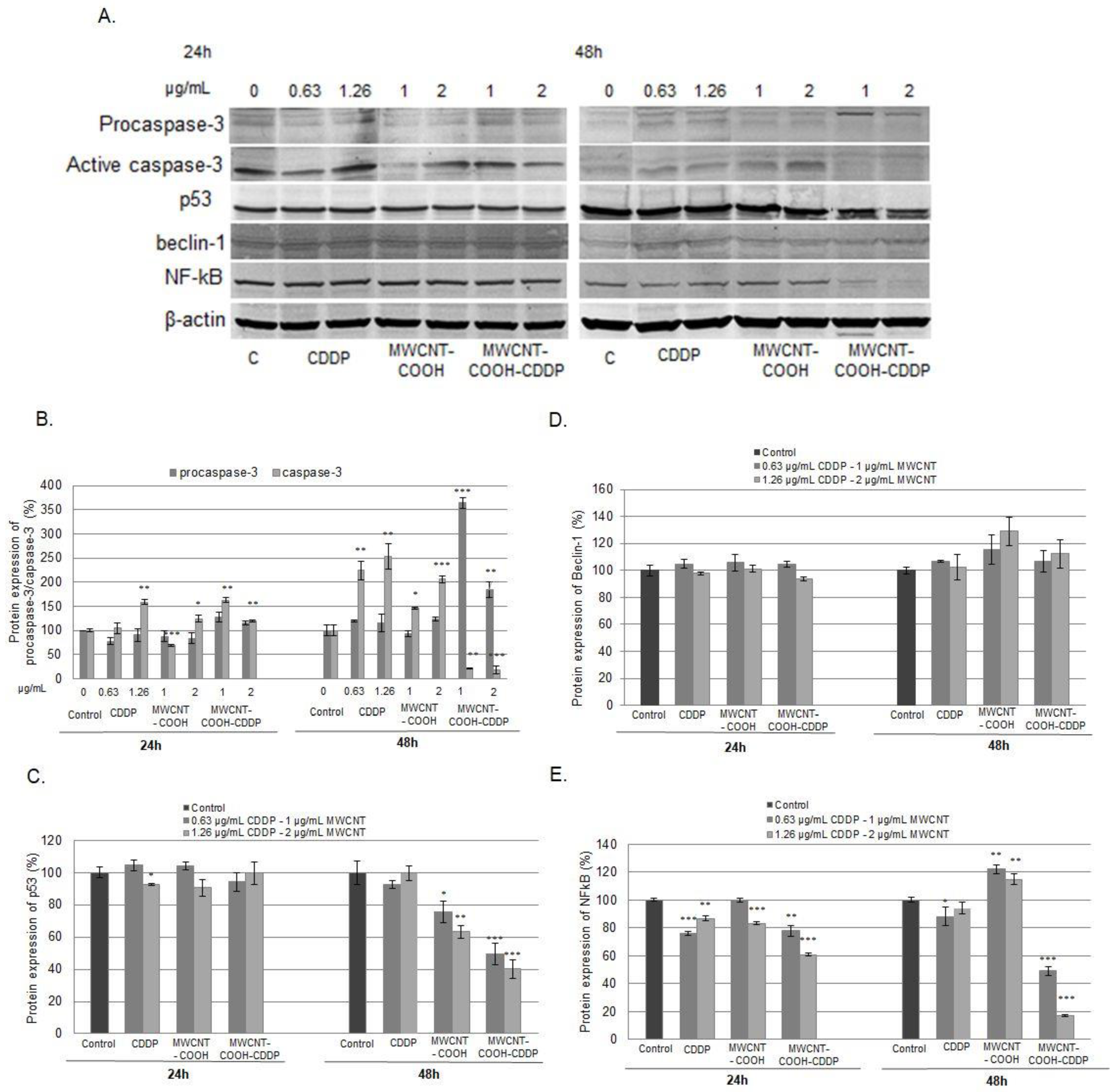
3.9. Cell Death Inhibition
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Prodana, M.; Ionita, D.; Ungureanu, C.; Bojin, D.; Demetrescu, I. Enhancing antibacterial effect of multiwalled carbon nanotubes using silver nanoparticles. Dig. J. Nanomater. Biostruct. 2011, 6, 549–556. [Google Scholar]
- Boncel, S.; Brzezinski, M.; Mrowiec-Bialon, J.; Janas, D.; Koziol, K.K.K.; Walczak, K.Z. Oxidised multi-wallcarbonnanotubes–(R)-polylactide composite with a covalent β-d-uridine filler-matrixlinker. Mater. Lett. 2013, 91, 50–54. [Google Scholar] [CrossRef]
- Hilder, T.A.; Hill, J.M. Carbon nanotubes as drug delivery nanocapsules. Curr. Appl. Phys. 2008, 8, 258–261. [Google Scholar] [CrossRef]
- Boncel, S.; Zając, P.; Koziol, K.K.K. Liberation of drugs from multi-wall carbon nanotube carriers. J. Control. Release 2013, 169, 126–140. [Google Scholar] [CrossRef] [PubMed]
- Shaffer, M.S.P.; Koziol, K. Polystyrene grafted multi-walled carbon nanotubes. Chem. Commun. 2002, 18, 2074–2075. [Google Scholar] [CrossRef]
- Karousis, N.; Tagmatarchis, N.; Tasis, D. Current progress on the chemical modification of carbon nanotubes. Chem. Rev. 2010, 110, 5366–5397. [Google Scholar] [CrossRef] [PubMed]
- Ganta, S.; Devalapally, H.; Shahiwala, A.; Amiji, M. A review of stimuli-responsive nanocarriers for drug and gene delivery. J. Control. Release 2008, 126, 187–204. [Google Scholar] [CrossRef] [PubMed]
- Kostarelos, K.; Lacerda, L.; Pastorin, G.; Wu, W.; Wieckowski, S.; Luangsivilay, J.; Godefroy, S.; Pantarotto, D.; Briand, J.P.; Muller, S.; et al. Cellular uptake of functionalized carbon nanotubes is independent of functional group and cell type. Nat. Nanotechnol. 2007, 2, 108–113. [Google Scholar] [CrossRef] [PubMed]
- Heister, E.; Neves, V.; Tilmaciu, C.; Lipert, K.; Beltran, V.S.; Coley, H.M.; Silva, S.R.P.; McFadden, J. Triple functionalisation of single-walled carbon nanotubes with doxorubicin, a monoclonal antibody, and a fluorescent marker for targeted cancer therapy. Carbon 2009, 47, 2152–2160. [Google Scholar] [CrossRef] [Green Version]
- Wong, B.S.; Yoong, S.L.; Jagusiak, A.; Panczyk, T.; Ho, H.K.; Ange, W.H.; Pastorin, G. Carbon nanotubes for delivery of small molecule drugs. Adv. Drug Deliv. Rev. 2013, 65, 1964–2015. [Google Scholar] [CrossRef] [PubMed]
- Mody, N.; Tekade, R.K.; Mehra, N.K.; Chopdey, P.; Jain, N.K. Dendrimer, Liposomes, Carbon Nanotubes and PLGA Nanoparticles: One Platform Assessment of Drug Delivery Potential. AAPS PharmSciTech 2014, 15, 388–399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aysola, K.; Desai, A.; Welch, C.; Xu, J.; Qin, Y.; Reddy, V.; Matthews, R.; Owens, C.; Okoli, J.; Beech, D.J.; et al. Triple negative breast cancer—An overview. Hereditary Genet. 2013, 2, 1–3. [Google Scholar] [CrossRef]
- Wahba, H.A.; El-Hadaad, H.A. Current approaches in treatment of triple-negative breast cancer. Cancer Biol. Med. 2015, 12, 106–116. [Google Scholar] [CrossRef] [PubMed]
- Wein, L.; Loi, S. Mechanisms of resistance of therpay in early-stage triple negative breast cancer (TNBC). Breast 2017, 34 (Suppl. 1), 27–30. [Google Scholar] [CrossRef]
- Balas, M.; Constanda, S.; Duma (Voiculet), A.; Prodana, M.; Hermenean, A.; Pop, S.; Demetrescu, I.; Dinischiotu, A. Fabrication and toxicity characterization of a hybrid material based on oxidized and aminated MWCNT loaded with carboplatin. Toxicol. In Vitro 2016, 37, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.G.P.; Baburao, C.; Pispati, V.; Pathipati, H.; Muthy, N.; Prassana, S.R.V.; Rathode, B.G. Carbon nanotubes. A novel drug delivery system. Int. J. Res. Pharm. Chem. 2012, 2, 523–532. [Google Scholar]
- Rastogi, V.; Yadav, P.; Bhattacharya, S.S.; Mishra, A.K.; Verma, N.; Verma, A.; Pandit, J.K. Carbon Nanotubes: An Emerging Drug Carrier for Targeting Cancer Cells. J. Drug Deliv. 2014, 2014, 670815. [Google Scholar] [CrossRef] [PubMed]
- Dasari, S.; Tchounwou, P.B. Cisplatin in cancer therapy: Molecular mechanisms of action. Eur. J. Pharmacol. 2014, 740, 364–378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siddik, Z.H. Cisplatin: Mode of cytotoxic action and molecular basis of resistance. Oncogene 2003, 22, 7265–7279. [Google Scholar] [CrossRef] [PubMed]
- Petrović, M.; Todorović, D. Biochemical and molecular mechanisms of action of cisplatin in cancer cells. FU Med. Biol. 2016, 18, 12–18. [Google Scholar]
- Florea, A.M.; Büsselberg, D. Cisplatin as an anti-tumor drug: Cellular mechanisms of activity, drug resistance and induced side effects. Cancers 2011, 3, 1351–1371. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Senovilla, L.; Vitale, I.; Michels, J.; Martins, I.; Kepp, O.; Castedo, M.; Kroemer, G. Molecular mechanisms of cisplatin resistance. Oncogene 2012, 31, 1869–1883. [Google Scholar] [CrossRef] [PubMed]
- De Souza, L.A.; Nogueira, C.A.S.; Ortega, P.F.R.; Lopes, J.F.; Calado, H.D.R.; Lavall, R.L.; Silva, G.G.; Dos Santos, H.F.; De Almeida, W.B. Inclusion complex between cisplatin and single-walled carbon nanotube: An integrated experimental and theoretical approach. Inorg. Chim. Acta 2016, 447, 38–44. [Google Scholar] [CrossRef]
- Montaser, A.; Golightly, D.W. Inductively Coupled Plasmas in Analytical Atomic Spectrometry, 2nd ed.; VCH Publishers: New York, NY, USA, 1992; p. 1040. ISBN 978-0-471-18811-7. [Google Scholar]
- Verbeke, J.S.; Detaevernier, M.R.; Denis, L.; Massart, D.L. The determination of platinum in biological fluids by means of graphite furnace atomic absorption spectroscopy. Clin. Chim. Acta 1981, 113, 329–333. [Google Scholar] [CrossRef]
- Jones, A.H. Determination of platinum and palladium in blood and urine by flameless atomic absorption spectroscopy. Anal. Chem. 1976, 48, 1472–14744. [Google Scholar] [CrossRef] [PubMed]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Habig, W.H.; Pabst, M.J.; Jakoby, W.B. Glutathione S-transferase. The first enzymatic step in mercapturic acid formation. J. Biol. Chem. 1974, 249, 7130–7139. [Google Scholar] [PubMed]
- Aviles, F.; Cauich-Rodriguez, J.V.; Tahl, M. Evaluation of mild acid oxidation treatments for MWCNT functionalization. Carbon 2009, 47, 2970–2975. [Google Scholar] [CrossRef]
- Gabano, E.; Ravera, M.; Cassino, C.; Bonetti, S.; Palmisano, G.; Osella, D. Stepwise assembly of platinum–folic acid conjugates. Inorg. Chim. Acta 2008, 361, 1447–1455. [Google Scholar] [CrossRef]
- Ajima, K.; Murakam, T.; Mizoguch, Y.; Tsuchida, K.; Ichihash, T.; Iijima, S.; Yudasaka, M. Enhancement of in vivo anticancer effects of cisplatin by incorporation inside single-wall carbon nanohorns. ACS Nano 2008, 2, 2057–2064. [Google Scholar] [CrossRef] [PubMed]
- Tripisciano, C.; Kraemer, K.; Taylor, A.; Borowiak-Palena, E. Single-wall carbon nanotubes based anticancer drug delivery system. Chem. Phys. Lett. 2009, 478, 200–205. [Google Scholar] [CrossRef]
- Ajima, K.; Yudasaka, M.; Murakami, T.; Maigné, A.; Shiba, K.; Iijima, S. Carbon nanohorns as anticancer drug carriers. Mol. Pharm. 2005, 2, 475–480. [Google Scholar] [CrossRef] [PubMed]
- Ajima, K.; Yudasaka, M.; Maigné, A.; Miyawaki, J.; Iijima, S. Effect of functional groups at hole edges on cisplatinrelease from inside single-wall carbon nanohorns. J. Phys. Chem. B 2006, 110, 5773–5778. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Xie, J.; Li, J.; Liu, F.; Wu, X.; Wang, Z. Cisplatin suppresses the growth and proliferation of breast and cervical cancer cell lines by inhibiting integrin β5-mediated glycolysis. Am. J. Cancer Res. 2016, 6, 1108–1117. [Google Scholar] [PubMed]
- Pauzi, A.Z.; Yeap, S.K.; Abu, N.; Lim, K.L.; Omar, A.R.; Aziz, S.A.; Chow, A.L.; Subramani, T.; Tan, S.G.; Alitheen, N.B. Combination of cisplatin and bromelain exerts synergistic cytotoxic effects against breast cancer cell line MDA-MB-231 in vitro. Chin. Med. 2016, 11, 46. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Wu, S.; Tan, L.; Tan, H.; Yu, B. Chitosan-functionalised single-walled carbon nanotube-mediated drug delivery of SNX-2112 in cancer cells. J. Biomater. Appl. 2016, 31, 379–386. [Google Scholar] [CrossRef] [PubMed]
- Zeidan, Y.H.; Jenkins, R.W.; Hannun, Y.A. Remodeling of cellular cytoskeleton by the acid sphingomyelinase/ceramide pathway. J. Cell Biol. 2008, 181, 335–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baribeau, S.; Chaudhry, P.; Parent, S.; Asselin, É. Resveratrol inhibits cisplatin-induced epithelial-to-mesenchymal transition in ovarian cancer cell lines. PLoS ONE 2014, 9, e86987. [Google Scholar] [CrossRef] [PubMed]
- Pacurari, M.; Qian, Y.; Fu, W.; Schwegler-Berry, D.; Ding, M.; Castranova, V.; Guo, N.L. Cell permeability, migration, and reactive oxygen species induced by multiwalled carbon nanotubes in human microvascular endothelial cells. J. Toxicol. Environ. Health A 2012, 75, 129–147. [Google Scholar] [CrossRef] [PubMed]
- Stańczyk, M.; Gromadzińska, J.; Wąsowicz, W. Roles of reactive oxygen species and selected antioxidants in regulation of cellular metabolism. Int. J. Occup. Med. Environ. Health 2005, 18, 15–26. [Google Scholar] [PubMed]
- Held, P. An Introduction to Reactive Oxygen Species; BioTek: Winuski, VT, USA, 2010. [Google Scholar]
- Liou, G.Y.; Storz, P. Reactive oxygen species in cancer. Free Radic. Res. 2010, 44, 479–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marullo, R.; Werner, E.; Degtyareva, N.; Moore, B.; Altavilla, G.; Ramalingam, S.S.; Doetsch, P.W. Cisplatin Induces a Mitochondrial-ROS Response That Contributes to Cytotoxicity Depending on Mitochondrial Redox Status and Bioenergetic Functions. PLoS ONE 2013, 8, e81162. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.M.; Kim, H.K.; Shim, W.; Anwar, M.A.; Kwon, J.W.; Kwon, H.K.; Kim, H.J.; Jeong, H.; Kim, H.M.; Hwang, D.; et al. Mechanism of cisplatin-induced cytotoxicity is correlated to impaired metabolism due to mitochondrial ROS generation. PLoS ONE 2015, 10, e0135083. [Google Scholar] [CrossRef] [PubMed]
- Gęgotek, A.; Cyuńczyk, M.; Łuczaj, W.; Bielawska, A.; Bielawski, K.; Skrzydlewska, E. The redox status of human breast cancer cell lines (MCF-7 and MDA-MB231) treated with novel dinuclear berenil-platinum(II) complexes. Pharmazie 2014, 69, 923–928. [Google Scholar] [PubMed]
- Itoh, T.; Terazawa, R.; Kojia, K.; Nakane, K.; Deguchi, T.; Ando, M.; Tsukamasa, Y.; Ito, M.; Nozawa, Y. Cisplatin induces production of reactive oxygen species via NADPH oxidase activation in human prostate cancer cells. Free Radic. Res. 2011, 45, 1033–1039. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Xing, G.; Chai, Z. Are carbon nanotubes safe? Nat. Nanotechnol. 2008, 3, 191–192. [Google Scholar] [CrossRef] [PubMed]
- Fu, P.P.; Xia, Q.; Hwang, H.M.; Ray, P.C.; Yu, H. Mechanisms of nanotoxicity: Generation of reactive oxygen species. J. Food Drug Anal. 2014, 22, 64–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muthu, M.S.; Abdulla, A.; Pandey, B.L. Major toxicities of carbon nanotubes induced by reactive oxygen species: Should we worry about the effects on the lungs, liver and normal cells? Nanomedicine 2013, 8, 863–866. [Google Scholar] [CrossRef] [PubMed]
- Ju, L.; Zhang, G.; Zhang, X.; Jia, Z.; Gao, X.; Jiang, Y.; Yan, C.; Duerksen-Hughes, P.J.; Chen, F.F.; Li, H.; et al. Proteomic Analysis of Cellular Response Induced by Multi-Walled Carbon Nanotubes Exposure in A549 Cells. PLoS ONE 2014, 9, e84974. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Stilwell, J.; Zhang, T.; Elboudwarej, O.; Jiang, H.; Selegue, J.P.; Cooke, P.A.; Gray, J.W.; Chen, F.F. Molecular characterization of the cytotoxic mechanism of multiwall carbon nanotubes and nano-onions on human skin fibroblast. Nano Lett. 2005, 5, 2448–2464. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Liu, B.; Zhu, R. Targeting ROS for Cancer Therapy. Chemo Open Access 2016, 5, 199. [Google Scholar] [CrossRef]
- Kim, D.; Park, G.B.; Hur, D.Y. Apoptotic signaling through reactive oxygen species in cancer cells. World J. Immunol. 2014, 4, 158–173. [Google Scholar] [CrossRef]
- Jansen, B.A.J.; Brouwer, J.; Reedijk, J. Glutathione induces cellular resistance against cationic dinuclear platinum anticancer drugs. J. Inorg. Biochem. 2002, 89, 197–202. [Google Scholar] [CrossRef]
- Godwin, A.K.; Meister, A.; O’Dwyer, P.J.; Huang, C.S.; Hamilton, T.C.; Anderson, M.E. High resistance to cisplatin in human ovarian cancer cell lines is associated with marked increase of glutathione synthesis. Proc. Natl. Acad. Sci. USA 1992, 89, 3070–3074. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.H.W.; Kuo, M.T. Role of glutathione in the regulation of cisplatin resistance in cancer chemotherapy. Met. Based Drugs 2010, 2010, 430939. [Google Scholar] [CrossRef] [PubMed]
- Sheehan, D.; Meade, G.; Foley, V.M.; Dowd, C.A. Structure, function and evolution of glutathione transferases: Implications for classification of non-mammalian members of an ancient enzyme superfamily. Biochem. J. 2001, 360, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Townsend, D.M.; Tew, K.D. The role of glutathione-S-transferase in anti-cancer drug resistance. Oncogene 2003, 22, 7369–7375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.H.; Wu, H.T.; Cheng, H.M.; Yen, T.J.; Lu, I.H.; Chang, H.C.; Jao, S.C.; Shing, T.K.; Li, W.S. Inhibition of glutathione S-transferase M1 by new gabosine analogues is essential for overcoming cisplatin resistance in lung cancer cells. J. Med. Chem. 2011, 54, 8574–8581. [Google Scholar] [CrossRef] [PubMed]
- Sajadimajd, S.; Khazaei, M. Oxidative Stress and Cancer: The role of Nrf2. Curr. Cancer Drug Targets 2018, 18, 538–557. [Google Scholar] [CrossRef] [PubMed]
- Bryan, H.K.; Olayanju, A.; Goldring, C.E.; Park, B.K. The Nrf2 cell defence pathway: Keap1-dependent and -independent mechanisms of regulation. Biochem. Pharmacol. 2013, 85, 705–717. [Google Scholar] [CrossRef] [PubMed]
- Kansanen, E.; Kuosmanen, S.M.; Leinonen, H.; Levonen, A.L. The Keap1-Nrf2 pathway: Mechanisms of activation and dysregulation in cancer. Redox Biol. 2013, 1, 45–49. [Google Scholar] [CrossRef] [PubMed]
- Lau, A.; Tian, W.; Whitman, S.A.; Zhang, D.D. The Predicted Molecular Weight of Nrf2: It Is What It Is Not. Antioxid. Redox Signal. 2013, 18, 91–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kemmerer, Z.A.; Ader, N.R.; Mulroy, S.S.; Eggler, A.L. Comparison of human Nrf2 antibodies: A tale of two proteins. Toxicol. Lett. 2015, 238, 83–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sporn, M.B.; Liby, K.T. NRF2 and cancer: The good, the bad and the importance of context. Nat. Rev. Cancer 2012, 12. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.M.; Manandhar, S.; Lee, H.R.; Park, H.M.; Kwak, M.K. Role of the Nrf2-antioxidant system in cytotoxicity mediated by anticancer cisplatin: Implication to cancer cell resistance. Cancer Lett. 2008, 260, 96–108. [Google Scholar] [CrossRef] [PubMed]
- Bao, L.J.; Jaramillo, M.C.; Zhang, Z.B.; Zheng, Y.X.; Yao, M.; Zhang, D.D.; Yi, X.F. Nrf2 induces cisplatin resistance through activation of autophagy in ovarian carcinoma. Int. J. Clin. Exp. Pathol. 2014, 7, 1502–1513. [Google Scholar] [PubMed]
- Benchimol, S. p53-dependent pathways of apoptosis. Cell Death Differ. 2001, 8, 1049–1051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuler, M.; Green, D.R. Mechanisms of p53-dependent apoptosis. Biochem. Soc. Trans. 2001, 29, 684–688. [Google Scholar] [CrossRef] [PubMed]
- Kang, R.; Zeh, H.J.; Lotze, M.T.; Tang, D. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ. 2011, 18, 571–580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.-Y.; White, E. Role of autophagy in cancer prevention. Cancer Prev. Res. 2011, 4, 973–983. [Google Scholar] [CrossRef] [PubMed]
- Roos, W.P.; Kaina, B. DNA damage-induced cell death: From specific DNA lesions to the DNA damage response and apoptosis. Cancer Lett. 2013, 332, 237–248. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Mani, A.M.; Wu, Z.-H. DNA damage-induced nuclear factor-kappa B activation and its role in cancer progression. J. Cancer Metastasis Treat. 2017, 3, 45–59. [Google Scholar] [CrossRef] [PubMed]
- Antoon, J.W.; White, M.D.; Slaughter, E.M.; Driver, J.L.; Khalili, H.S.; Elliott, S.; Smith, C.D.; Burow, M.E.; Beckman, B.S. Targeting NFkB mediated breast cancer chemoresistance through selective inhibition of sphingosine kinase-2. Cancer Biol. Ther. 2011, 11, 678–689. [Google Scholar] [CrossRef] [PubMed]
- Ito, Y.; Kikuchi, E.; Tanaka, N.; Kosaka, T.; Suzuki, E.; Mizuno, R.; Shinojima, T.; Miyajima, A.; Umezawa, K.; Oya, M. Down-regulation of NF kappa B activation is an effective therapeutic modality in acquired platinum-resistant bladder cancer. BMC Cancer 2015, 15, 324. [Google Scholar] [CrossRef] [PubMed]
- Pendleton, C.S. Mechanisms of Cisplatin Resistance in Triple Negative Breast Cancer. Master’s Thesis, Vanderbilt University, Nashville, TN, USA, 2014. [Google Scholar]
- Gohr, K.; Hamacher, A.; Engelke, L.H.; Kassack, M.U. Inhibition of PI3K/Akt/mTOR overcomes cisplatin resistance in the triple negative breast cancer cell line HCC38. BMC Cancer 2017, 17, 711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Wang, H.; Zhao, L.; Fan, S.; Yang, Z.; Gao, F.; Chen, L.; Xiao, G.G.; Molnár, J.; Wang, Q. Down-regulation of P-glycoprotein is associated with resistance to cisplatin and VP-16 in human lung cancer cell lines. Anticancer Res. 2010, 30, 3593–3598. [Google Scholar] [PubMed]
- Dickens, E.; Ahmed, S. Principles of cancer treatment by chemotherapy. Surgery 2018, 36, 134–138. [Google Scholar] [CrossRef]
- Gorsic, L.K.; Stark, A.L.; Wheeler, H.E.; Wong, S.S.; Im, H.K.; Dolan, M.E. EPS8Inhibition Increases Cisplatin Sensitivity in Lung Cancer Cells. PLoS ONE 2013, 8, e82220. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.J.; Zhang, W.; Wang, Q.; Yang, Z.J.; Liao, S.B.; Li, L. FOLR1 increases sensitivity to cisplatin treatment in ovarian cancer cells. J. Ovarian Res. 2018, 11, 15. [Google Scholar] [CrossRef] [PubMed]













© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Badea, M.A.; Prodana, M.; Dinischiotu, A.; Crihana, C.; Ionita, D.; Balas, M. Cisplatin Loaded Multiwalled Carbon Nanotubes Induce Resistance in Triple Negative Breast Cancer Cells. Pharmaceutics 2018, 10, 228. https://doi.org/10.3390/pharmaceutics10040228
Badea MA, Prodana M, Dinischiotu A, Crihana C, Ionita D, Balas M. Cisplatin Loaded Multiwalled Carbon Nanotubes Induce Resistance in Triple Negative Breast Cancer Cells. Pharmaceutics. 2018; 10(4):228. https://doi.org/10.3390/pharmaceutics10040228
Chicago/Turabian StyleBadea, Madalina Andreea, Mariana Prodana, Anca Dinischiotu, Carmen Crihana, Daniela Ionita, and Mihaela Balas. 2018. "Cisplatin Loaded Multiwalled Carbon Nanotubes Induce Resistance in Triple Negative Breast Cancer Cells" Pharmaceutics 10, no. 4: 228. https://doi.org/10.3390/pharmaceutics10040228