Nose-to-Brain Delivery of Antiviral Drugs: A Way to Overcome Their Active Efflux?

Abstract
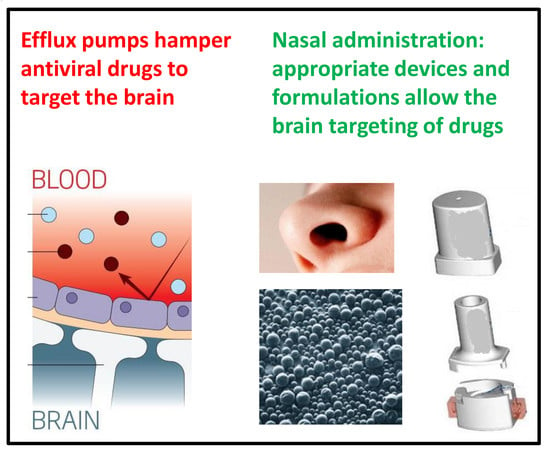
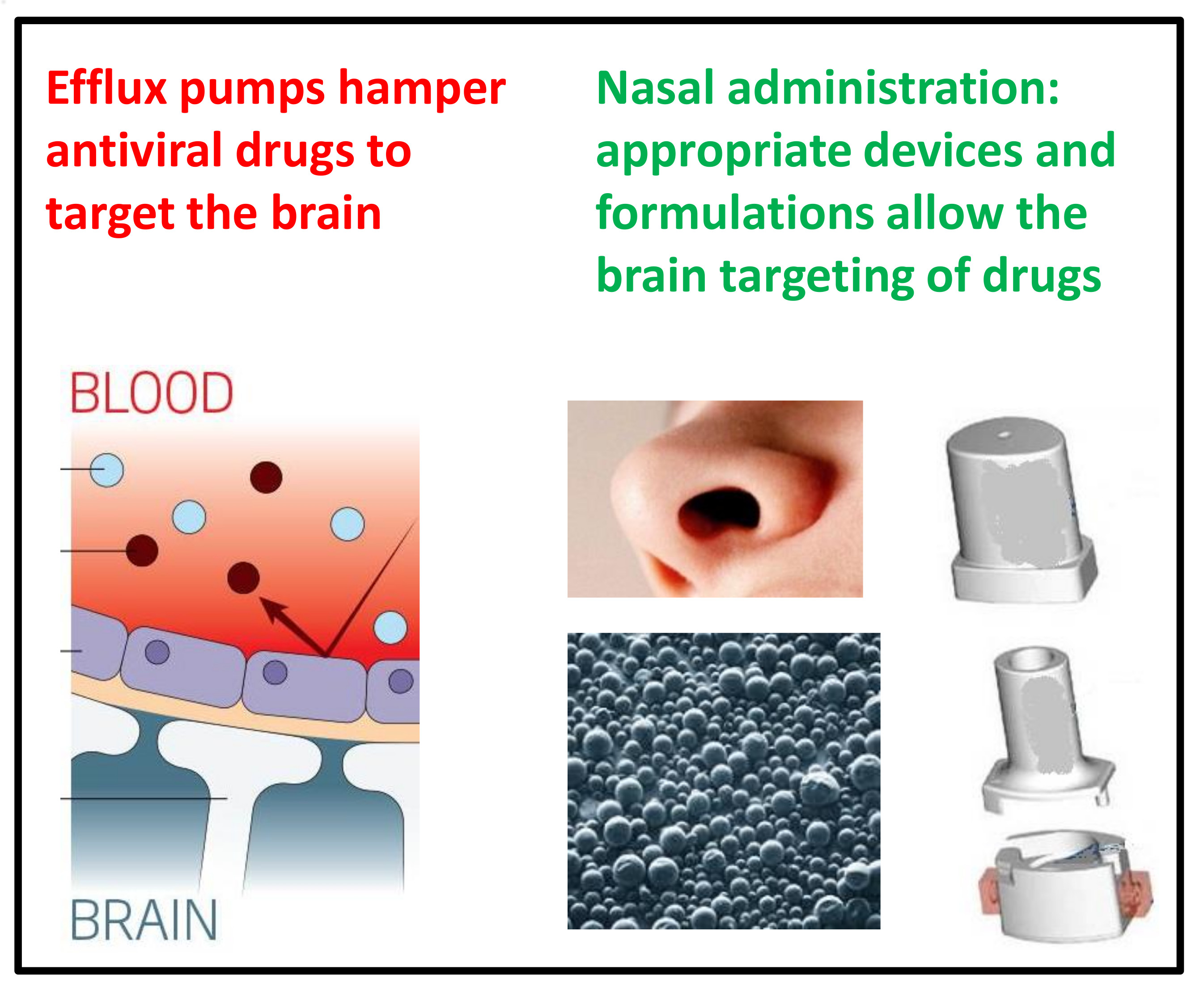
:
1. Viruses Can Have Important Neurotropic Effects
2. Active Efflux Transporters Do Not Allow the Antiviral Drugs to Reach the Sanctuaries of Viruses
- The ATP-binding cassette (ABC) gene family of active transporters requiring ATP hydrolysis for their efflux activity; and
- The solute carrier (SLC) gene family of energy-independent or secondary active efflux transporters.
3. Antiviral Drugs Can Enhance the Expression of Active Efflux Transporters
4. AET Inhibitors: Promising in Vitro Results Not Confirmed by Clinical Trials
5. Prodrugs of Antiviral Drugs: New Proposals against the AET Activity
6. Micro- and Nano-Particulate Systems: Can These Innovative Formulations Target the Antiviral Drugs in the Central Nervous System?
7. Nasal Administration: A Promising Strategy for Antiviral Drug Uptake in the Brain
8. What Strategies Are Currently Related to Nasal Administration of Antiviral Drugs?
8.1. An Innovative Device for the Nasal Administration of Antiviral Drugs
8.2. Design of Innovative Nasal Formulations for Antiviral Drugs
8.3. Nasal Formulations and Brain Targeting of Antiviral Drugs
8.3.1. Nasal Formulations for Zidovudine Administration
8.3.2. Nasal Formulations for the Administration of a Prodrug of Zidovudine
8.3.3. Can Nasal Administration of Insulin be Useful against AIDS Neurotoxicity?
9. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Calisher, C.H. Medically important arboviruses of the United States and Canada. Clin. Microbiol. Rev. 1994, 7, 89–116. [Google Scholar] [CrossRef] [PubMed]
- Huneycutt, B.S.; Bi, Z.; Aoki, C.J.; Reiss, C.S. Central neuropathogenesis of vesicular stomatitis virus infection of immunodeficient mice. J. Virol. 1993, 67, 6698–6706. [Google Scholar] [PubMed]
- Van den Pol, A.N.; Dalton, K.P.; Rose, J.K. Relative neurotropism of a recombinant rhabdovirus expressing a green fluorescent envelope glycoprotein. J. Virol. 2002, 76, 1309–1327. [Google Scholar] [CrossRef] [PubMed]
- Van den Pol, A.N.; Davis, J.N. Highly attenuated recombinant vesicular stomatitis virus VSV-12’GFP displays immunogenic and oncolytic activity. J. Virol. 2013, 87, 1019–1034. [Google Scholar] [CrossRef] [PubMed]
- Tyler, K.L. Update on herpes simplex encephalitis. Rev. Neurol. Dis. 2004, 1, 169–178. [Google Scholar] [PubMed]
- Whitley, R.J.; Kimberlin, D.W. Herpes simplex encephalitis: Children and adolescents. Semin. Pediatr. Infect. Dis. 2005, 16, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Aronson, J.F.; Grieder, F.B.; Davis, N.L.; Charles, P.C.; Knott, T.; Brown, K.; Johnston, R.E. A single-site mutant and revertants arising in vivo define early steps in the pathogenesis of Venezuelan equine encephalitis virus. Virology 2000, 270, 111–123. [Google Scholar] [CrossRef] [PubMed]
- Charles, P.C.; Trgovcich, J.; Davis, N.L.; Johnston, R.E. Immunopathogenesis and immune modulation of Venezuelan equine encephalitis virus-induced disease in the mouse. Virology 2001, 284, 190–202. [Google Scholar] [CrossRef] [PubMed]
- Davis, N.L.; Grieder, F.B.; Smith, J.F.; Greenwald, G.F.; Valenski, M.L.; Sellon, D.C.; Charles, P.C.; Johnston, R.E. A molecular genetic approach to the study of Venezuelan equine encephalitis virus pathogenesis. Arch. Virol. Suppl. 1994, 9, 99–109. [Google Scholar] [PubMed]
- Pomerantz, R.J. Reservoirs, sanctuaries and residual disease: The hiding spots of HIV-1. HIV Clin. Trials 2003, 4, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Lambotte, O.; Deiva, K.; Tardieu, M. HIV-1 persistence, viral reservoir and the central nervous system in the HAART era. Brain Pathol. 2003, 13, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Aquaro, S.; Svicher, V.; Schols, D.; Pollicita, M.; Antinori, A.; Balzarini, J.; Perno, C.F. Mechanisms underlying activity of antiretroviral drugs in HIV-1-infected macrophages: New therapeutic strategies. J. Leukoc. Biol. 2006, 80, 1103–1110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaul, M. HIV’s double strike at the brain: Neuronal toxicity and compromised neurogenesis. Front. Biosci. 2008, 13, 2484–2494. [Google Scholar] [CrossRef] [PubMed]
- Kolson, D.L.; Gonzalez-Scarano, F. HIV and HIV dementia. J. Clin. Investig. 2000, 106, 11–13. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, P.H.; Smith, D.G.; Satchell, C.; Cooper, D.A.; Brew, B. Evidence for independent development of resistance to HIV-1 reverse transcriptase inhibitors in the cerebrospinal fluid. AIDS 2000, 14, 1949–1954. [Google Scholar] [CrossRef] [PubMed]
- Gray, F.; Scaravilli, F.; Everall, I.; Chretien, F.; An, S.; Boche, D.; Adle-Biassette, H.; Wingertsmann, L.; Durigon, M.; Hurtrel, B.; et al. Neuropathology of early HIV-1 infection. Brain Pathol. 1996, 6, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Panel on Antiretroviral Guidelines for Adults and Adolescents (2017). Guidelines for the Use of Antiretroviral Agents in Adults and Adolescents Living with HIV. Department of Health and Human Services. Available online: http://www.aidsinfo.nih.gov/ContentFiles/AdultandAdolescentGL.pdf (accessed on 21 March 2018).
- Moore, J.P.; Kitchen, S.G.; Pugach, P.; Zack, J.A. The CCR5 and CXCR4 coreceptors—Central to understanding the transmission and pathogenesis of human immunodeficiency virus type 1 infection. AIDS Res. Hum. Retroviruses 2004, 20, 111–126. [Google Scholar] [CrossRef] [PubMed]
- Dorr, P.; Westby, M.; Dobbs, S.; Griffin, P.; Irvine, B.; Macartney, M.; Mori, J.; Rickett, G.; Smith-Burchnell, C.; Napier, C.; et al. Maraviroc (UK-427, 857), a potent, orally bioavailable, and selective small-molecule inhibitor of chemokine receptor CCR5 with broad-spectrum anti-human immunodeficiency virus type 1 activity. Antimicrob. Agents Chemother. 2005, 49, 4721–4732. [Google Scholar] [CrossRef] [PubMed]
- Reese, M.J.; Savina, P.M.; Generaux, G.T.; Tracey, H.; Humphreys, J.E.; Kanaoka, E.; Webster, L.O.; Harmon, K.A.; Clarke, J.D.; Polli, J.W. In vitro investigations into the roles of drug transporters and metabolizing enzymes in the disposition and drug interactions of dolutegravir, a HIV integrase inhibitor. Drug Metab. Dispos. 2013, 41, 353–361. [Google Scholar] [CrossRef] [PubMed]
- Boyd, M.; Reiss, P. The long-term consequences of antiretroviral therapy: A review. J. HIV Ther. 2006, 11, 26–35. [Google Scholar] [PubMed]
- Chaudhary, P.M.; Mechetner, E.B.; Roninson, I.B. Expression and activity of the multidrug resistance P-glycoprotein in human peripheral blood lymphocytes. Blood 1992, 80, 2735–2739. [Google Scholar] [PubMed]
- Neyfakh, A.A.; Serpinskaya, A.S.; Chervonsky, A.V.; Apasov, S.G.; Kazarov, A.R. Multidrug-resistance phenotype of a subpopulation of T-lymphocytes without drug selection. Exp. Cell Res. 1989, 185, 496–505. [Google Scholar] [CrossRef]
- Janneh, O.; Jones, E.; Chandler, B.; Owen, A.; Khoo, S.H. Inhibition of P-glycoprotein and multidrug resistance-associated proteins modulates the intracellular concentration of lopinavir in cultured CD4 T cells and primary human lymphocytes. J. Antimicrob. Chemother. 2007, 60, 987–993. [Google Scholar] [CrossRef] [PubMed]
- Dalpiaz, A.; Fogagnolo, M.; Ferraro, L.; Capuzzo, A.; Pavan, B.; Rassu, G.; Salis, A.; Giunchedi, P.; Gavini, E. Nasal chitosan microparticles target a zidovudine prodrug to brain HIV sanctuaries. Antivir. Res. 2015, 123, 146–157. [Google Scholar] [CrossRef] [PubMed]
- Namanja, H.A.; Emmert, D.; Davis, D.A.; Campos, C.; Miller, D.S.; Hrycyna, C.A.; Chmielewski, J. Toward eradicating HIV reservoirs in the brain: Inhibiting P-glycoprotein at the blood-brain barrier with prodrug abacavir dimers. J. Am. Chem. Soc. 2012, 134, 2976–2980. [Google Scholar] [CrossRef] [PubMed]
- Pavan, B.; Dalpiaz, A. Prodrugs and endogenous transporters: Are they suitable tools for drug targeting into the central nervous system? Curr. Pharm. Des. 2011, 17, 3560–3576. [Google Scholar] [CrossRef] [PubMed]
- Pavan, B.; Paganetto, G.; Rossi, D.; Dalpiaz, A. Multidrug resistance in cancer or inefficacy of neuroactive agents: Innovative strategies to inhibit or circumvent the active efflux transporters selectively. Drug Discov. Today 2014, 19, 1563–1571. [Google Scholar] [CrossRef] [PubMed]
- Pardridge, W.M. The blood–Brain barrier: Bottleneck in brain drug development. NeuroRX 2005, 2, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Pardridge, W.M. Blood–Brain barrier delivery. Drug Discov. Today 2007, 12, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Pardridge, W.M. Drug targeting to the brain. Pharm. Res. 2007, 24, 1733–1744. [Google Scholar] [CrossRef] [PubMed]
- Wijnholds, J.; Mol, C.A.; van Deemter, L.; de Haas, M.; Scheffer, G.L.; Baas, F.; Beijnen, J.H.; Scheper, R.J.; Hatse, S.; De Clercq, E.; et al. Multidrug-resistance protein 5 is a multispecific organic anion transporter able to transport nucleotide analogs. Proc. Natl. Acad. Sci. USA 2000, 97, 7476–7481. [Google Scholar] [CrossRef] [PubMed]
- Jorajuria, S.; Dereuddre-Bosquet, N.; Becher, F.; Martin, S.; Porcheray, F.; Garrigues, A.; Mabondzo, A.; Benech, H.; Grassi, J.; Orlowski, S.; et al. ATP binding cassette multidrug transporters limit the anti-HIV activity of zidovudine and indinavir in infected human macrophages. Antivir. Ther. 2004, 9, 519–528. [Google Scholar] [PubMed]
- Gupta, A.; Zhang, Y.; Unadkat, J.D.; Mao, Q. HIV protease inhibitors are inhibitors but not substrates of the human breast cancer resistance protein (BCRP/ABCG2). J. Pharmacol. Exp. Ther. 2004, 310, 334–341. [Google Scholar] [CrossRef] [PubMed]
- Ronaldson, P.T.; Lee, G.; Dallas, S.; Bendayan, R. Involvement of P-glycoprotein in the transport of saquinavir and indinavir in rat brain microvessel endothelial and microglia cell lines. Pharm. Res. 2004, 21, 811–818. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Sinko, P.J. P-glycoprotein and mutlidrug resistance-associated proteins limit the brain uptake of saquinavir in mice. J. Pharmacol. Exp. Ther. 2005, 312, 1249–1256. [Google Scholar] [CrossRef] [PubMed]
- Bachmeier, C.J.; Spitzenberger, T.J.; Elmquist, W.F.; Miller, D.W. Quantitative assessment of HIV-1 protease inhibitor interactions with drug efflux transporters in the blood-brain barrier. Pharm. Res. 2005, 22, 1259–1268. [Google Scholar] [CrossRef] [PubMed]
- Eilers, M.; Roy, U.; Mondal, D. MRP (ABCC) transporters-mediated efflux of anti-HIV drugs, saquinavir and zidovudine, from human endothelial cells. Exp. Biol. Med. 2008, 233, 1149–1160. [Google Scholar] [CrossRef] [PubMed]
- Dallas, S.; Miller, D.S.; Bendayan, R. Multidrug resistance-associated proteins: Expression and function in the central nervous system. Pharmacol. Rev. 2006, 58, 140–161. [Google Scholar] [CrossRef] [PubMed]
- Sampath, J.; Adachi, M.; Hatse, S.; Naesens, L.; Balzarini, J.; Flatley, R.M.; Matherly, L.H.; Schuetz, J.D. Role of MRP4 and MRP5 in biology and chemotherapy. AAPS Pharm. Sci. 2002, 4, E14. [Google Scholar] [CrossRef] [PubMed]
- Janneh, O.; Owen, A.; Chandler, B.; Hartkoorn, R.C.; Hart, C.A.; Bray, P.G.; Ward, S.A.; Back, D.J.; Khoo, S.H. Modulation of the intracellular accumulation of saquinavir in peripheral blood mononuclear cells by inhibitors of MRP1, MRP2, P-gp and BCRP. AIDS 2005, 19, 2097–2102. [Google Scholar] [CrossRef] [PubMed]
- Meaden, E.R.; Hoggard, P.G.; Newton, P.; Tjia, J.F.; Aldam, D.; Cornforth, D.; Lloyd, J.; Williams, I.; Back, D.J.; Khoo, S.H. P-glycoprotein and MRP1 expression and reduced ritonavir and saquinavir accumulation in HIV-infected individuals. J. Antimicrob. Chemother. 2002, 50, 583–588. [Google Scholar] [CrossRef] [PubMed]
- Varatharajan, L.; Thomas, S.A. The transport of anti-HIV drugs across blood–CNS interfaces: Summary of current knowledge and recommendations for further research. Antiv. Res. 2009, 82, A99–A109. [Google Scholar] [CrossRef] [PubMed]
- Sosnik, A. Reversal of multidrug resistance by the inhibition of ATP-binding cassette pumps employing “Generally Recognized As Safe” (GRAS) nanopharmaceuticals: A review. Adv. Drug Deliv. Rev. 2013, 65, 1828–1851. [Google Scholar] [CrossRef] [PubMed]
- Walker, D.K.; Abel, S.; Comby, P.; Muirhead, G.J.; Nedderman, A.N.; Smith, D.A. Species differences in the disposition of the CCR5 antagonist, UK-427, 857, a new potential treatment for HIV. Drug Metab. Dispos. 2005, 33, 587–595. [Google Scholar] [CrossRef] [PubMed]
- Walker, D.K.; Bowers, S.J.; Mitchell, R.J.; Potchoiba, M.J.; Schroeder, C.M.; Small, H.F. Preclinical assessment of the distribution of maraviroc to potential human immunodeficiency virus (HIV) sanctuary sites in the central nervous system (CNS) and gut-associated lymphoid tissue (GALT). Xenobiotica 2008, 38, 1330–1339. [Google Scholar] [CrossRef] [PubMed]
- Ramanathan, S.; Abel, S.; Tweedy, S.; West, S.; Hui, J.; Kearney, B.P. Pharmacokinetic interaction of ritonavir-boosted elvitegravir and maraviroc. J. Acquir. Immune Defic. Syndr. 2010, 53, 209–214. [Google Scholar] [CrossRef] [PubMed]
- Ponte-Sucre, A. Availability and applications of ATP-binding cassette (ABC) transporter blockers. Appl. Microbiol. Biotechnol. 2007, 76, 279–286. [Google Scholar] [CrossRef] [PubMed]
- Owen, A.; Janneh, O.; Hartkoorn, R.C.; Chandler, B.; Bray, P.G.; Martin, P.; Ward, S.A.; Hart, C.A.; Khoo, S.H.; Back, D.J. In vitro synergy and enhanced murine brain penetration of saquinavir coadministered with mefloquine. J. Pharmacol. Exp. Ther. 2005, 314, 1202–1209. [Google Scholar] [CrossRef] [PubMed]
- Kruh, G.D.; Belinsky, M.G. The MRP family of drug efflux pumps. Oncogene 2003, 22, 7537–7552. [Google Scholar] [CrossRef] [PubMed]
- Roy, U.; Bulot, C.; Honer zu Bentrup, K.; Mondal, D. Specific increase in MDR1 mediated drug-efflux in human brain endothelial cells following co-exposure to HIV-1 and saquinavir. PLoS ONE 2013, 8, e75374. [Google Scholar] [CrossRef] [PubMed]
- Perloff, M.D.; von Moltke, L.L.; Greenblatt, D.J. Ritonavir and dexamethasone induce expression of CYP3A and P-glycoprotein in rats. Xenobiotica 2004, 34, 133–150. [Google Scholar] [CrossRef] [PubMed]
- Perloff, M.D.; von Moltke, L.L.; Fahey, J.M.; Greenblatt, D.J. Induction of P-glycoprotein expression and activity by ritonavir in bovine brain microvessel endothelial cells. J. Pharm. Pharmacol. 2007, 59, 947–953. [Google Scholar] [CrossRef] [PubMed]
- Zastre, J.A.; Chan, G.N.Y.; Ronaldson, P.T.; Ramaswamy, M.; Couraud, P.O.; Romero, I.A.; Weksler, B.; Bendayan, M.; Bendayan, R. Up-regulation of P-glycoprotein by HIV protease inhibitors in a human brain microvessel endothelial cell line. J. Neurosci. Res. 2009, 87, 1023–1036. [Google Scholar] [CrossRef] [PubMed]
- Urquhart, B.L.; Tirona, R.G.; Kim, R.B. Nuclear receptors and the regulation of drug-metabolizing enzymes and drug transporters: Implications for interindividual variability in response to drugs. J. Clin. Pharmacol. 2007, 47, 566–578. [Google Scholar] [CrossRef] [PubMed]
- Bauer, B.; Hartz, A.M.S.; Fricker, G.; Miller, D.S. Pregnane X receptor up-regulation of P-glycoprotein expression and transport function at the blood-brain barrier. Mol. Pharmacol. 2004, 66, 413–419. [Google Scholar] [CrossRef] [PubMed]
- Bauer, B.; Yang, X.; Hartz, A.M.S.; Olson, E.R.; Zhao, R.; Kalvass, J.C.; Pollack, G.M.; Miller, D.S. In vivo activation of human pregnane X receptor tightens the blood-brain barrier to methadone through P-glycoprotein up-regulation. Mol. Pharmacol. 2006, 70, 1212–1219. [Google Scholar] [CrossRef] [PubMed]
- Ott, M.; Fricker, G.; Bauer, B. Pregnane X receptor (PXR) regulates P-glycoprotein at the blood-brain barrier: Functional similarities between pig and human PXR. J. Pharmacol. Exp. Ther. 2009, 329, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Sykes, D.B.; Miller, D.S. Constitutive androstane receptormediated up-regulation of ATP-driven xenobiotic efflux transporters at the blood-brain barrier. Mol. Pharmacol. 2010, 78, 376–383. [Google Scholar] [CrossRef] [PubMed]
- Lemmen, J.; Tozakidis, I.E.P.; Bele, P.; Galla, H.J. Constitutive androstane receptor upregulates Abcb1 and Abcg2 at the blood-brain barrier after CITCO activation. Brain Res. 2013, 1501, 68–80. [Google Scholar] [CrossRef] [PubMed]
- Chan, G.N.; Hoque, M.T.; Cummins, C.L.; Bendayan, R. Regulation of P-glycoprotein by orphan nuclear receptors in human brain microvessel endothelial cells. J. Neurochem. 2011, 118, 163–175. [Google Scholar] [CrossRef] [PubMed]
- Dussault, I.; Lin, M.; Hollister, K.; Wang, E.H.; Synold, T.W.; Forman, B.M. Peptide mimetic HIV protease inhibitors are ligands for the orphan receptor SXR. J. Biol. Chem. 2001, 276, 33309–33312. [Google Scholar] [CrossRef] [PubMed]
- Svärd, J.; Spiers, J.P.; Mulcahy, F.; Hennessy, M. Nuclear receptor mediated induction of CYP450 by antiretrovirals: Functional consequences of NR1I2 (PXR) polymorphisms and differential prevalence in whites and sub-Saharan Africans. J. Acquir. Immune Defic. Syndr. 2010, 55, 536–549. [Google Scholar] [CrossRef] [PubMed]
- Chan, G.N.; Patel, R.; Cummins, C.L.; Bendayan, R. Induction of P-glycoprotein by antiretroviral drugs in human brain microvessel endothelial cells. Antimicrob. Agents Chemother. 2013, 57, 4481–4488. [Google Scholar] [CrossRef] [PubMed]
- Shukla, S.; Ohnuma, S.; Ambudkar, S.V. Improving cancer chemotherapy with modulators of ABC drug transporters. Curr. Drug Targets 2011, 12, 621–630. [Google Scholar] [CrossRef] [PubMed]
- Gillet, J.P.; Efferth, T.; Remacle, J. Chemotherapy-induced resistance by ATP-binding cassette transporter genes. Biochim. Biophys. Acta 2007, 1775, 237–262. [Google Scholar] [CrossRef] [PubMed]
- Xia, C.Q.; Smith, P.G. Drug efflux transporter and multidrug resistance in acute leukemia: Therapeutic impact and novel approaches to mediation. Mol. Pharmacol. 2012, 82, 1008–1021. [Google Scholar] [CrossRef] [PubMed]
- Begley, D.J. Delivery of therapeutic agents to central nervous system: The problems and the possibilities. Pharmacol. Ther. 2004, 104, 29–45. [Google Scholar] [CrossRef] [PubMed]
- Aller, S.G.; Yu, J.; Ward, A.; Weng, Y.; Chittaboina, S.; Zhuo, R.; Harrell, P.M.; Trinh, Y.T.; Zhang, Q.; Urbatsch, I.L.; et al. Structure of P-glycoprotein reveals a molecular basis for poly-specific drug binding. Science 2009, 323, 1718–1722. [Google Scholar] [CrossRef] [PubMed]
- Namanja, H.A.; Emmert, D.; Hrycyna, C.A.; Chmielewski, J. Homodimers of the antiviral abacavir as modulators of P-glycoprotein transport in cell culture: Probing tether length. Medchemcomm 2013, 4, 1344–1349. [Google Scholar] [CrossRef] [PubMed]
- Dalpiaz, A.; Paganetto, G.; Pavan, B.; Fogagnolo, M.; Medici, A.; Beggiato, S.; Perrone, D. Zidovudine and ursodeoxycholic acid conjugation: Design of a new prodrug potentially able to bypass the active efflux transport systems of the central nervous system. Mol. Pharm. 2012, 9, 957–968. [Google Scholar] [CrossRef] [PubMed]
- Dalpiaz, A.; Contado, C.; Mari, L.; Perrone, D.; Pavan, B.; Paganetto, G.; Hanuskovà, M.; Vighi, E.; Leo, E. Development and characterization of PLGA nanoparticles as delivery systems of a prodrug of zidovudine obtained by its conjugation with ursodeoxycholic acid. Drug Deliv. 2014, 21, 221–1232. [Google Scholar] [CrossRef] [PubMed]
- Dalpiaz, A.; Ferraro, L.; Perrone, D.; Leo, E.; Iannuccelli, V.; Pavan, B.; Paganetto, G.; Beggiato, S.; Scalia, S. Brain uptake of a Zidovudine prodrug after nasal administration of solid lipid microparticles. Mol. Pharm. 2014, 11, 1550–1561. [Google Scholar] [CrossRef] [PubMed]
- Zink, M.C. Translational research models and novel adjunctive therapies for neuroAIDS. J. Neuroim. Pharmacol. 2007, 2, 14–19. [Google Scholar] [CrossRef] [PubMed]
- Dou, H.; Morehead, J.; Destache, C.J.; Kingsley, J.D.; Shlyakhtenko, L.; Zhou, Y.; Chaubal, M.; Werling, J.; Kipp, J.; Rabinow, B.E.; et al. Laboratory investigations for the morphologic, pharmacokinetic, and anti-retroviral properties of indinavir nanoparticles in human monocyte-derived macrophages. Virology 2007, 358, 148–158. [Google Scholar] [CrossRef] [PubMed]
- Gorantla, S.; Dou, H.; Boska, M.; Destache, C.J.; Nelson, J.; Poluektova, L.; Rabinow, B.E.; Gendelman, H.E.; Mosley, R.L. Quantitative magnetic resonance and SPECT imaging for macrophage tissue migration and nanoformulated drug delivery. J. Leukoc. Biol. 2006, 80, 1165–1174. [Google Scholar] [CrossRef] [PubMed]
- Gao, H. Progress and perspectives on targeting nanoparticles for brain drug delivery. Acta Pharm. Sin. B 2016, 6, 268–286. [Google Scholar] [CrossRef] [PubMed]
- Manfredini, S.; Pavan, B.; Vertuani, S.; Scaglianti, M.; Compagnone, D.; Biondi, C.; Scatturin, A.; Tanganelli, S.; Ferraro, L.; Prasad, P.; et al. Design, synthesis and activity of ascorbic acid prodrugs of nipecotic, kynurenic and diclophenamic acids, liable to increase neurotropic activity. J. Med. Chem. 2002, 45, 559–562. [Google Scholar] [CrossRef] [PubMed]
- Illum, L. Is nose-to-brain transport of drugs in man a reality? J. Pharm. Pharmacol. 2004, 56, 3–17. [Google Scholar] [CrossRef] [PubMed]
- Johnson, N.J.; Hanson, L.R.; Frey, W.H. Trigeminal pathways deliver a low molecular weight drug from the nose to the brain and orofacial structures. Mol. Pharm. 2010, 7, 884–893. [Google Scholar] [CrossRef] [PubMed]
- Casettari, L.; Illum, L. Chitosan in nasal delivery systems for therapeutic drugs. J. Control. Release 2014, 190, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Hanson, L.R.; Frey, W.H., 2nd. Strategies for intranasal delivery of therapeutics for the prevention and treatment of neuroAIDS. J. Neuroimmune Pharmacol. 2007, 2, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Boivin, N.; Sergerie, Y.; Rivest, S.; Boivin, G. Effect of pretreatment with toll-like receptor agonists in a mouse model of herpes simplex virus type 1 encephalitis. J. Infect. Dis. 2008, 198, 664–672. [Google Scholar] [CrossRef] [PubMed]
- Elia, G.; Belloli, C.; Cirone, F.; Lucente, M.S.; Caruso, M.; Martella, V.; Decaro, N.; Buonavoglia, C.; Ormas, P. In vitro efficacy of ribavirin against canine distemper virus. Antivir. Res. 2008, 77, 108–113. [Google Scholar] [CrossRef] [PubMed]
- Colombo, G.; Lorenzini, L.; Zironi, E.; Galligioni, V.; Sonvico, F.; Balducci, A.G.; Pagliuca, G.; Giuliani, A.; Calzà, L.; Scagliarini, A. Brain distribution of ribavirin after intranasal administration. Antivir. Res. 2011, 92, 408–414. [Google Scholar] [CrossRef] [PubMed]
- Giuliani, A.; Balducci, A.G.; Zironi, E.; Colombo, G.; Bortolotti, F.; Lorenzini, L.; Galligioni, V.; Pagliuca, G.; Scagliarini, A.; Calzà, L.; et al. In vivo nose-to-brain delivery of the hydrophilic antiviral ribavirin by microparticle agglomerates. Drug Deliv. 2018, 25, 376–387. [Google Scholar] [CrossRef] [PubMed]
- Mistry, A.; Stolnik, S.; Illum, L. Nanoparticles for direct nose-to-brain delivery of drugs. Int. J. Pharm. 2009, 379, 146–157. [Google Scholar] [CrossRef] [PubMed]
- Hoekman, J.D.; Ho, R.J. Effects of localized hydrophilic mannitol and hydrophobic nelfinavir administration targeted to olfactory epithelium on brain distribution. AAPS PharmSciTech. 2011, 12, 534–543. [Google Scholar] [CrossRef] [PubMed]
- Stutzle, M.; Flamm, J.; Carle, S.; Schindowski, K. Nose-to-Brain delivery of insulin for Alzheimer’s disease. ADMET DMPK 2015, 3, 190–202. [Google Scholar] [CrossRef]
- Djupesland, P.G.; Messina, J.C.; Mahmoud, R.A. The nasal approach to delivering treatment for brain diseases: An anatomic, physiologic, and delivery technology overview. Ther. Deliv. 2014, 5, 709–733. [Google Scholar] [CrossRef] [PubMed]
- Qu, W.; Li, N.; Yu, R.; Zuo, W.; Fu, T.; Fei, W.; Hou, Y.; Liu, Y.; Yang, J. Cationic DDA/TDB liposome as a mucosal vaccine adjuvant for uptake by dendritic cells in vitro induces potent humoural immunity. Artif. Cells Nanomed. Biotechnol. 2018, 15, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Hulisz, D. Efficacy of zinc against common cold viruses: An overview. J. Am. Pharm. Assoc. 2004, 44, 594–603. [Google Scholar] [CrossRef]
- Seremeta, K.P.; Chiappetta, D.A.; Sosnik, A. Poly(ε-caprolactone), Eudragit® RS 100 and poly(ε-caprolactone)/Eudragit® RS 100 blend submicron particles for the sustained release of the antiretroviral efavirenz. Colloids Surf. B Biointerfaces. 2013, 102, 441–449. [Google Scholar] [CrossRef] [PubMed]
- Lehr, C.M.; Bouwstra, J.A.; Schacht, E.H.; Junginger, H.E. In vitro evaluation of mucoadhesive properties of chitosan and some other natural polymers. Int. J. Pharm. 1992, 78, 43–48. [Google Scholar] [CrossRef]
- Nazar, H.; Fatouros, D.G.; van der Merwe, S.M.; Bouropoulos, N.; Avgouropoulos, G.; Tsibouklis, J.; Roldo, M. Thermosensitive hydrogels for nasal drug delivery: The formulation and characterisation of systems based on N-trimethyl chitosan chloride. Eur. J. Pharm. Biopharm. 2011, 77, 225–232. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Maitani, Y.; Lowman, A.M.; Takayama, K.; Peppas, N.A.; Nagai, T. Uptake and release of budesonide from mucoadhesive, pH-sensitive copolymers and their application to nasal delivery. J. Control. Release 1999, 61, 329–335. [Google Scholar] [CrossRef]
- Soane, R.J.; Hinchcliffe, M.; Davis, S.S.; Illum, L. Clearance characteristics of chitosan based formulations in the sheep nasal cavity. Int. J. Pharm. 2001, 217, 183–191. [Google Scholar] [CrossRef]
- Zaki, N.M.; Awad, G.A.; Mortada, N.D.; Abd Elhady, S.S. Enhanced bioavailability of metoclopramide HCl by intranasal administration of a mucoadhesive in situ gel with modulated rheological and mucociliary transport properties. Eur. J. Pharm. Sci. 2007, 32, 296–307. [Google Scholar] [CrossRef] [PubMed]
- Lungare, S.; Bowen, J.; Badhan, R. Development and Evaluation of a Novel Intranasal Spray for the Delivery of Amantadine. J. Pharm. Sci. 2016, 105, 1209–1220. [Google Scholar] [CrossRef] [PubMed]
- Paul, W.; Sharma, C.P. Chitosan, a drug carrier for the 21st century: A review. S.T.P. Pharm. Sci. 2000, 10, 5–22. [Google Scholar]
- Vllasaliu, D.; Exposito-Harris, R.; Heras, A.; Casettari, L.; Garnett, M.; Illum, L.; Stolnik, S. Tight junction modulation by chitosan nanoparticles: Comparison with chitosan solution. Int. J. Pharm. 2010, 400, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Bertram, U.; Bodmeier, R. In situ gelling, bioadhesive nasal inserts for extended drug delivery: In vitro characterization of a new nasal dosage form. Eur. J. Pharm. Sci. 2006, 27, 62–71. [Google Scholar] [CrossRef] [PubMed]
- Roos, A.; Creton, C.; Novikov, M.B.; Feldstein, M.M. Viscoelasticity and tack of poly(vinyl pyrrolidone)–poly(ethylene glycol) blends. J. Polym. Sci. Part B Polym. Phys. 2002, 40, 2395–2409. [Google Scholar] [CrossRef]
- Alsarra, I.A.; Hamed, A.Y.; Mahrous, G.M.; El Maghraby, G.M.; Al-Robayan, A.A.; Alanazi, F.K. Mucoadhesive polymeric hydrogels for nasal delivery of acyclovir. Drug Dev. Ind. Pharm. 2009, 35, 352–362. [Google Scholar] [CrossRef] [PubMed]
- Krishan, M.; Gudelsky, G.A.; Desai, P.B.; Genter, M.B. Manipulation of olfactory tight junctions using papaverine to enhance intranasal delivery of gemcitabine to the brain. Drug Deliv. 2014, 21, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Mahajan, H.S.; Mahajan, M.S.; Nerkar, P.P.; Agrawal, A. Nanoemulsion-based intranasal drug delivery system of saquinavir mesylate for brain targeting. Drug Deliv. 2014, 21, 148–154. [Google Scholar] [CrossRef] [PubMed]
- Pardridge, W.M. Blood-brain barrier drug targeting: The future of brain drug development. Mol. Interv. 2003, 3, 90–105. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.L.; Van Belle, K.; Sawchuk, R.J. Distributional transport kinetics of zidovudine between plasma and brain extracellular fluid/cerebrospinal fluid in the rabbit: Investigation of the inhibitory effect of probenecid utilizing microdialysis. J. Pharmacol. Exp. Ther. 1993, 264, 899–909. [Google Scholar] [PubMed]
- Takasawa, K.; Terasaki, T.; Suzuki, H.; Sugiyama, Y. In vivo evidence for carrier-mediated efflux transport of 3′-azido-3′-deoxythymidine and 2′, 3′-dideoxyinosine across the blood-brain barrier via a probenecid-sensitive transport system. J. Pharmacol. Exp. Ther. 1997, 281, 369–375. [Google Scholar] [PubMed]
- Wang, Y.; Sawchuk, R.J. Zidovudine transport in the rabbit brain during intravenous and intracerebroventricular infusion. J. Pharm. Sci. 1995, 7, 871–876. [Google Scholar] [CrossRef]
- Wong, S.L.; Wang, Y.; Sawchuk, R.J. Analysis of zidovudine distribution to specific regions in rabbit brain using microdialysis. Pharm. Res. 1992, 9, 332–338. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, A.L.; Naif, H.; Saksena, N.; Lynch, G.; Chang, J.; Li, S.; Jozwiak, R.; Alali, M.; Wang, B.; Fear, W.; et al. HIV infection of macrophages and pathogenesis of AIDS dementia complex: Interaction of the host cell and viral genotype. J. Leukocyte Biol. 1997, 62, 117–125. [Google Scholar] [CrossRef] [PubMed]
- Ghersi-Egea, J.F.; Finnegan, W.; Chen, J.L.; Fenstermacher, J.D. Rapid distribution of intraventricularly administered sucrose into cerebrospinal fluid cisterns via subarachnoid velae in rat. Neuroscience 1996, 75, 1271–1288. [Google Scholar] [CrossRef]
- Seki, T.; Sato, N.; Hasegawa, T.; Kawaguchi, T.; Juni, K. Nasal absorption of zidovudine and its transport to cerebrospinal fluid in rats. Pharm. Bull. 1994, 17, 1135–1137. [Google Scholar] [CrossRef]
- Gill, P.S.; Rarick, M.; Brynes, R.K.; Causey, D.; Loureiro, C.; Levine, A. Azidothymidine associated with bone marrow failure in the acquired immunodeficiency syndrome (AIDS). Ann. Int. Med. 1987, 107, 502–505. [Google Scholar] [CrossRef] [PubMed]
- Ved, P.M.; Kim, K. Poly(ethylene oxide/propylene oxide) copolymer thermo-reversible gelling system for the enhancement of intranasal zidovudine delivery to the brain. Int. J. Pharm. 2011, 411, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Al-Ghananeem, A.M.; Smith, M.; Coronel, M.L.; Tran, H. Advances in brain targeting and drug delivery of anti-HIV therapeutic agents. Expert Opin. Drug Deliv. 2013, 10, 973–985. [Google Scholar] [CrossRef] [PubMed]
- Mamik, M.K.; Asahchop, E.L.; Chan, W.F.; Zhu, Y.; Branton, W.G.; McKenzie, B.A.; Cohen, E.A.; Power, C. Insulin treatment prevents neuroinflammation and neuronal injury with restored neurobehavioral function in models of HIV/AIDS neurodegeneration. J. Neurosci. 2016, 36, 10683–10695. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Transporter | Name | Gene Symbol | Substrates | Antiviral Substrates | Inhibitors |
|---|---|---|---|---|---|
| P-glycoprotein | P-gp | ABCB1 | amphipatic cations and organic compounds | saquinavir, ritonavir, lopinavir, amprenavir, nelfinavir, indinavir, abacavir, dolutegravir | cyclosporine-A, verapamil, mefloquine |
| Multidrug Resistance Protein | MRP-1 | ABCC1 | hydrophilic anion compounds, large molecules | saquinavir, ritonavir, lopinavir | paclitaxel, probenecid, MK-571 |
| MRP-4 MRP-5 | ABCC4 ABCC5 | small polar compounds, nucleoside analogues | Zidovudine, didanosine | ||
| Breast-Cancer-Resistance Protein | BRCP | ABCG2 | partially overlap with those of P-gp | zidovudine, lamivudine, abacavir, zalcitabine, stavudine, efavirenz, dolutegravir | ritonavir, saquinavir, nelfinavir |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dalpiaz, A.; Pavan, B. Nose-to-Brain Delivery of Antiviral Drugs: A Way to Overcome Their Active Efflux? Pharmaceutics 2018, 10, 39. https://doi.org/10.3390/pharmaceutics10020039
Dalpiaz A, Pavan B. Nose-to-Brain Delivery of Antiviral Drugs: A Way to Overcome Their Active Efflux? Pharmaceutics. 2018; 10(2):39. https://doi.org/10.3390/pharmaceutics10020039
Chicago/Turabian StyleDalpiaz, Alessandro, and Barbara Pavan. 2018. "Nose-to-Brain Delivery of Antiviral Drugs: A Way to Overcome Their Active Efflux?" Pharmaceutics 10, no. 2: 39. https://doi.org/10.3390/pharmaceutics10020039