Risperidone Controlled Release Microspheres Based on Poly(Lactic Acid)-Poly(Propylene Adipate) Novel Polymer Blends Appropriate for Long Acting Injectable Formulations


 ,
,  , and
, and 
Abstract
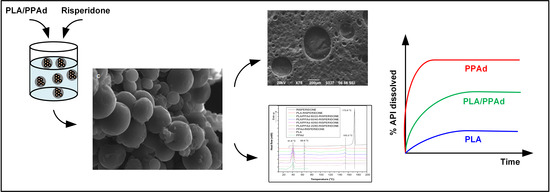
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Synthesis of Poly(Propylene Adipate) (PPAd) Aliphatic Polyesters
2.3. Poly(Lactic Acid)-Poly(Propylene Adipate) (PLA/PPAd) Blend Preparation
2.4. Characterization of Synthesized Polymers
2.4.1. Intrinsic Viscosity
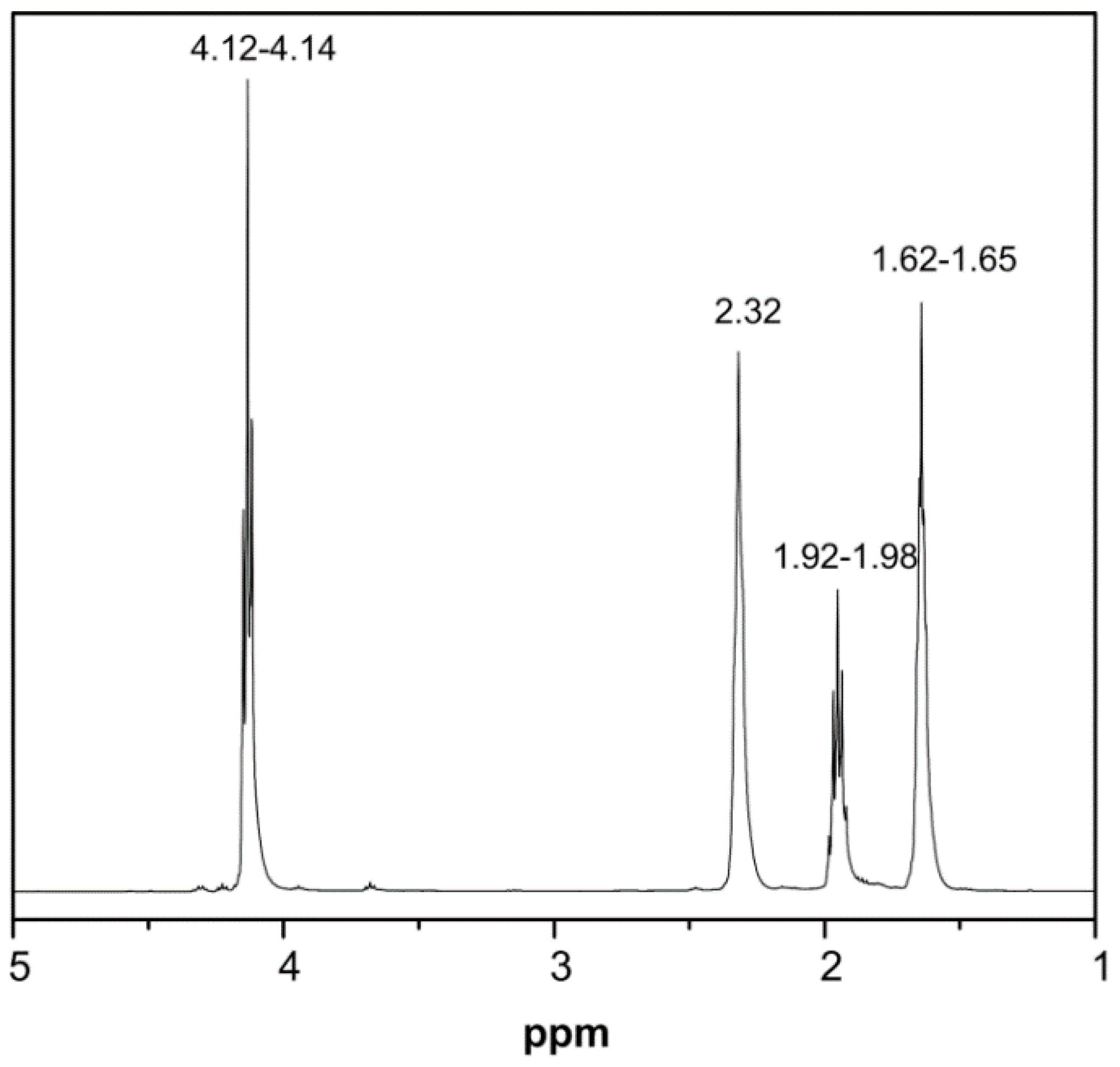
2.4.2. Nuclear Magnetic Resonance (NMR)
2.4.3. Gel permeation Chromatography
2.4.4. Wide Angle X-ray Scattering
2.4.5. Differential Scanning Calorimetry (DSC)
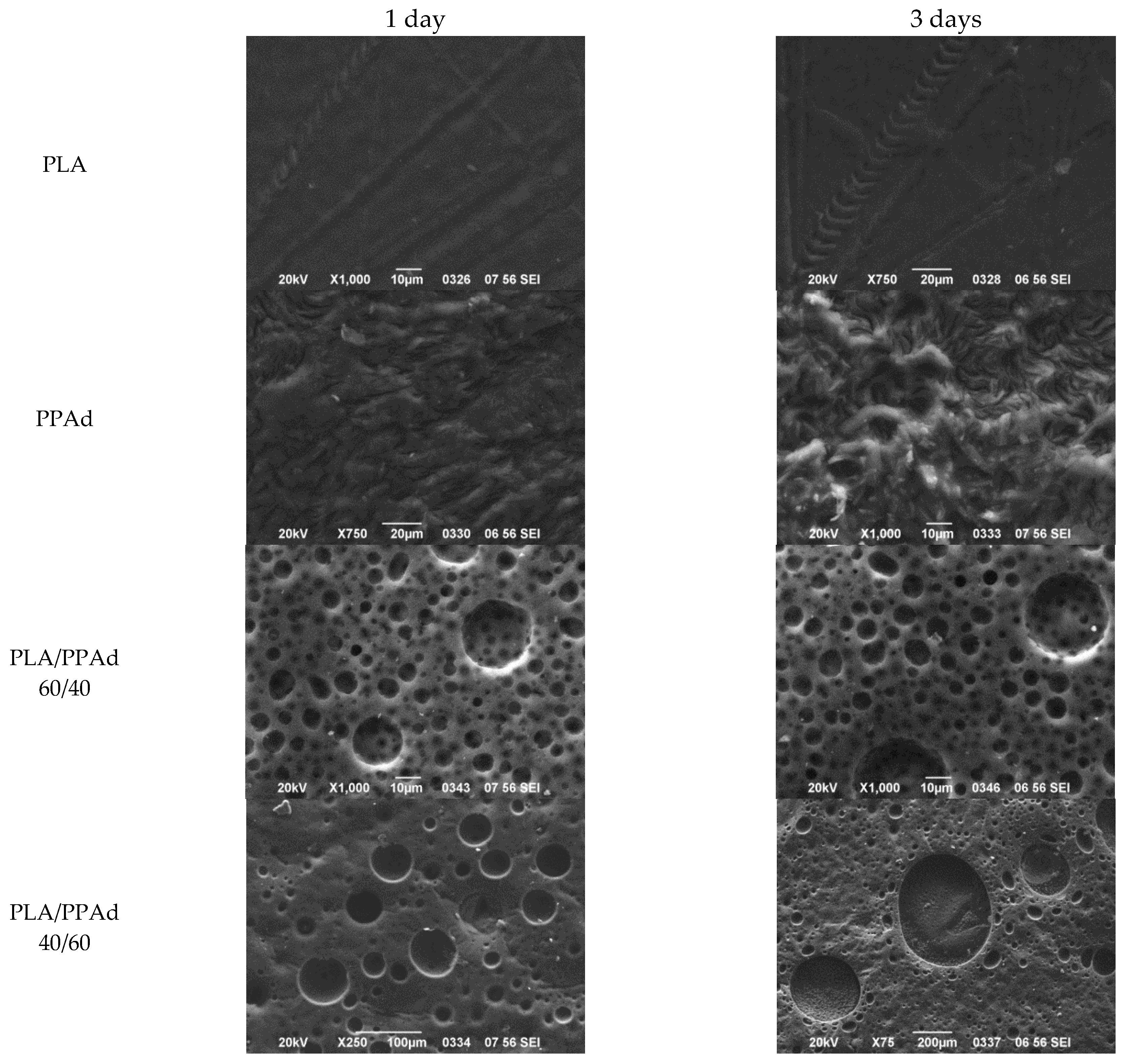
2.4.6. Enzymatic Hydrolysis
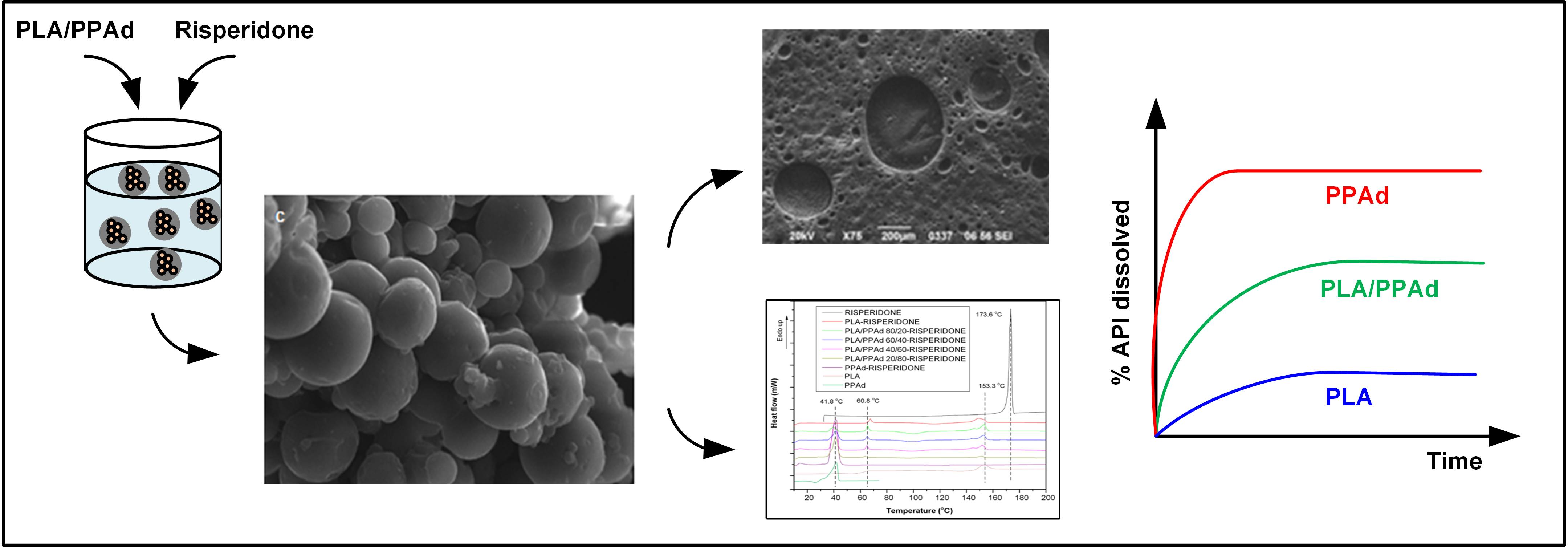
2.4.7. Scanning Electron Microscopy (SEM)
2.5. Preparation of Risperidone Microspheres
2.6. Characterization Techniques
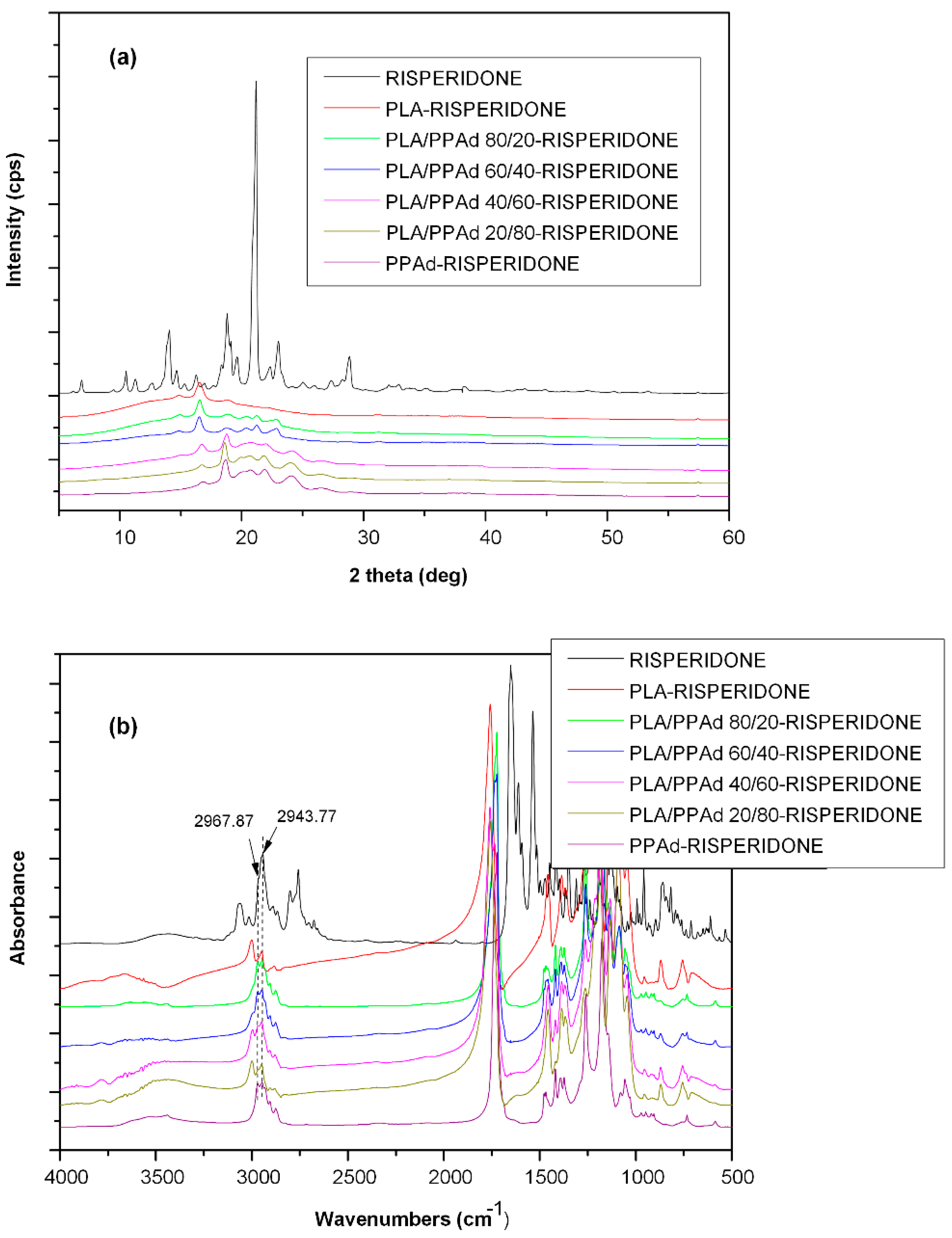
2.6.1. Fourier-Transformed Infrared (FTIR) Spectroscopy
2.6.2. Drug Loading, Yield, and Entrapment Efficiency (EE)
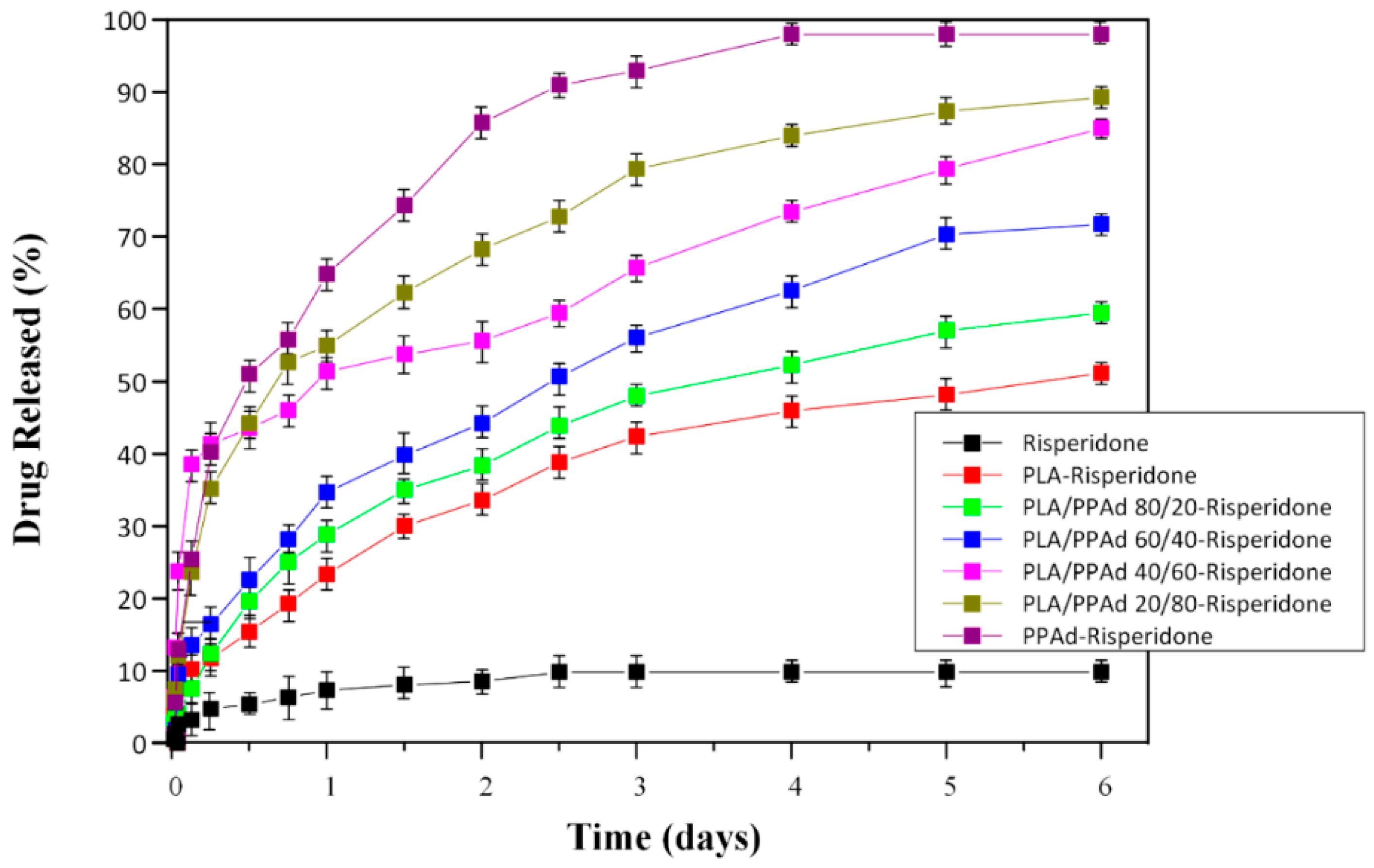
2.6.3. In-Vitro Drug Release
2.6.4. Statistical Moments
2.6.5. HPLC Method
3. Results and Discussion
3.1. Characterization of Prepared Polyesters
3.1.1. Synthesis and Characterization of PPAd
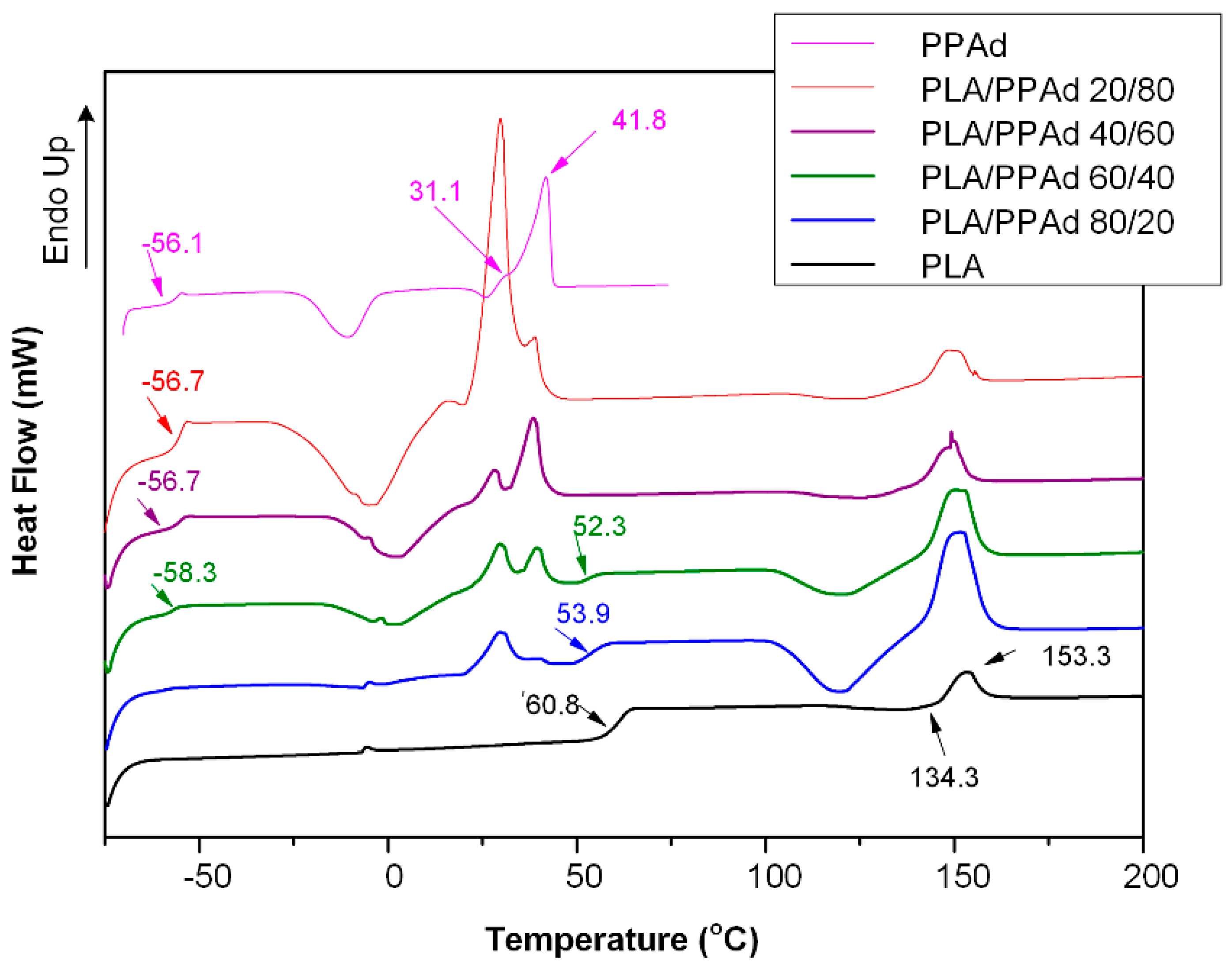
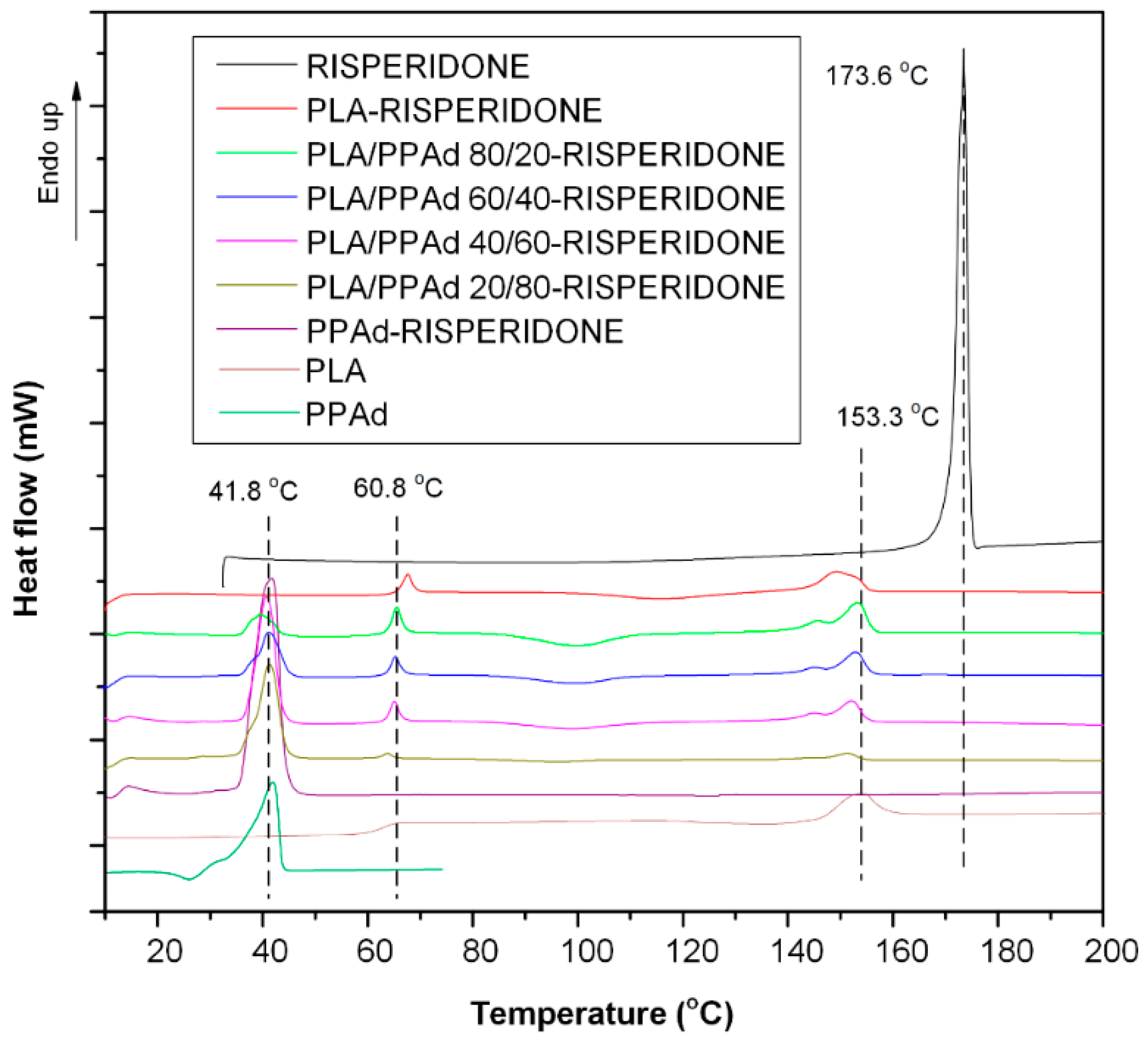
3.1.2. Thermal Analysis
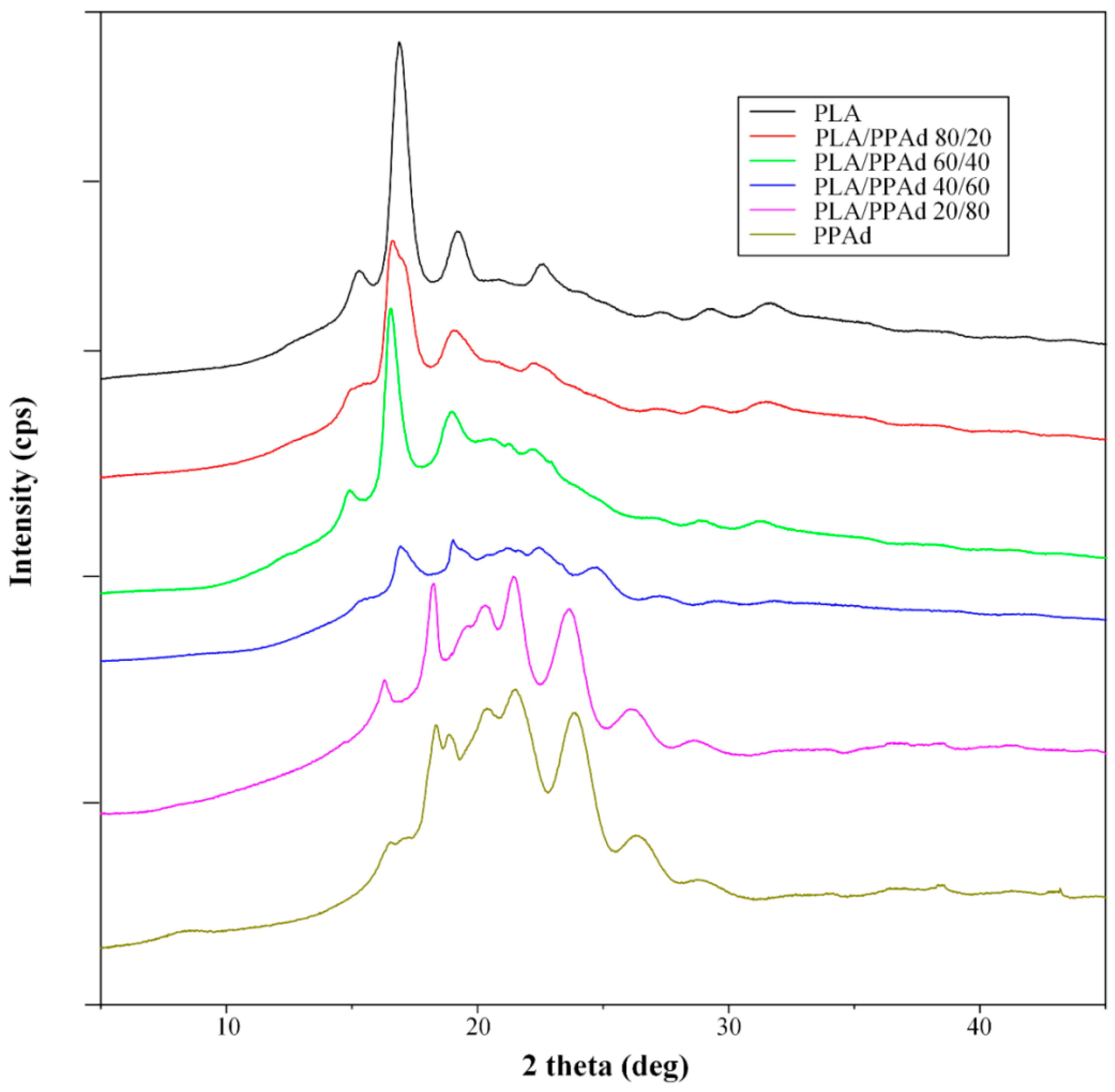
3.1.3. X-ray Diffraction Studies
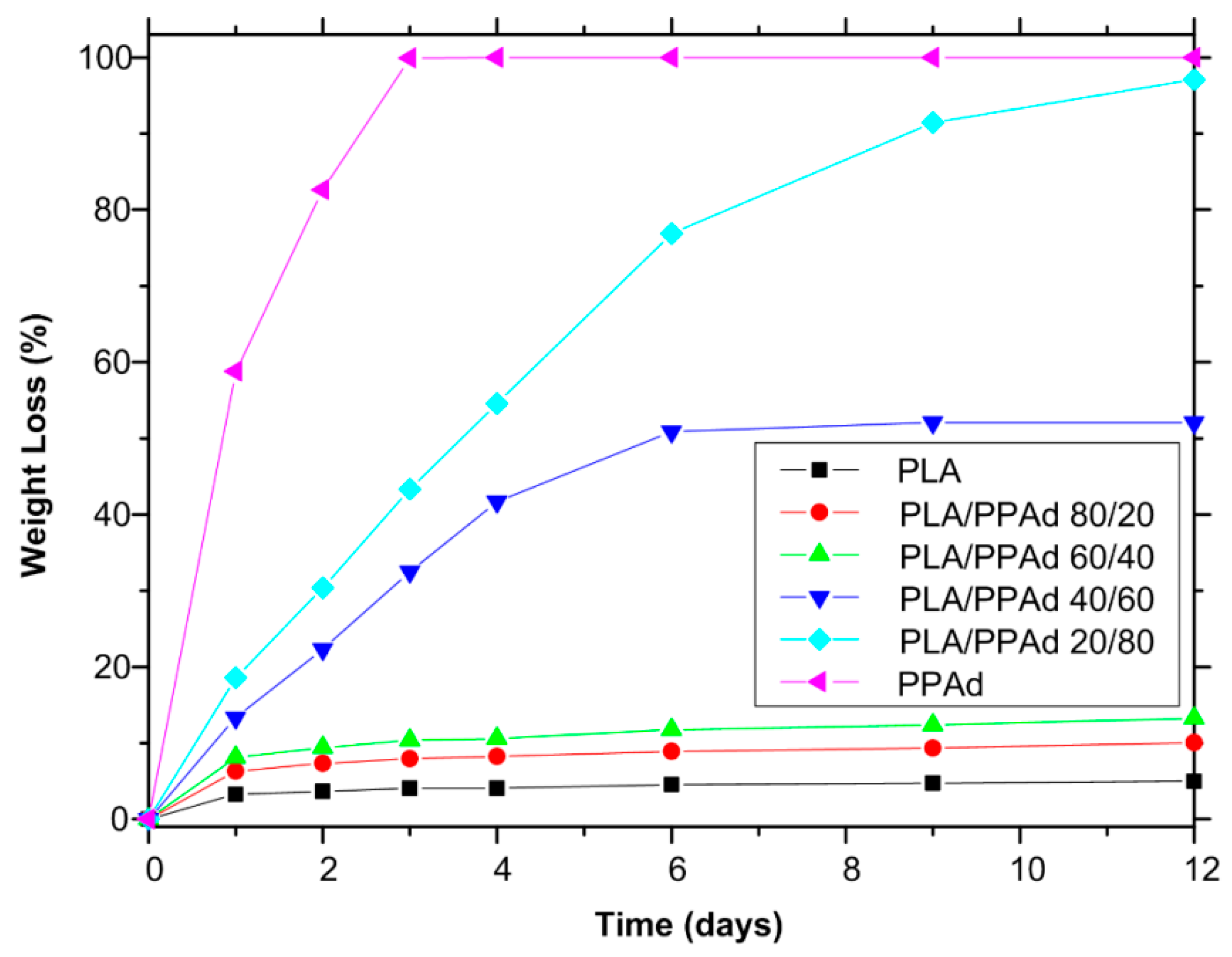
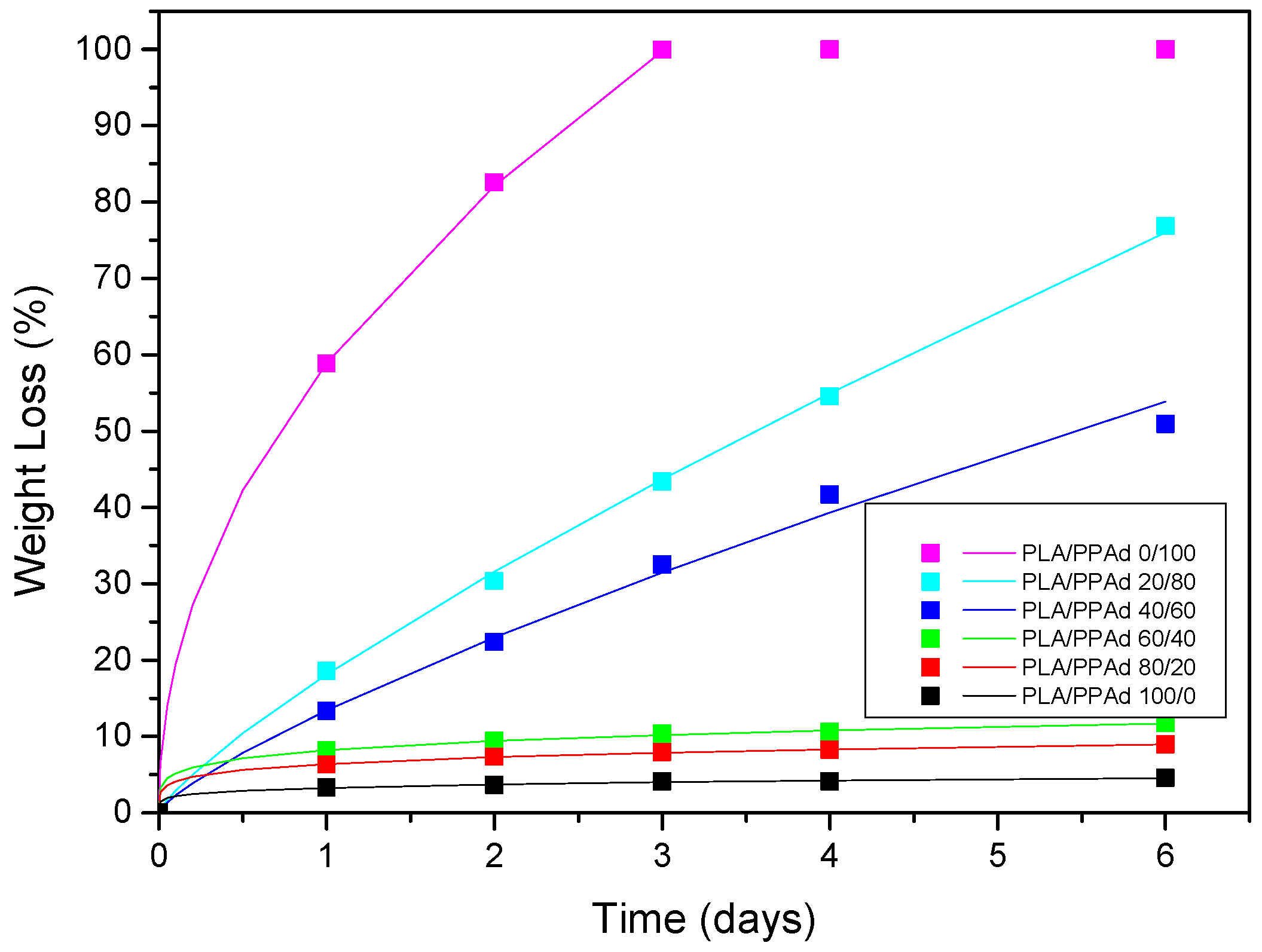
3.1.4. Hydrolysis Rate
3.2. Characterization of Risperidone Drug Formulations
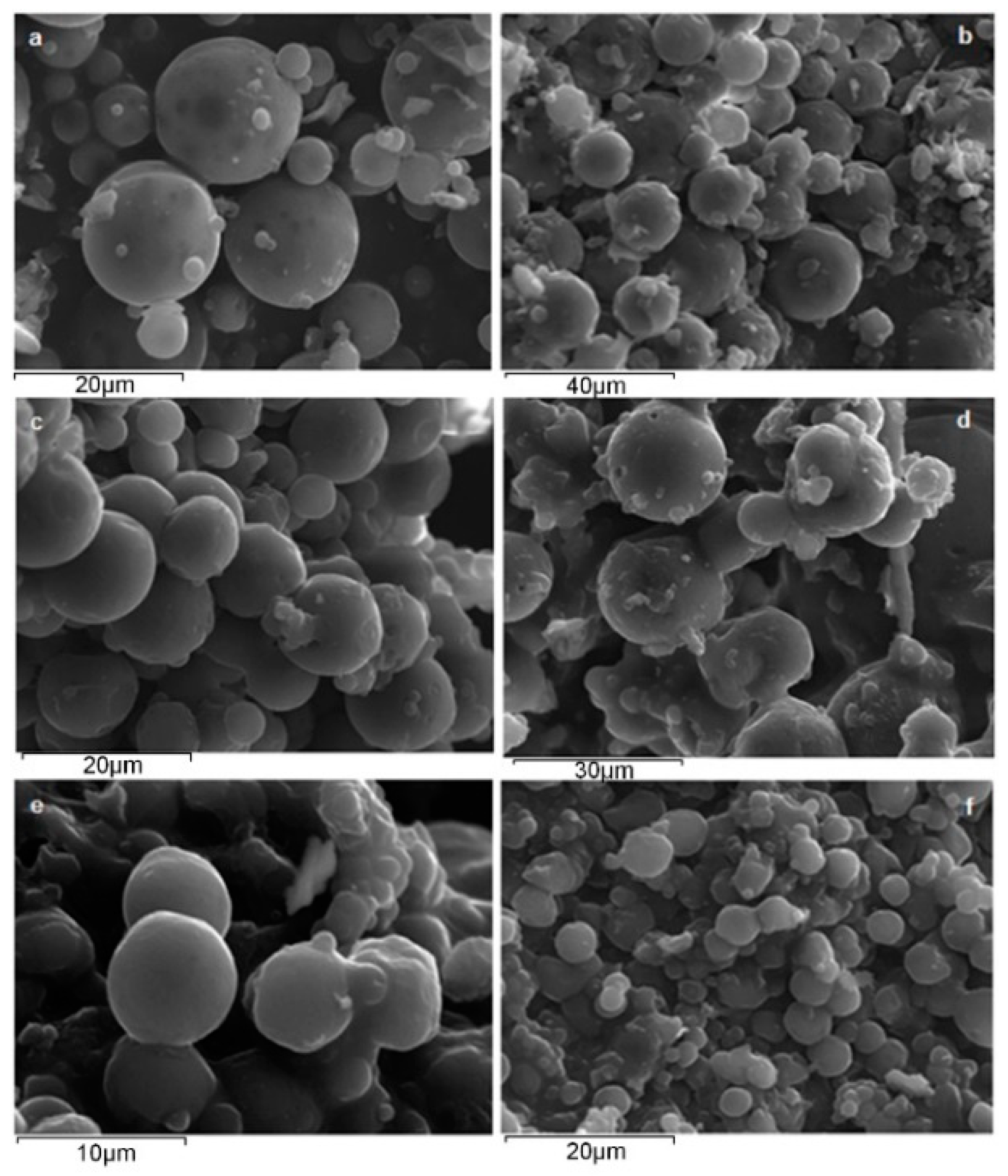
3.2.1. Microsphere Morphology
3.2.2. API Physical State Characterization
3.2.3. Drug Loading, Yield and % EE
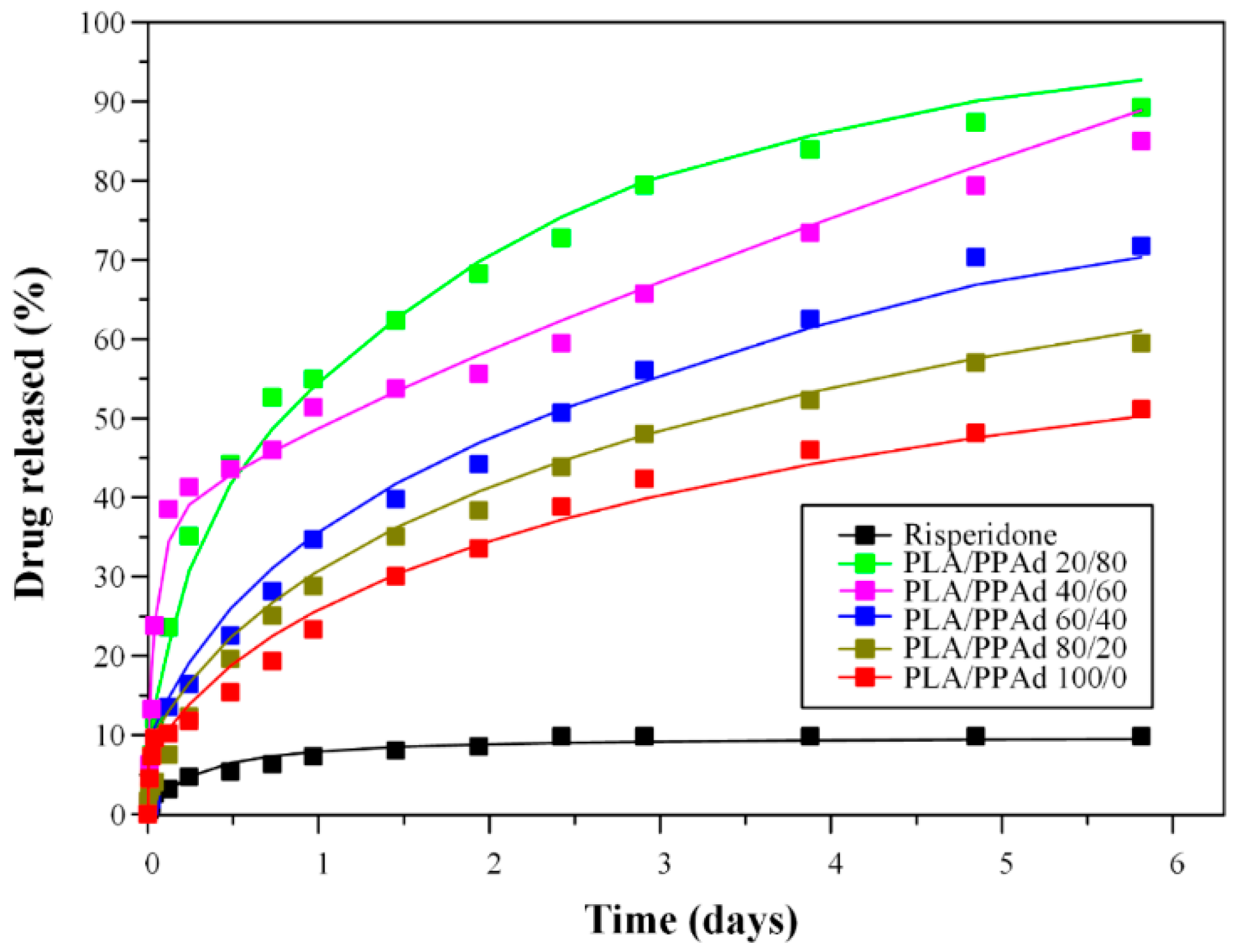
3.2.4. Dissolution Studies Results
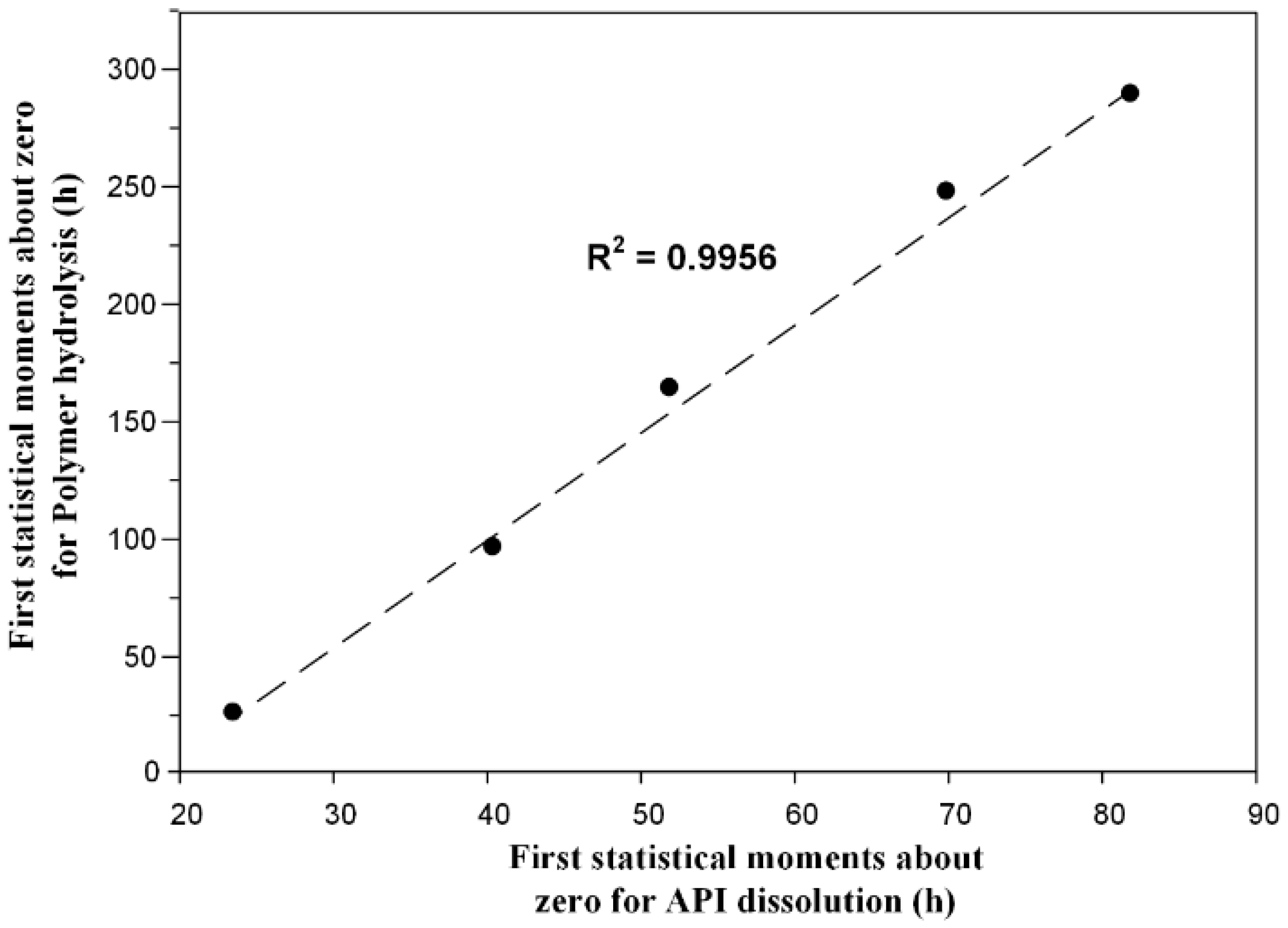
3.2.5. Statistical Moment Analysis
3.2.6. Release Data Modeling
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Baweja, R.; Sedky, K.; Lippmann, S. Long-acting antipsychotic medications. Curr. Drug Targets 2012, 13, 555–560. [Google Scholar] [CrossRef] [PubMed]
- Schwendeman, S.P.; Shah, R.B.; Bailey, B.A.; Schwendeman, A.S. Injectable controlled release depots for large molecules. J. Control. Release 2014, 190, 240–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wan, F.; Yang, M. Design of PLGA-based depot delivery systems for biopharmaceuticals prepared by spray drying. Int. Pharm. 2016, 498, 82–95. [Google Scholar] [CrossRef] [PubMed]
- Fleischhacker, W.W. Second-generation antipsychotic long-acting injections: Systematic review. Br. J. Psychiatry 2009, 52, S29–S36. [Google Scholar] [CrossRef] [PubMed]
- Chaurasia, S.; Mounika, K.; Bakshi, V.; Prasad, V. 3-month parenteral PLGA microsphere formulations of risperidone: Fabrication, characterization and neuropharmacological assessments. Mater. Sci. Eng. C Mater. Biol. Appl. 2017, 75, 1496–1505. [Google Scholar] [CrossRef] [PubMed]
- Remenar, J.F. Making the Leap from Daily Oral Dosing to Long-Acting Injectables: Lessons from the Antipsychotics. Mol. Pharm. 2014, 11, 1739–1749. [Google Scholar] [CrossRef] [PubMed]
- Tian, W.; Schulze, S.; Brandl, M.; Winter, G. Vesicular phospholipid gel-based depot formulations for pharmaceutical proteins: Development and in vitro evaluation. J. Control. Release 2010, 142, 319–325. [Google Scholar] [CrossRef] [PubMed]
- Dai, C.; Wang, B.; Zhao, H. Microencapsulation peptide and protein drugs delivery system. Colloids Surfaces B 2005, 41, 117–120. [Google Scholar] [CrossRef] [PubMed]
- Sinha, V.R.; Trehan, A. Biodegradable microspheres for protein delivery. J. Control. Release 2003, 90, 261–280. [Google Scholar] [CrossRef]
- Del Gaudio, C.; Crognale, V.; Serino, G.; Galloni, P.; Audenino, A.; Ribatti, D.; Morbiducci, U. Natural polymeric microspheres for modulated drug delivery. Mater. Sci. Eng. C Mater. Biol. Appl. 2017, 75, 408–417. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Gaete, C.; Retamal, M.; Chavez, C.; Bustos, P.; Godoy, R.; Torres-Vergara, P. Development, characterization and in vitro evaluation of biodegradable rhein-loaded microparticles for treatment of osteoarthritis. Eur. J. Pharm. Sci. 2017, 96, 390–397. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Gong, C.; Gou, M.; Fu, S.; Guo, Q.; Shi, S.; Luo, F.; Guo, G.; Qiu, L.; Qian, Z. Biodegradable poly(epsilon-caprolactone)-poly(ethylene glycol) copolymers as drug delivery system. Int. Pharm. 2009, 381, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Paciello, A.; Amalfitano, G.; Garziano, A.; Urciuolo, F.; Netti, P.A. Hemoglobin-Conjugated Gelatin Microsphere as a Smart Oxygen Releasing Biomaterial. Adv. Healthc. Mater. 2016, 5, 2655–2666. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Chen, X.; Fu, H.; Wen, X.; Ma, C.; Zhang, J.; Wu, C.; Huang, Y.; Pan, X.; Wu, C. Formation Mechanism and In Vitro Evaluation of Risperidone-Containing PLGA Microspheres Fabricated by Ultrafine Particle Processing System. J. Pharm. Sci. 2017, 106, 3363–3371. [Google Scholar] [CrossRef] [PubMed]
- Siafaka, P.I.; Barmpalexis, P.; Lazaridou, M.; Papageorgiou, G.Z.; Koutris, E.; Karavas, E.; Kostoglou, M.; Bikiaris, D.N. Controlled release formulations of risperidone antipsychotic drug in novel aliphatic polyester carriers: Data analysis and modelling. Eur. J. Pharm. Biopharm. 2015, 94, 473–484. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, A.S. The origins and evolution of “controlled” drug delivery systems. J. Control. Release 2008, 132, 153–163. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Jin, T. Polymer-Based Sustained-Release Dosage Forms for Protein Drugs, Challenges, and Recent Advances. AAPS PharmSciTech 2008, 9, 1218–1229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitragotri, S.; Burke, P.A.; Langer, R. Overcoming the challenges in administering biopharmaceuticals: Formulation and delivery strategies. Nat. Rev. Drug Discov. 2014, 13, 655–672. [Google Scholar] [CrossRef] [PubMed]
- Mao, S.; Guo, C.; Shi, Y.; Li, L.C. Recent advances in polymeric microspheres for parenteral drug delivery—Part 1. Expert Opin. Drug Deliv. 2012, 9, 1161–1176. [Google Scholar] [CrossRef] [PubMed]
- Gasmi, H.; Siepmann, F.; Hamoudi, M.C.; Danede, F.; Verin, J.; Willart, J.F.; Siepmann, J. Towards a better understanding of the different release phases from PLGA microparticles: Dexamethasone-loaded systems. Int. Pharm. 2016, 514, 189–199. [Google Scholar] [CrossRef] [PubMed]
- Casalini, T.; Rossi, F.; Lazzari, S.; Perale, G.; Masi, M. Mathematical modeling of PLGA microparticles: From polymer degradation to drug release. Mol. Pharm. 2014, 11, 4036–4048. [Google Scholar] [CrossRef] [PubMed]
- Park, E.J.; Amatya, S.; Kim, M.S.; Park, J.H.; Seol, E.; Lee, H.; Shin, Y.H.; Na, D.H. Long-acting injectable formulations of antipsychotic drugs for the treatment of schizophrenia. Arch. Pharm. Res. 2013, 36, 651–659. [Google Scholar] [CrossRef] [PubMed]
- Janssen, P.A.; Niemegeers, C.J.; Awouters, F.; Schellekens, K.H.; Megens, A.A.; Meert, T.F. Pharmacology of risperidone (R 64 766), a new antipsychotic with serotonin-S2 and dopamine-D2 antagonistic properties. J. Pharmacol. Exp. Ther. 1988, 244, 685–693. [Google Scholar] [PubMed]
- Cheng, F.; Jones, P.B. Drug treatments for schizophrenia: Pragmatism in trial design shows lack of progress in drug design. Epidemiol. Psychiatr. Sci. 2013, 22, 223–233. [Google Scholar] [CrossRef] [PubMed]
- Ereshefsky, L.; Mascarenas, C.A. Comparison of the effects of different routes of antipsychotic administration on pharmacokinetics and pharmacodynamics. J. Clin. Psychiatry 2003, 64 (Suppl. 16), 18–23. [Google Scholar] [PubMed]
- Yerragunta, B.; Jogala, S.; Chinnala, K.M.; Aukunuru, J. Development of a novel 3-month drug releasing risperidone microspheres. J. Pharm. Bioallied Sci. 2015, 7, 37–44. [Google Scholar] [PubMed]
- Su, Z.; Sun, F.; Shi, Y.; Jiang, C.; Meng, Q.; Teng, L.; Li, Y. Effects of formulation parameters on encapsulation efficiency and release behavior of risperidone poly(d,l-lactide-co-glycolide) microsphere. Chem. Pharm. Bull. 2009, 57, 1251–1256. [Google Scholar] [CrossRef] [PubMed]
- Su, Z.X.; Shi, Y.N.; Teng, L.S.; Li, X.; Wang, L.X.; Meng, Q.F.; Teng, L.R.; Li, Y.X. Biodegradable poly(d,l-lactide-co-glycolide) (PLGA) microspheres for sustained release of risperidone: Zero-order release formulation. Pharm. Dev. Technol. 2011, 16, 377–384. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Liu, Y.; Yuan, W.; Wu, F.; Su, J.; Jin, T. Effect of bases with different solubility on the release behavior of risperidone loaded PLGA microspheres. Colloids Surfaces B 2011, 86, 206–211. [Google Scholar] [CrossRef] [PubMed]
- Karavelidis, V.; Giliopoulos, D.; Karavas, E.; Bikiaris, D. Nanoencapsulation of a water soluble drug in biocompatible polyesters. Effect of polyesters melting point and glass transition temperature on drug release behavior. Eur. J. Pharm. Sci. 2010, 41, 636–643. [Google Scholar] [CrossRef] [PubMed]
- Karavelidis, V.; Bikiaris, D.; Avgoustakis, K. New thermosensitive nanoparticles prepared by biocompatible pegylated aliphatic polyester block copolymers for local cancer treatment. J. Pharm. Pharmacol. 2015, 67, 215–230. [Google Scholar] [CrossRef] [PubMed]
- Solomon, O.F.; Ciutǎ, I.Z. Détermination de la viscosité intrinsèque de solutions de polymères par une simple détermination de la viscosité. J. Appl. Polym. Sci. 1962, 6, 683–686. [Google Scholar] [CrossRef]
- Barmpalexis, P.; Kachrimanis, K.; Malamataris, S. Statistical moments in modelling of swelling, erosion and drug release of hydrophilic matrix-tablets. Int. Pharm. 2018, 540, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Nanaki, S.G.; Pantopoulos, K.; Bikiaris, D.N. Synthesis of biocompatible poly(varepsilon-caprolactone)-block-poly(propylene adipate) copolymers appropriate for drug nanoencapsulation in the form of core-shell nanoparticles. Int. J. Nanomed. 2011, 6, 2981–2995. [Google Scholar]
- Beslikas, T.; Gigis, I.; Goulios, V.; Christoforides, J.; Papageorgiou, G.Z.; Bikiaris, D.N. Crystallization study and comparative in vitro-in vivo hydrolysis of PLA reinforcement ligament. Int. J. Mol. Sci. 2011, 12, 6597–6618. [Google Scholar] [CrossRef] [PubMed]
- Papageorgiou, G.; Beslikas, T.; Gigis, J.; Christoforides, J.; Bikiaris, D.N. Crystallization and enzymatic hydrolysis of PLA grade for orthopedics. Adv. Polym. Technol. 2010, 29, 280–299. [Google Scholar] [CrossRef]
- Bikiaris, D.N.; Nianias, N.P.; Karagiannidou, E.G.; Docoslis, A. Effect of different nanoparticles on the properties and enzymatic hydrolysis mechanism of aliphatic polyesters. Polym. Degrad. Stab. 2012, 97, 2077–2089. [Google Scholar] [CrossRef]
- Qiu, Z.; Ikehara, T.; Nishi, T. Poly(hydroxybutyrate)/poly(butylene succinate) blends: Miscibility and nonisothermal crystallization. Polymer 2003, 44, 2503–2508. [Google Scholar] [CrossRef]
- Gan, Z.; Abe, H.; Kurokawa, H.; Doi, Y. Solid-State Microstructures, Thermal Properties, and Crystallization of Biodegradable Poly(butylene succinate) (PBS) and Its Copolyesters. Biomacromolecules 2001, 2, 605–613. [Google Scholar] [CrossRef] [PubMed]
- Daniel, J.S.P.; Veronez, I.P.; Rodrigues, L.L.; Trevisan, M.G.; Garcia, J.S. Risperidone—Solid-state characterization and pharmaceutical compatibility using thermal and non-thermal techniques. Thermochim. Acta 2013, 568, 148–155. [Google Scholar] [CrossRef]
- Pappa, C.; Nanaki, S.; Giliopoulos, D.; Triantafyllidis, K.; Kostoglou, M.; Avgeropoulos, A.; Bikiaris, D. Nanostructured Composites of Sodium Montmorillonite Clay and PEO Used in Dissolution Improvement of Aprepitant Drug by Melt Mixing. Appl. Sci. 2018, 8, 786. [Google Scholar] [CrossRef]
- Nanaki, S.; Tseklima, M.; Terzopoulou, Z.; Nerantzaki, M.; Giliopoulos, D.J.; Triantafyllidis, K.; Kostoglou, M.; Bikiaris, D.N. Use of mesoporous cellular foam (MCF) in preparation of polymeric microspheres for long acting injectable release formulations of paliperidone antipsychotic drug. Eur. J. Pharm. Biopharm. 2017, 117, 77–90. [Google Scholar] [CrossRef] [PubMed]
- Nanaki, S.; Siafaka, P.I.; Zachariadou, D.; Nerantzaki, M.; Giliopoulos, D.J.; Triantafyllidis, K.S.; Kostoglou, M.; Nikolakaki, E.; Bikiaris, D.N. PLGA/SBA-15 mesoporous silica composite microparticles loaded with paclitaxel for local chemotherapy. Eur. J. Pharm. Sci. 2017, 99, 32–44. [Google Scholar] [CrossRef] [PubMed]
- Iordanskii, A.L.; Zaikov, G.E.; Berlin, A.A. Diffusion kinetics of hydrolysis of biodegradable polymers. Weight loss and control of the release of low molecular weight substances. Polym. Sci. Ser. D 2015, 8, 211–218. [Google Scholar] [CrossRef]
- Tien, C. Adsorption Calculations and Modeling; Butterworth-Heinemann: Woburn, MA, USA, 1994. [Google Scholar]
- Korsmeyer, R.W.; Gurny, R.; Doelker, E.; Buri, P.; Peppas, N.A. Mechanisms of solute release from porous hydrophilic polymers. Int. Pharm. 1983, 15, 25–35. [Google Scholar] [CrossRef]
- Crank, J. The Mathematics of Diffusion; Clarendon Press: Oxford, UK, 1979. [Google Scholar]
- Georgiadis, M.C.; Kostoglou, M. On the optimization of drug release from multi-laminated polymer matrix devices. J. Control. Release 2001, 77, 273–285. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Particle Size Range (μm) | Microparticles Yield (%) | Drug Loading (%) | Entrapment Efficiency (%) |
|---|---|---|---|---|
| PLA–risperidone | 3–17 | 78.34 ± 2.1 | 9.84 ± 1.7 | 36.51 ± 2.2 |
| PLA/PPAd 80/20-risperidone | 3–15 | 79.56 ± 2.1 | 11.24 ± 3.1 | 38.46 ± 3.1 |
| PLA/PPAd 60/40-risperidone | 3–14 | 82.37 ± 1.9 | 12.87 ± 3.0 | 41.87 ± 3.2 |
| PLA/PPAd 40/60-risperidone | 3–15 | 81.48 ± 2.9 | 14.21 ± 2.5 | 40.42 ± 2.6 |
| PLA/PPAd 20/80-risperidone | 2–10 | 80.54 ± 3.0 | 10.07 ± 3.1 | 39.17 ± 4.1 |
| PPAd-risperidone | 2–8 | 82.17 ± 2.0 | 12.49 ± 1.9 | 42.82 ± 3.0 |
| Polyester Type | First Statistical Moment about Zero for: | |
|---|---|---|
| Dissolution (h) | Hydrolysis (h) | |
| PLA | 90.25 | 300.72 |
| PLA/PPAd 80/20 | 81.85 | 300.24 |
| PLA/PPAd 60/40 | 69.90 | 256.80 |
| PLA/PPAd 40/60 | 51.89 | 169.92 |
| PLA/PPAd 20/80 | 40.39 | 99.36 |
| PPAd | 23.50 | 26.16 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nanaki, S.; Barmpalexis, P.; Iatrou, A.; Christodoulou, E.; Kostoglou, M.; Bikiaris, D.N. Risperidone Controlled Release Microspheres Based on Poly(Lactic Acid)-Poly(Propylene Adipate) Novel Polymer Blends Appropriate for Long Acting Injectable Formulations. Pharmaceutics 2018, 10, 130. https://doi.org/10.3390/pharmaceutics10030130
Nanaki S, Barmpalexis P, Iatrou A, Christodoulou E, Kostoglou M, Bikiaris DN. Risperidone Controlled Release Microspheres Based on Poly(Lactic Acid)-Poly(Propylene Adipate) Novel Polymer Blends Appropriate for Long Acting Injectable Formulations. Pharmaceutics. 2018; 10(3):130. https://doi.org/10.3390/pharmaceutics10030130
Chicago/Turabian StyleNanaki, Stavroula, Panagiotis Barmpalexis, Alexandros Iatrou, Evi Christodoulou, Margaritis Kostoglou, and Dimitrios N. Bikiaris. 2018. "Risperidone Controlled Release Microspheres Based on Poly(Lactic Acid)-Poly(Propylene Adipate) Novel Polymer Blends Appropriate for Long Acting Injectable Formulations" Pharmaceutics 10, no. 3: 130. https://doi.org/10.3390/pharmaceutics10030130