1. Introduction
Inferring phylogenies is one of the key problems in bioinformatics, and its importance has increased more and more in recent years, as the availability of genetic data extracted from cancer samples has allowed researchers to examine how somatic mutations occur and, most importantly, accumulate during the progression of cancer. Furthermore, recent improvements in targeted therapies for cancer treatment require an accurate inference of the clonal evolution and progression of the disease to provide effective therapy [
1,
2].
Given the importance of the problem [
3], several methods [
4,
5,
6,
7,
8,
9,
10,
11,
12,
13,
14,
15,
16,
17,
18,
19,
20] designed to infer cancer progression from next-generation sequencing data have been published. The field has also recently witnessed the development of pipelines [
21] and preprocessing steps.
Cancer cells proliferate in a highly constrained environment under very strong evolutionary pressure—for example, due to low levels of oxygen and the reaction of the immune system. This leads to frequently observed mutations that are very unusual in healthy cells [
22], such as copy number aberrations, gene duplications, and ploidy changes. Some tools try to incorporate these mutational events when inferring tumoral evolutions (see, for example, [
8,
23]). While the predictions should become more accurate with such events included, the models become more complicated. Because the focus of this paper is a computational comparison, we do not consider these events, only the acquisition and loss of mutations.
On the other hand, single-cell sequencing data have provided clear evidence that mutation losses and (less frequently) mutation recurrences are common [
24,
25]. In fact, allowing only mutation accumulation corresponds to the directed perfect phylogeny problem on binary characters, which has a linear-time algorithm [
26], while more general models (where mutation losses or recurrences are also allowed) result in NP-complete problems, with a much larger solution space. The perfect phylogeny model has found several applications in bioinformatics, most notably in haplotyping [
27,
28,
29,
30], and has been deeply investigated. For example, the combinatorial properties of perfect phylogenies are instrumental in the development of efficient algorithms, including the graph-theoretical characterization of genotype matrices that admit a tree representation [
27,
28,
30,
31].
In this paper, we focus on the Dollo-
k model [
5,
32,
33,
34], which is a generalization of the perfect phylogeny model that incorporates the potential loss of characters. Under the Dollo-
k model, each mutation can only be acquired once and lost at most
k times in the entire evolutionary process. The restriction on the number of losses is motivated by the fact that even if character losses are possible, they are not frequent [
24,
35]. Variants of the Dollo-
k model are considered in some recent tools, such as TRaIT [
36], SiFit [
37] and SPhyR [
15], SASC [
6], and ScisTree [
38].
Notice that we are considering the
binary case, where each mutation is either present or missing. The phylogeny inference problem under the Dollo-
k model is NP complete [
39], motivating the use of specialized metaheuristic strategies [
6,
15] which work well in practice even with fairly large instances. The fact that practical cases have incomplete or noisy information makes the use of heuristics more relevant [
40,
41]. Nonetheless, at least an exact solution for the problem, based on ILP, has been proposed [
4], but it is too slow to be used in practice. To mitigate this problem, that tool has a hill climbing final step to guarantee that a local optimum is returned.
Given the success of nature-inspired metaheuristics in attacking various combinatorial problems and of a Simulated Annealing approach [
6,
42,
43] to attacking the tumor phylogeny inference problem, we further investigate some additional approaches for inferring tumor phylogenies under the Dollo-
k model, with the main goal of understanding which metaheuristics might be amenable to being the basis of future tools. More precisely, we investigate Particle Swarm Optimization [
44], Genetic Programming [
45], and Variable Neighbourhood Programming [
46]: the first two are population-based, while the third is trajectory-based. We test the proposed methods on six different synthetic datasets with different levels of complexity and evaluate their performance according to three different quality metrics, including one specifically designed for the evaluation of cancer progression [
47]. The results show that Particle Swarm Optimization is roughly comparable with Simulated Annealing, while the results obtained via the other methods do not achieve the same level of accuracy.
2. Preliminaries
In this paper, we consider the computational problem of inferring a tumor phylogeny from single cell data in the presence of partial information. The input of the problem is an ternary matrix that represents the presence/absence of m mutations in n cells. The entries of I can assume three potential values: an entry indicates that cell i does not have mutation j, indicates the presence of mutation j in cell i, and a ? indicates uncertainty about the presence/absence of mutation j in cell i. The uncertainty in mutation presence within a cell is typically caused by insufficient coverage during sequencing.
Moreover, our models account for potential false positive and false negative entries in the input matrix, which are parameterized with a false positive/negative rate value. Specifically, let
E denote the final
output matrix, which is the binary matrix without errors and noise estimated by the algorithm. We denote
as the false negative rate of mutation
j, and
as the false positive rate, similarly to [
11,
15,
37,
48]. Hence, for each entry
, the following holds:
|
| |
| . |
Notice that, due to the nature of the experimental measure, the false positive rate is usually low (around
), while the false negative rate can be considerably higher (even
[
11,
37,
48]. Furthermore, each mutation can have a different false negative rate; for instance, a highly expressed gene will have significantly higher coverage than an under-expressed gene.
Similar to previous reconstruction methods ([
6]), the objective of the reconstruction problem is to compute a matrix that maximizes the likelihood of the observed matrix
I [
48], assuming we know the probability rates of false positives, false negatives, and missing entries. This is conducted under the hypothesis that mutations in samples are acquired and lost following a specific evolutionary model.
A tumor phylogeny T on a set C of m mutations and n cells is defined as a rooted tree whose internal nodes are labeled by the acquisitions or losses of mutations from C, while the leaves are labeled by the cells.
The state of a node x is defined as the set of mutations c such that: (1) there is an ancestor of x where c is acquired, and (2) for each ancestor y of x where c is lost, there is a node of the path from y to x where c is acquired. Therefore, the state of each node x in the tree T can be represented as a binary vector of length m, called genotype profile and denoted by , where if and only if the node x has the mutation j and 0 otherwise.
Given a binary matrix
E, we say that a tree
T encodes E if there exists a mapping
of the rows (cells) of
E to the leaves of
T such that
for each row
i of
E, where
denotes the image of row
i through the mapping
. Informally,
is the node in the phylogenetic tree corresponding to the node where the cell
i is attached. It is important to note that, given an input matrix
I, the pair
fully characterizes the output matrix
E. Consequently, the reconstruction problem can be formulated as finding a tree
T that maximizes the following objective function:
To simplify the computation of the likelihood, we make a slight abuse of notation by assuming that
. Furthermore, given a tumor phylogeny
T, we can define the mapping
as the one that maximizes the objective function. This means that
can be computed directly from
T, with
T being the only variable in the optimization problem [
6].
The Dollo-k Model
In this paper, we employ the Dollo-
k evolutionary model, where each mutation can be acquired exactly once and lost at most
k times throughout the entire tumor phylogeny tree [
4]. The Dollo-0 and Dollo-1 models correspond to the perfect [
26] and persistent [
34] phylogeny models, respectively. The phylogeny reconstruction problem under a Dollo-
k model is NP-hard [
39] for any
.
Experimental evidence suggests that only a small number of mutation losses occur during the evolutionary process of each tumor [
24]. To account for this observation, we introduce an additional constraint to our models, limiting the total number of mutation losses throughout the progression to a maximum of
d occurrences. Alongside
k,
d is a parameter provided by the user.
In most of the cases studied, we set and . Furthermore, in our experiments, when the number of mutations is not too small, setting effectively implies that , making the parameter k redundant. However, we choose to retain it as it ensures that certain degenerate trees will never be computed.
3. Methods
3.1. Particle Swarm Optimization
The main idea of Particle Swarm Optimization [
49] (PSO) revolves around maintaining a population of
n feasible solutions (called particles). These particles move within the search space under the influence of the best overall solutions encountered by the entire population and the individuals themselves. Each particle is characterized by its
position and its
velocity. The population moves synchronously, with all individuals updating their positions and velocities simultaneously, until either convergence is achieved or a specified timeout is reached. The general outline of PSO is provided by Algorithm 1.
| Algorithm 1: Particle Swarm Optimization |
![Algorithms 16 00333 i001]() |
In our approach, each phylogeny is represented as a particle, with its topology serving as its position. Initially, positions are generated as random binary trees representing Perfect Phylogeny. At each iteration, particles can move using some of the following operations on a tree T and a node i, where denotes the parent of i in T:
Subtree Prune and Reattach: Given two internal nodes, , that are not ancestors of each other, we prune the subtree of T rooted in u by removing the edge . We then reattach this pruned subtree as a new child of v by adding the edge ;
Add a deletion: Given two nodes such that v is an ancestor of u, we insert a node in T to represent the loss of mutation v. The new node becomes the parent of u. Note that this operation is only performed if the resulting tree satisfies the desired phylogeny model. For the Dollo-k model, we must check that the mutation v has been lost in the tree at most times.
Remove a deletion: Given a node labeled as a loss, we simply remove it from the tree. All children of u are added as children of , then the node u is deleted.
Swap node labels: Given two internal nodes , the labels of u and v are swapped. If this operation renders a previously added loss invalid—because a mutation c is lost in a node , but the node where the mutation c is acquired is no longer an ancestor of —we remove the deletion .
Subtree transplant: This operation requires an additional tree Q which does not necessarily contain the acquisition of all characters of T. The transplant involves: (1) deleting the subtree of T rooted at i (i.e., removing i and all its descendants), (2) making the root of Q a new child of (i.e., attaching Q to T), and (3) adjusting the resulting tree to ensure it is a Dollo-k phylogeny for the input mutations. The adjustment consists of two parts: (a) contracting a node corresponding to the loss of mutation c for each mutation c that has been lost more than k times, until c is lost k times in the tree, and (b) randomly adding a node for each input mutation that is not acquired in the tree. The resulting tree is guaranteed to be a Dollo-k phylogeny and a feasible solution to our problem.
To guide the update of particle positions, we introduce a new notion of distance. This distance, represented by Equation (
1), is experimentally assessed and must be computed very rapidly to avoid degrading the performance of the method. This distance is computed by constructing a graph
G whose vertex set is the union of vertices of two trees,
and
. Each edge
of
G connects a vertex
v of
with a vertex
w of
. The edge is weighted with the number of mutations that are shared by the subtree of
rooted at
v and the subtree of
rooted at
w. To determine the distance between two trees, we compute a maximum weight matching
M of the graph
G. This matching establishes a one-to-one correspondence between the vertices of the trees, maximizing the overall number of shared mutations. To transform a matching into a suitable distance measure, we subtract the weight of
M from the maximum number of mutations in either input tree, resulting in the following distance formulation:
This notion of distance is utilized to compute the velocity of the particles. The idea is that particles that are far from the local or the global optimum should have higher speed to encourage exploration. This higher velocity is determined by swapping subtrees within the tree, where the size of the swapped subtree is equal to the velocity. In other words, the higher the velocity of a particle, the larger the portion of the tree that gets replaced or modified during the optimization process.
3.2. Genetic Programming
Genetic programming (GP), introduced by Koza in 1992 [
50], is population-based metaheuristics that has been successfully applied in various problem domains, including bioinformatics [
51,
52]. It is inspired by Genetic Algorithm (GA) and mimics natural selection and reproduction processes [
53]. Both GP and GA imitate some spontaneous optimization processes in natural selection and reproduction. At each iteration (generation), GP manipulates a set (population) of encoded solutions (individuals). These individuals are evaluated using a fitness function, which represents the objective function. Based on their fitness, good individuals are selected to produce new solutions (offspring) through operators such as crossover and mutation. The offspring then replace some individuals in the current population. Extensive computational experience on several optimization problems shows that GA often produces high-quality solutions in a reasonable time [
54,
55]. We refer the interested reader to [
56].
In this section, we present a GP approach (Algorithm 2) where each individual represents a feasible solution—a Dollo-k phylogeny. The same approach can be viewed as a GA, where the main difference between the two approaches is the representation of a solution. An individual in GA can be a string, while for GP, each individual is Dollo-k phylogeny.
In our implementation, a population size of 20 is used for smaller instances and 200 for larger instances. The halting criterion is a timeout of 3000 s on all instances. To maintain population diversity and avoid premature convergence, all individuals in the population are unique, meaning any duplicates are discarded.
| Algorithm 2: Genetic Programming |
![Algorithms 16 00333 i002]() |
3.2.1. Representation and Initialization
The initialization process of the GP approach ensures that only feasible solutions (Dollo-k phylogenies) are generated, similar to the PSO approach. The initialization starts by generating a random tree with a specific number of nodes, equal to the number of mutations (m), where no character is lost. This guarantees a fast and correct initialization, because each mutation is assigned one-to-one to a node. It is important to note that the initialization process allows for the exploration of the entire search space of feasible solutions. Any initial feasible solution can reach any other feasible solution in the search space through crossover and mutation operations. This property ensures that the GP approach has the capability to explore and search the space of Dollo-k phylogenies effectively.
3.2.2. Fitness Calculation
The fitness of an individual in the GP approach is primarily determined by its log-likelihood value, as described in
Section 2. However, to prioritize simpler and smaller phylogenies over larger and more complicated ones, we consider some additional factors, such as the number of nodes
and the depth of the tree
T in the definition of fitness. The fitness equation for an individual is as follows:
This definition of fitness helps prevent the occurrence of a phenomenon called
bloat [
57], which is the progressive growth of solutions, generation after generation, leading to larger and more complex individuals without a significant improvement in fitness.
3.2.3. Selection
The selection operator in GP determines which individuals will produce offspring in the next generation, based on their fitness. More precisely, individuals with higher fitness have a higher probability of being selected. Additionally, we employ an elitist strategy, where a few (in our case, one) highly fit individuals are directly passed to the new generation, while the selection process is applied to the remaining individuals to determine the rest of the population in the new generation. The individuals directly passed to the new generations are referred to as elite, while the others are classified as non-elite. The main effect of the elitist strategy is an emphasis on the exploitation phase. However, both elite and non-elite individuals play an equal role in the subsequent phases of the GP algorithm.
The selection procedure has a huge impact on the balance between the exploration phase, which focuses on finding new solutions, and the exploitation phase, which concentrates on the portion of the search space that has already been visited in the exploration phase. Finding an optimal balance between exploration and exploitation is crucial for the effectiveness of the GP algorithm.
The literature on selection operators [
53] for GA and GP is rich and includes roulette wheel selection, ranking selection, and tournament selection. In this paper, we employ FGTS (fine-grained tournament selection [
54]), which is an enhanced version of the standard tournament selection.
In the standard tournament selection, a tournament round is conducted for each non-elite individual that needs to be selected for the next generation. In each tournament round, a random subset of non-elite individuals is chosen, and the fittest individual among them is selected as the winner. The winner is then passed on to the next generation, and a new tournament round begins. This process continues until the selection is complete. Notice that an individual can participate in multiple tournaments within a generation, which allows for fair competition among individuals and provides opportunities for selection based on their fitness in different contexts.
The tournament size
is typically an integer value that determines the balance between exploration and exploitation. Choosing an appropriate value for
can be challenging, as smaller sizes lead to slower convergence, while larger sizes result in faster convergence. To overcome this problem, FGTS introduces a rational (i.e., not necessarily integral) parameter
, which represents the desired
average tournament size. In FGTS, tournaments can have different sizes, but the average size is maintained close to
. In our implementation, we have set
. The pseudocode for the selection operator is Algorithm 3.
| Algorithm 3: Fine-grained tournament selection (FGTS) |
![Algorithms 16 00333 i003]() |
3.2.4. Crossover
The crossover operator, also known as sexual recombination, introduces some variation in the population by combining genetic material from the parent individuals [
56]. In the context of GP [
50] for our problem, the crossover operator selects a random node—the crossover point—in each parent tree
and
. The crossover
fragment for a parent
is the subtree rooted at the crossover point. The offspring is created by replacing the crossover fragment of
with the crossover fragment of
—this operation can be seen as an extension to a pair of trees of the Subtree Prune and Regraft operation of the PSO. Additionally, a symmetric offspring is created by replacing the crossover fragment of
with the crossover fragment of
.
Designing a crossover operator for our problem is challenging because we need to guarantee that all offspring generated are feasible solutions. The crossover procedure involves the use of the function to create two offspring from the parents. The mating procedure must maintain the validity of the individuals in the population. The crossover procedure (Algorithm 4) starts by randomly selecting a mutation and checking if it can identify a valid crossover point in each tree. In other words, the crossover operation should result in two valid tumor phylogenies. If the check is successful, the offspring are created, and the procedure terminates. If the check fails, another mutation is selected, and the procedure is iterated.
It is important to note that the crossover operation is performed with a certain probability, known as the crossover probability, set to
in our approach.
| Algorithm 4: Crossover |
![Algorithms 16 00333 i004]() |
3.2.5. Mutation
Mutation is an asexual operator that operates on a single individual by modifying a specific mutation point. Many variants of the standard GP mutation have been presented in the literature.
In our approach, we have chosen one of the simplest mutation operations: the insertion of a new child of a given node and the deletion of a node—deleting a node x involves adding all its children as new children of the parent of the node x, then removing the node itself.
During the mutation process, each node of the individual is selected with a mutation probability that is smaller than the crossover probability. In our approach, the mutation probability is set to . If a node is selected for mutation, there is an equal probability of either inserting a new child or deleting the node. This choice helps maintain the number of nodes in the individual and prevents excessive growth (bloat). If the resulting individual after a mutation is not a feasible solution, the mutation is discarded and the individual remains unchanged.
3.2.6. Additional Info about GP Algorithm and Implementation
We have implemented the GP algorithm in Python 3, using the DEAP library [
58] for genetic programming, anytree for tree-like structures manipulation, and bitstring for the efficient manipulation of bit-arrays.
3.3. Variable Neighbourhood Programming
Genetic Programming and Particle Swarm Optimization are two population-based metaheuristics that explore the search space to find a near-optimal solution. However, they often converge slowly to a such a solution [
59]. To address this issue, local search-based algorithms—where we apply a sequence of local changes to the current solution—have been developed. Variable Neighbourhood Programming (VNP) [
60] is such a technique. VNP is inspired by automatic programming solution representation and Variable Neighbourhood Search (VNS) metaheuristics, combining the concept of neighborhood change with local search methods to ensure the discovery of local optima. Since VNP is based on non-binary representations, it should be more suited to problems on tree-like objects.
The main idea of VNS [
46] and VNP is to change neighborhoods as the search progresses and incorporate local search methods to guarantee the identification of local optima. We refer the interested reader to [
61]. The neighborhood structure plays a crucial role in defining the search space, both during the descent to local minima and in the escape from the valleys which contain them.
A fundamental ingredient of VNS and VNP is to define a suitable neighborhood structure for the solution space. In our case, we define four different neighborhoods for a given tumor phylogeny T. The first neighborhood, denoted by , consists of all valid Dollo-k phylogenies that differ from T in a subtree whose root is a leaf, or a parent of a leaf, or a grandparent of a leaf. In other words, the root of such subtree has distance of at most 2 to a leaf. The second and third neighborhoods, and respectively, consist of all valid Dollo-k phylogenies that differ from T in a subtree whose root has a maximum distance of 5 (resp. 8) to a leaf. Finally, the last neighborhood, denoted as , consists of all valid Dollo-k phylogenies. Notice that the cardinality of the neighborhoods decreases with increasing index (that is, for each ). To facilitate the presentation of the algorithm, we introduce the function , where , , and . Each set consists of all valid Dollo-k phylogenies that differ from T only in nodes with a distance at most to a leaf in T.
The main idea of VNP is alternating exploration of the current neighborhood through a local search, and the shaking, where we move to another neighborhood and different parts of the solution space can be explored. The pseudocode of VNP is given in Algorithm 5.
| Algorithm 5: Variable Neighbourhood Programming |
![Algorithms 16 00333 i005]() |
The VNP follows a main loop that is iterated until a halting condition is met. Usually, this condition includes a limit on the number of iterations and a timeout for the overall execution time. In our case, the timeout is set to 3000 s for all instances. An important invariant of the algorithm is that
T always is the best solution visited so far. The main loop consists of two phases: the shaking phase and the local search. In the shaking phase, a new solution
is generated by exploring the current neighborhood, while the local search produces a solution
. If
is better than
T, then
becomes the new incumbent and the next search begins at the first neighborhood
. However, if
does not improve upon
T, the algorithm progresses to the next neighborhood in the sequence to attempt further improvement. If the last neighborhood
is explored and a solution better than the incumbent is found, the search restarts from the first neighborhood. Notice that the initial solution is a feasible solution computed as in
Section 3.1 and that the fitness of a solution is computed as described in
Section 3.2.2.
3.3.1. Shaking
The shaking phase in the VNP serves to introduce diversity and explore the current neighborhood
in search of a feasible solution
. The shaking procedure typically involves the random extraction of a feasible solution. In our case, the definition of neighborhood classes, as described earlier, is tailored for the phylogeny sizes we expect to manage. Larger datasets would require a different definition of neighborhoods; for example, changing the subtree depth or introducing new classes.
| Algorithm 6: Shaking |
Data: s: current neighbourhood class. T: current solution. Result: : a feasible solution
- 1
; - 2
a random vertex of that has distance at most to a leaf - 3
the subtree of rooted at r - 4
is a random tree with the same edge labels as - 5
is obtained from by replacing with - 6
Contract in all edges of labeled by a mutation loss such that none of its ancestors is labeled - 7
return
|
In Algorithm 6, the shaking procedure selects a subtree from the current solution T and permutes its edges. The selected subtree is denoted as , and its edges are randomly rearranged to generate a new solution . Since is a permutation of the edges of , the set of mutation acquisitions and losses is preserved. The only possible case when is not valid only can happen when some mutation loss precedes a mutation acquisition. Line 6 of the algorithm explicitly removes such losses. The resulting tree is, therefore, a valid Dollo-k phylogeny.
The shaking procedure is designed to guarantee that all solutions in the search space can be potentially reached. This means that there are no feasible solutions that cannot be reached through the shaking procedure.
3.3.2. Local Search
Local search is performed on a shaken solution
to potentially find an improved solution
. Local search strategies commonly used in VNS and VNP include best improvement (highest descent) and first improvement (first descent). In our implementation, we have chosen the first improvement strategy for the local search. This means that the local search terminates as soon as the first improvement is found. The pseudocode for the local search with the first improvement strategy is presented in Algorithm 7.
| Algorithm 7: Local search, first improvement strategy |
![Algorithms 16 00333 i006]() |
3.3.3. Additional Info about VNP Algorithm and Implementation
The VNP algorithm is implemented as a Python 3 program using the anytree library for tree-like structures manipulation and the bitstring library for efficient bit-array manipulation.
5. Discussion
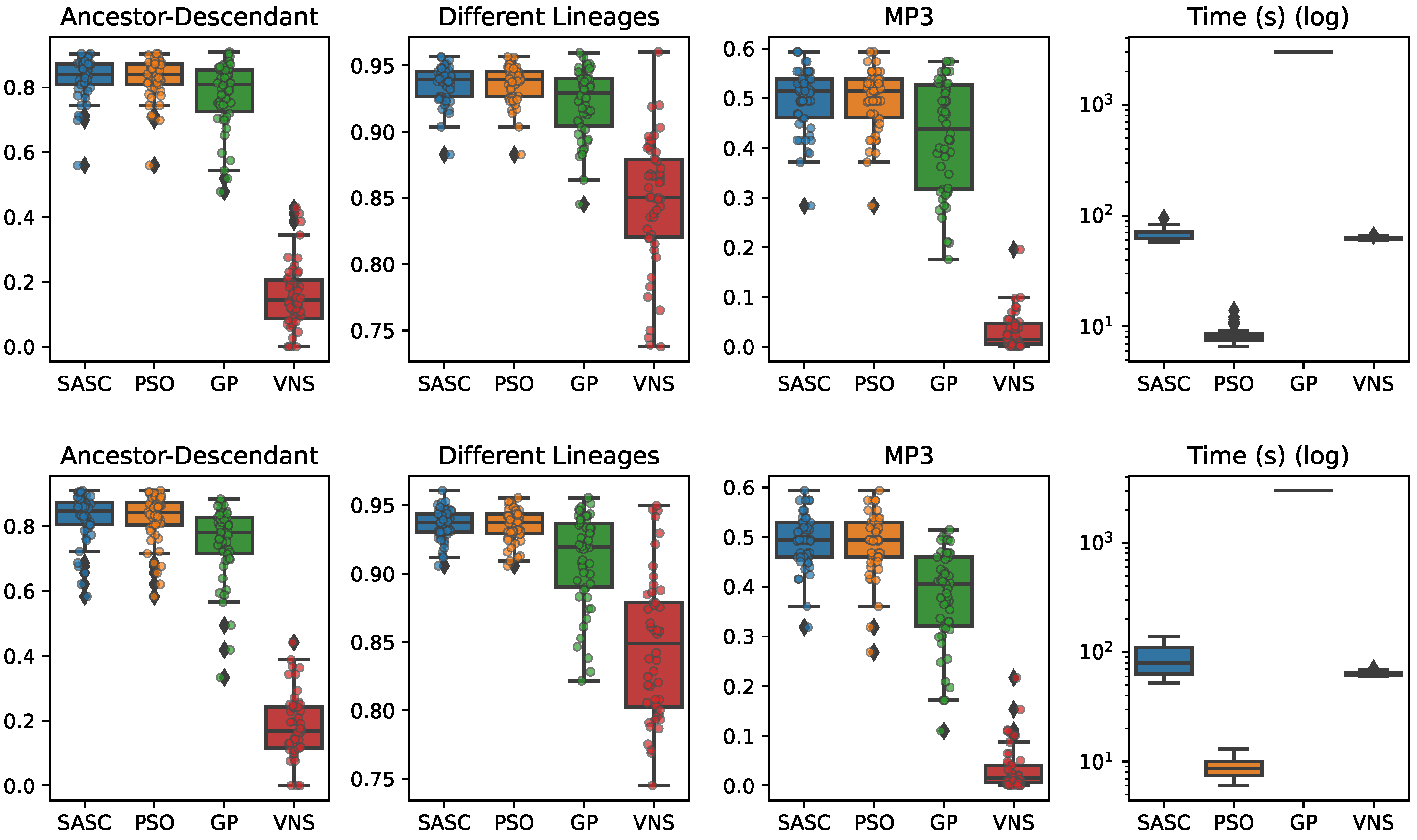
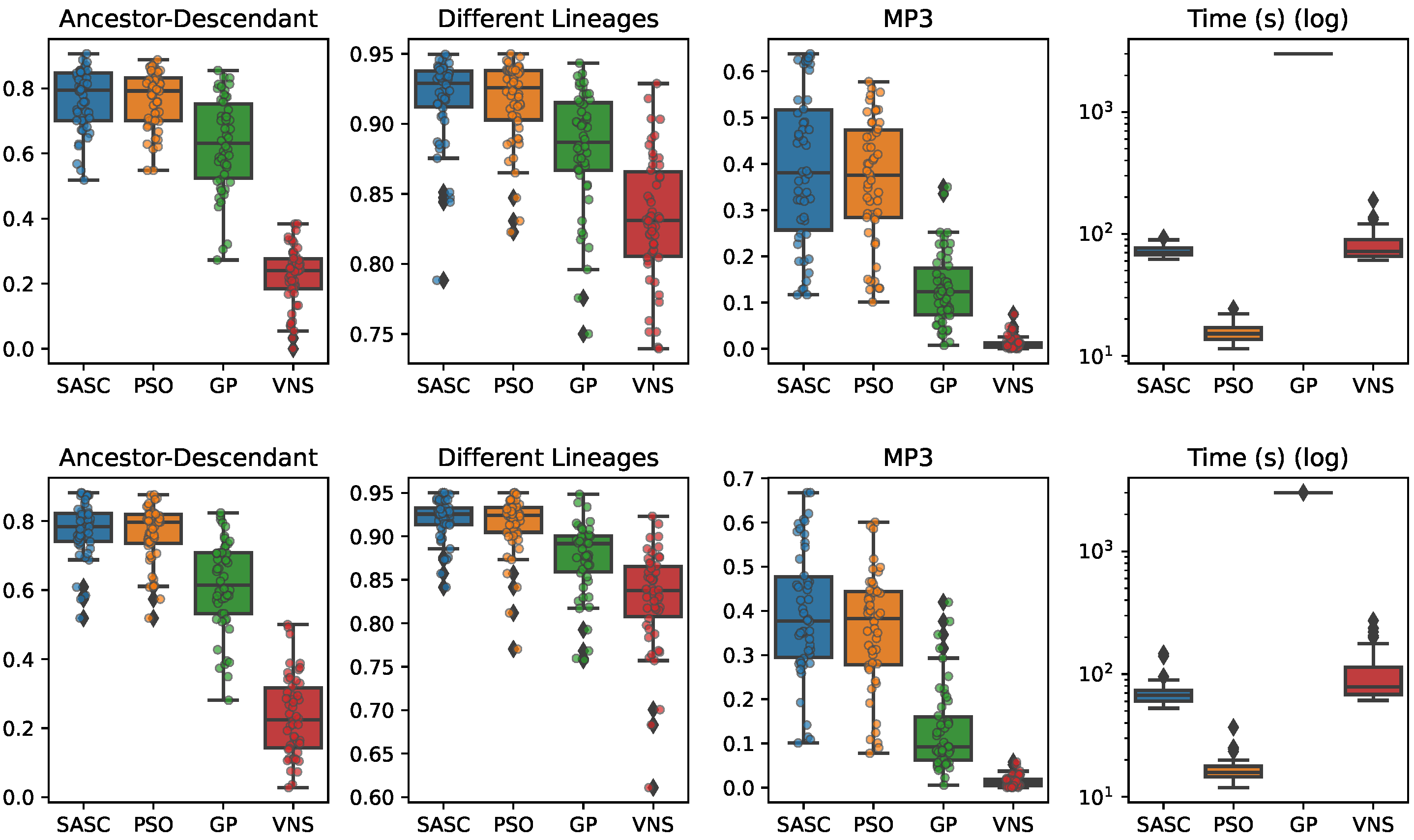
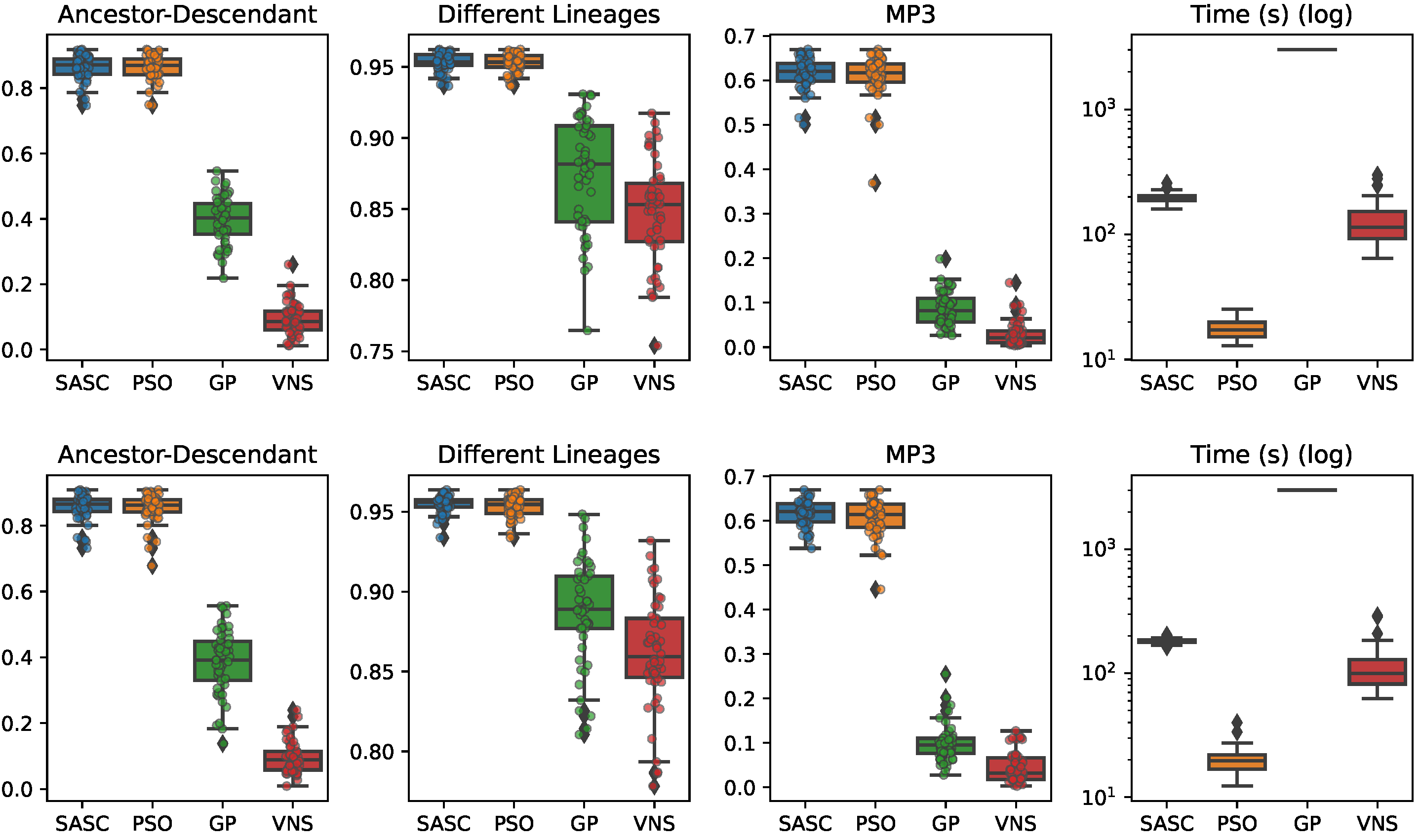
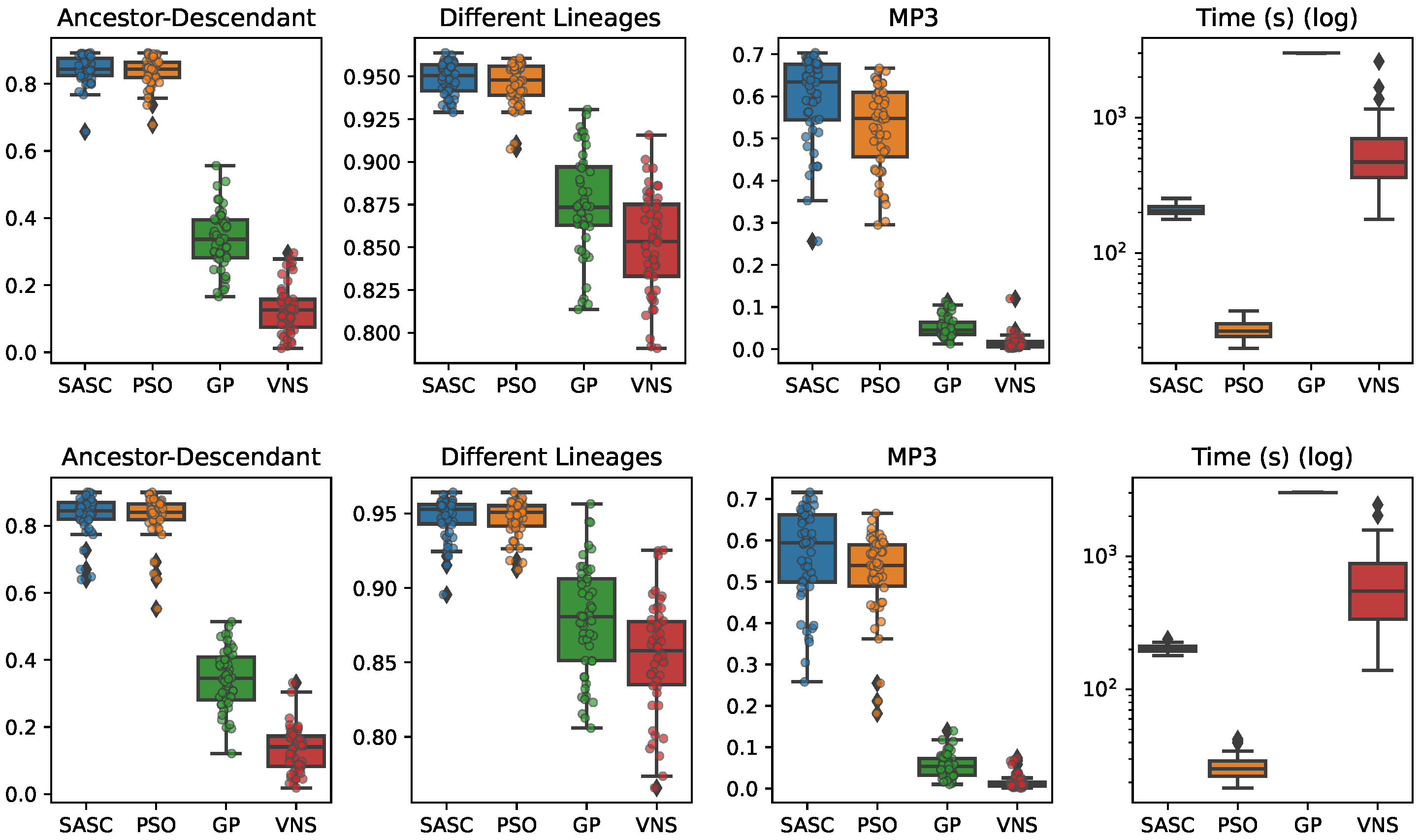
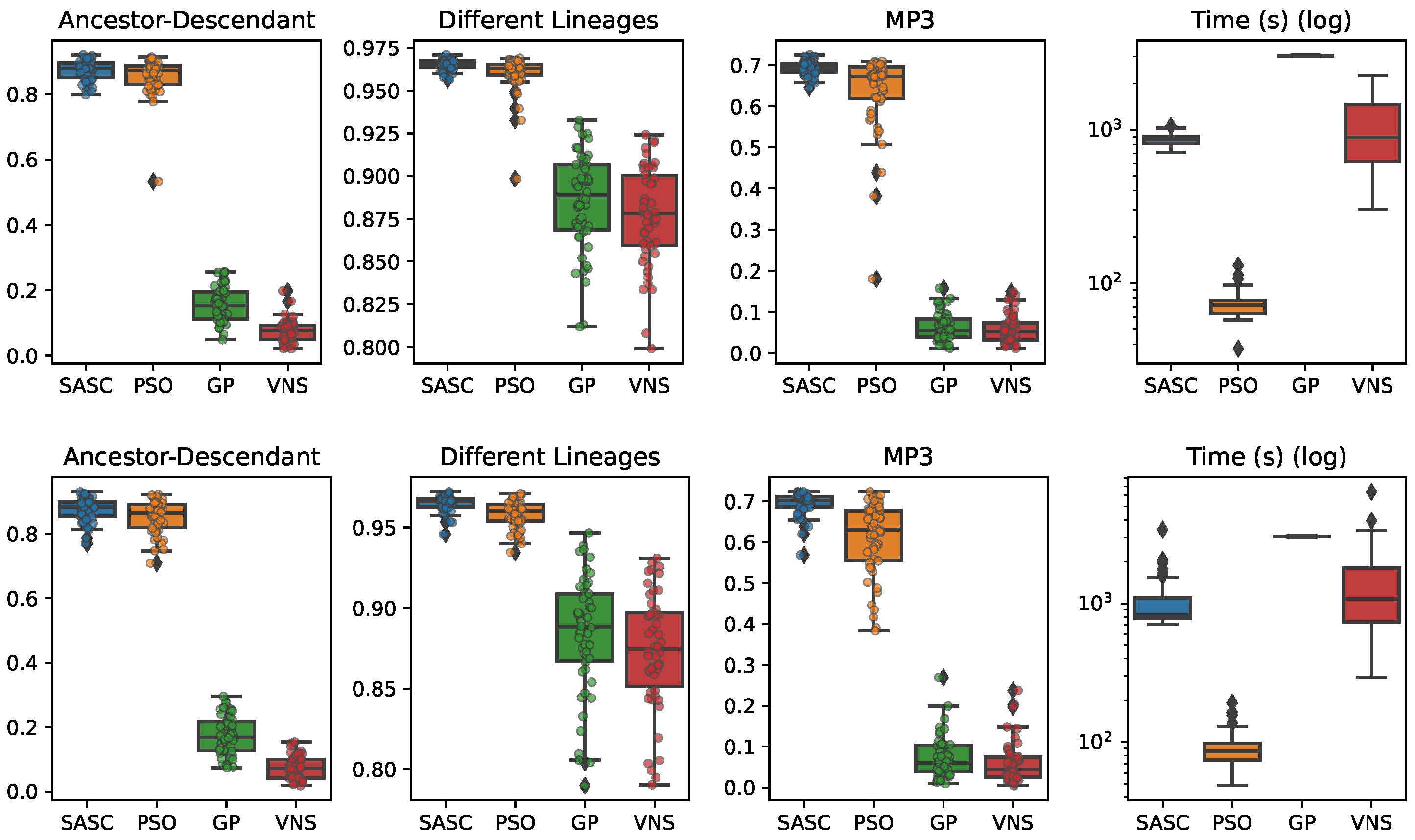
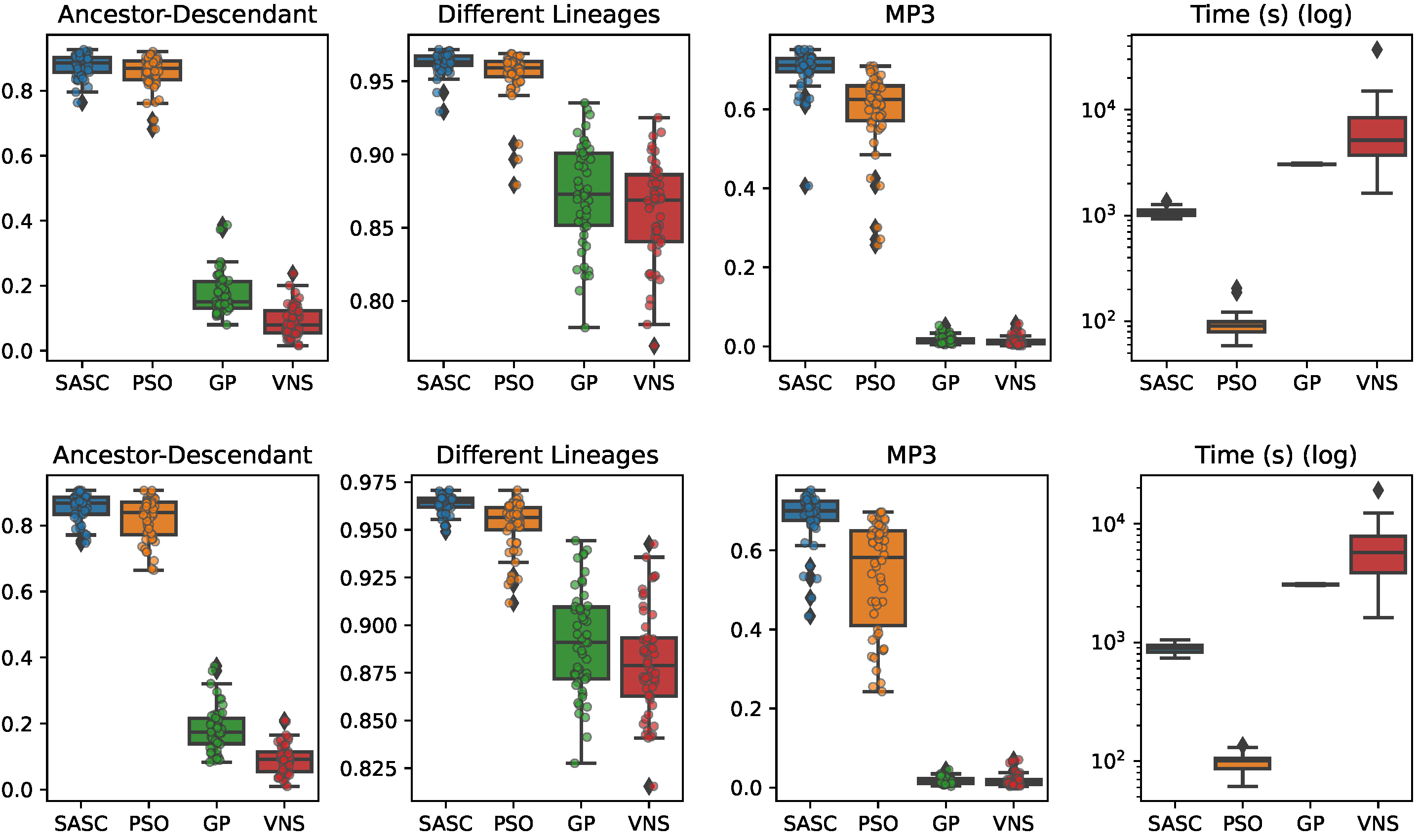
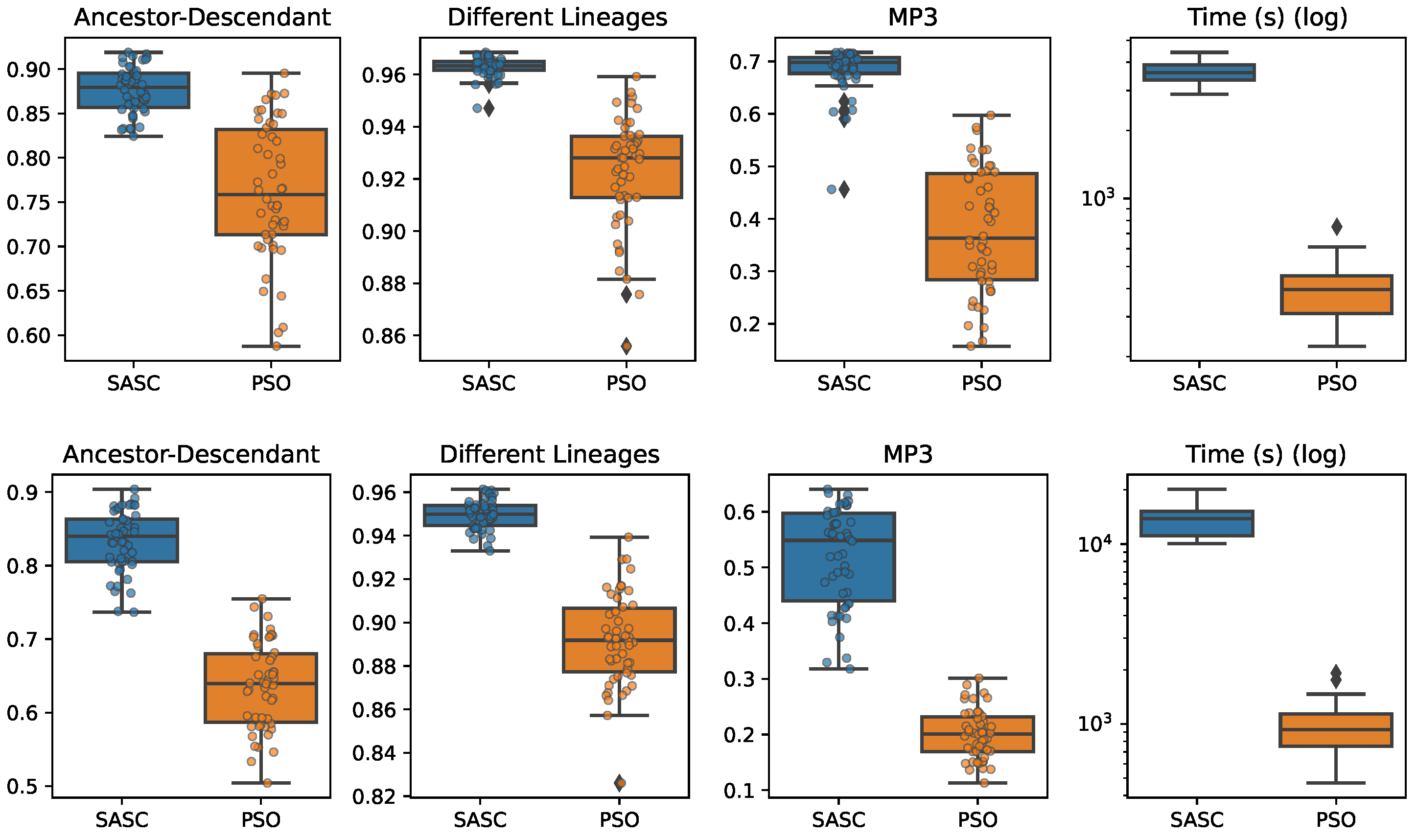
In this paper, we have presented and experimentally assessed three different metaheuristics (PSO, GP, and VNP) to infer tumor phylogenies under the Dollo model. We have compared these methods to the state-of-the-art Simulated Annealing approach (SASC).
Among the three metaheuristics, PSO showed the most promising results, although it was not able to outperform SASC. PSO demonstrated accuracy comparable to SASC in most experiments and measures, but its performance dropped as the complexity of the problem increased.
The main challenges faced by these methods stem from the fact that trees are not inherently a numeric space, which poses difficulties in defining distance, velocity, and direction on trees. Consequently, the design of appropriate definitions for these parameters is a crucial area for future research. These definitions should be computationally efficient and capable of capturing the properties that the PSO approach can exploit. Successful developments in this research direction have the potential to significantly enhance the accuracy of PSO. Moreover, we expect the parallelism capabilities of PSO to allow us to exploit many-cores infrastructures to allow an even larger exploration of the search space, leading to higher-accuracy tumor phylogenies.

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}