1. Introduction
Glass fibers (GF) are the most common reinforcement for polymeric matrix composites. Their principal advantages are the relationship between their low cost, high tensile strength, high chemical resistance, and insulating properties. The disadvantages are low tensile modulus, relatively high specific gravity, sensitivity to abrasion during handling, low fatigue resistance, and high hardness. E-glass and S-glass are the types of fibers more commonly used in the fiber-reinforced plastic industry. E-glass fibers have the lowest cost of all commercially available reinforcing GFs, which is the reason for their widespread use in the fiber-reinforced plastic industry. S-glass, originally developed for aircraft components and missile casings, has the highest tensile strength among all fibers in use. However, the compositional difference and higher manufacturing cost make it more expensive than E-glass [
1,
2,
3].
The manufacturing process for GFs includes several steps. Various ingredients in the glass formulation are first dry mixed and melted in a refractory furnace at about 1,370 °C. The molten glass is exuded through a number of orifices contained in a platinum bushing and rapidly drawn into filaments of approximately 10 µm diameter. A protective coating (“size”) is then applied on individual filaments before they are gathered together into strands and wound on a drum. The size is a mixture of lubricants, which prevent abrasion between the filaments, antistatic agents, which reduce static friction between the filaments and a binder, which packs the filaments together into a strand. It may also contain small percentages of a coupling agent that promotes adhesion between fibers and the specific matrix for which it is formulated [
1].
Fiber-reinforced composite materials consist of fibers with high strength and modulus embedded in a matrix. Both fibers and matrix retain their physical and chemical identities and the new material has a combination of properties that cannot be achieved with either of the constituents acting alone. In general, fibers are the main load-carrying members, while the matrix functions are: to transfer stresses between the fibers, to provide a barrier against an adverse environment, and to protect the fiber surface from mechanical abrasion [
1,
3].
Polymer composites have been used in a wide range of industrial applications like aeronautic, naval, construction, sporting goods, home appliances, furniture,
etc. They have been on the market for over fifty years, and have generally been used to replace materials such as wood, aluminum and steel. Their advantages over traditional materials include greater mechanical strength, lighter weight, better dimensional stability, higher dielectric strength and corrosion resistance, and flexibility to improve the designs [
4]. The major advantage of these composites that helped them enter into such a variety of markets is their high specific property, which is greater than that of metals and ceramics. Nevertheless, the tailoring of well-bonded and/or durable interphases between the matrix and reinforcement remains a critical issue in this kind of material. This factor is critical with thermoplastic polymer matrices such as polyethylene (PE), polypropylene (PP), polyvinylchloride (PVC), polystyrene (PS) and polyamide (PA).
The effectiveness of reinforcement essentially depends on the adhesion between matrix and fiber, so this is a key factor in determining the final properties of the composite material, particularly its mechanical properties [
1,
5,
6,
7]. The fiber-matrix adhesion is confined to a region or “third phase” known as interphase, where stress-transfer occurs. The interphase is defined as the tridimensional region, located between the fiber and the matrix. It is considered as a transition region or third phase with its own characteristics, corresponding neither to fiber properties nor to matrix ones [
8]. Wu postulated that the extent of molecular or local segmental diffusion across the interface determines the structure of the interfacial zone, which critically affects the mechanical strength of an adhesive bond. Negligible diffusion will give a sharp interface with weak adhesion. In this case, high adhesive strength can be expected only when strong polar interactions or chemical bonds exist across it. On the other hand, if the interphase is relatively thick and gradual as a result of extensive diffusion, a high adhesive strength will result just by the effect of dispersion forces [
9].
3. Fiber In-Situ Polymerization as a Route of Adhesion Improvement
PP polarity is so low then the adhesion with GF is bad. As it was shown above, a lot of effort was done in order to enhance the compatibility between both surfaces, but improvement obtained was low. Also its viscosity in the molten state is so high that good penetrability in GF mats is impeded. The best possible adhesion is given by chemical anchorage between the polymer and fiber. Following this idea, PP was polymerized and chemically bonded on the glass surface [
36,
37] in our group. This reaction involves a metallocenic catalysis starting with propylene gas. In this sense, when polymerization is performed onto the glass-fiber mat, the penetrability of this gas is so high that PP “prepreg” mats could be obtained [
38].
The experimental reaction route involves an initial contact with methylaluminoxane (MAO) and hydroxy-α-olefin (9-decen-1-ol) to generate the anchorage points on the fiber surface, followed by a propylene polymerization catalyzed by EtInd
2ZrCl
2 (metallocene)/MAO. The reaction was carried out onto GFs of about 25 μm nominal diameter, provided by Vetrotex Argentina. It should be noted that, because the extruding glass process takes place at very high temperatures on the surface, hydrolysis used for all surface modifications, silanization, titanization, and so on, and will also be used as anchor points for the proposed direct copolymerization in this study.
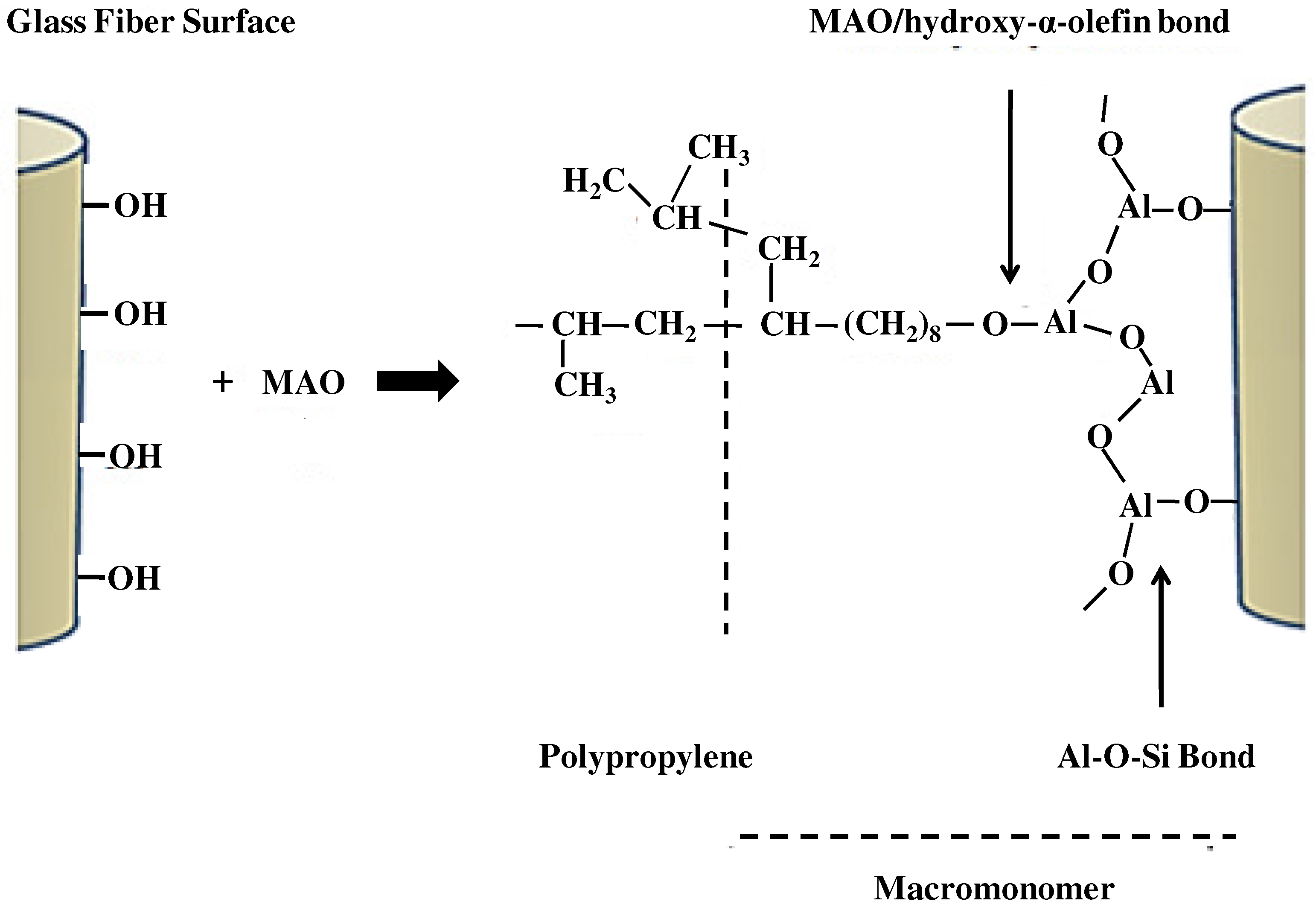
Figure 1 shows the reaction scheme proposed for the PP grafting molecules onto glass surface.
Figure 1.
Scheme proposed for the polypropylene (PP) grafting molecules onto glass surface.
Figure 1.
Scheme proposed for the polypropylene (PP) grafting molecules onto glass surface.
The graft reaction of PP onto GFs includes two steps:
- -
Glass-MAO pretreatment. The first step is MAO addition in order to produce an appropriate concentration of CH
3 groups (from the MAO), which subsequently react with the compounds to be copolymerized (the hydroxy-α-olefin) through the OH groups. The glass reacts with MAO through the OH groups anchored on its surface, releasing methane (CH
4) and generating a bond Al-O-Si (glass). The methyl group on the (glass) Si-O-Al-CH
3 reacts with the OH of the hydroxy-α-olefin, releasing CH
4. The final anchorage on the surface, previous to the propylene polymerization reaction, is AlO
3, with one of the O of the AlO
3 linked to an alkyl group and the other two to Si or Al. The addition of a hydroxy-α-olefin to supply the necessary vinyl-bonds to initiate the propylene polymerization is performed. It was demonstrated that short hydroxy-α-olefins (5-hexen-1-ol) [
39] tend to act as Lewis bases; however the use of long-chain hydroxy-α-olefins (9-decen-1-ol,10-undecen-1-ol) is preferred to assure a low level of poisoning [
40].
- -
Reaction of the glass surface with hydroxy-α-olefin. After the MAO reaction to the glass surface, hydroxy-α-olefin is added. The hydroxy-α-olefin reacts with the MAO-CH
3 groups with evolution of methane and the setting of terminal vinyl groups to the glass. Then, a copolymerization reaction can be initiated from these macrocomonomers. The propylene (CH
3–CH=CH
2) molecules are attached via catalytic polymerization using EtInd
2ZrCl
2 (metallocene)/MAO. The MAO is included in the metallocenic catalyst to alkylate the metallocene, and to generate and stabilize the cationic active zirconocene. As a result, PP chains grow by copolymerization of propylene with the vinyl group of the Al-O(CH
2)n-C=CH
2, remaining chemically bonded to the glass. Finally, the polymer precipitates by acidified ethanol aggregate. Details of the experimental procedure are described in previous works [
36,
38]. This reaction scheme (
Figure 1) was further proved by consistent evidence obtained by combining scanning electron microscopy techniques (SEM) with X-ray energy dispersive analysis (EDX).
The hydroxy-α-olefin concentration used in each case was calculated in order to ensure the activity of the metallocene catalyst. The metallocene concentration remained constant. Therefore, the reaction variables were selected aimed to obtain a high level of PP coverage on the glass surface by chemical bonding between them.
Table 1 summarizes the nomenclature of all reactions performed, where the concentrations in percentage were calculated following the calculations explained in reference [
37]. The experimental methodology, as well as the techniques used for characterization, are schematically described in
Figure 2.
Table 1.
Nomenclature of all reactions performed.
Table 1.
Nomenclature of all reactions performed.
| Name | Hydroxy-α-olefin [μL] | Total MAO [mL] |
|---|
| F0 | ---- | ---- |
| F0.5% | 50 | 10 |
| F1% | 100 | 10 |
| F1.5% | 150 | 12 |
| F2% | 200 | 12 |
Figure 2.
Experimental methodology.
Figure 2.
Experimental methodology.
In order to determine the reaction occurrence, a comparative study was performed on fiber surfaces after strong PP extraction. In a previous work, screening experiments were performed using extremely low and high hydroxy-α-olefin concentrations, 0.4 and 4%, respectively. In these studies, it was demonstrated that propylene effectively copolymerizes onto fiber surfaces and the final morphology depends on the hydroxy-α-olefin amount used during the reaction [
36,
41].
4. Morphological Aspects of in Situ Polymerized Fiber Glass
Figure 3 shows the scanning electron micrograph of GFs without any treatment and its corresponding EDX spectrum, which is used as standard. Peaks of Si, O, Al and Ca for the E-glass fiber chemical composition can be seen. Note that the peak of C is undetectable, so it is not possible to establish an initial C/Si ratio. A complete morphological study was performed on all reaction products in order to optimize the alcohol concentration that maximizes the PP-glass adhesion.
Figure 4,
Figure 5,
Figure 6 and
Figure 7 show the fiber surfaces after
in-situ polymerization for all hydroxy-α-olefin concentrations. The EDX spectra are also included to verify the nature of the species attached to the fibers; and to allow a rough estimation of their relative amounts. In all cases, the polymers attached to the fibers appear as white particles in the SEM micrographs. From the EDX spectra, it is clear that the C peak increases notably after polymerization.
Figure 3.
Scanning electron micrograph (SEM) of the sample F0 (2,000×) with its corresponding EDX spectrum.
Figure 3.
Scanning electron micrograph (SEM) of the sample F0 (2,000×) with its corresponding EDX spectrum.
Figure 4.
SEM micrographs for sample F0.5% and its corresponding EDX spectrum. (a) 600× and (b) 2,000×.
Figure 4.
SEM micrographs for sample F0.5% and its corresponding EDX spectrum. (a) 600× and (b) 2,000×.
Figure 5.
SEM micrographs for sample F1% and its corresponding EDX spectrum. (a) 600×; (b) 6,000×; (c) and (d) 10,000×.
Figure 5.
SEM micrographs for sample F1% and its corresponding EDX spectrum. (a) 600×; (b) 6,000×; (c) and (d) 10,000×.
Figure 6.
SEM micrographs for sample F1.5% and its corresponding EDX spectrum. (a) 200×; (b) 600×; (c) 2,000× and (d) 6,000×.
Figure 6.
SEM micrographs for sample F1.5% and its corresponding EDX spectrum. (a) 200×; (b) 600×; (c) 2,000× and (d) 6,000×.
Figure 7.
SEM micrographs for sample F2% and its corresponding EDX spectrum. (a) 200×, (b) 2,000× and (c) 6,000×.
Figure 7.
SEM micrographs for sample F2% and its corresponding EDX spectrum. (a) 200×, (b) 2,000× and (c) 6,000×.
For all
in-situ polymerizations, the EDX spectra show large C peaks and obviously O, Al, Si and Ca. The C peak corresponds to PP adhered to the glass surface. Note that in some cases, when the amount of C is very large (F1% in
Figure 5) the typical peaks from the fiber (Si, Al, O) are relatively reduced, as expected. In low magnification, the micrographs show that the degree of coverage for different concentrations of hydroxy-α-olefin used is different.
In extreme conditions (F0.5% and F2%) the coverage is not homogeneous, presenting zones with larger polymer agglomerates. At high magnifications, it is evident that the polymer onto fibers presents different morphologies. These crystalline forms are typical of the crystallization during polymerization of polyolefins, particularly PP. Wunderlich shows similar results for PP, where the difference between the spherical macroscopic crystalline forms attached to the polymer is first generated and then crystallized, nucleated by a particle of the catalyst. The smaller rod-shaped particles are those where the crystallization occurs as the polymer is formed [
42,
43].
Taking into account that the objective is the improvement of adhesion, a homogeneous polymer layer on the fibers is needed. F1% and F1.5% samples show the highest degree of coverage while F0.5% and F2% samples have agglomerated polymer. The greater the amount of hydroxy-α-olefin, the greater the number of anchor points and the lower the PP chain length. Aaltonen
et al. [
44,
45], report that an increase in the concentration of hydroxy-α-olefin reduces the rate of consumption of propylene when other polymerization conditions are kept constant..
From these results it appears that the length of the PP chains decreases when the concentration of hydroxy-α-olefin increases. These observations also agree with the EDX results. Comparing the EDX spectra, it is clear that in all polymerizations the C peak appears, but its relative intensity is much greater for conditions F1% and F1.5% that for F0.5% and F2% samples.
Thermal characterization of the polymer grafted onto fibers was performed by Differential Scanning Calorimetry (DSC) and Thermogravimetrical analysis (TGA).
Figure 8 shows the DSC thermograms of the PP grafted onto fibers with different percentages of hydroxy-α-olefin and compared with untreated fibers. Low melting temperatures (110–122 °C) in PP copolymerized onto fibers is typical of γ-PP metallocene polymerization. TGA gives the evidence that molecules grafted onto the fibers at the optimal condition are PP ones. The thermograms obtained allow the evaluation of the mass changes that occur at different temperatures, indicating the transformations taking place at all times.
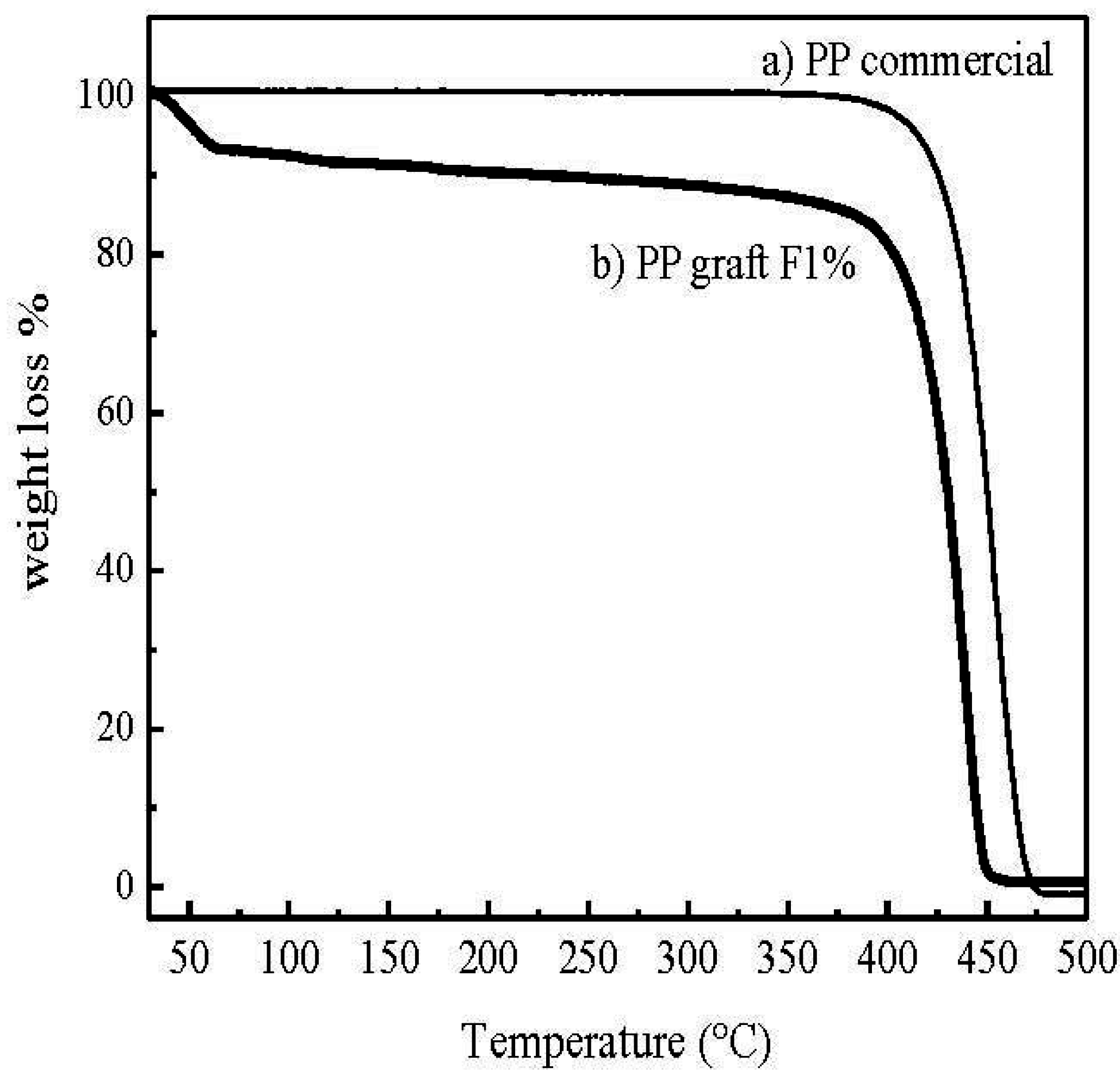
Figure 9 shows the weight loss as a function of temperature. Degradation of PP can be observed at 425 °C whereas commercial PP degrades at approximately 450 °C.
Figure 8.
Differential Scanning Calorimetry (DSC) thermograms of the polymer grafted onto the GFs with different hydroxy-α-olefin concentrations.
Figure 8.
Differential Scanning Calorimetry (DSC) thermograms of the polymer grafted onto the GFs with different hydroxy-α-olefin concentrations.
Figure 9.
Thermogravimetrical analysis (TGA) thermograms for the sample (a) PP commercial and (b) PP graft MF1%.
Figure 9.
Thermogravimetrical analysis (TGA) thermograms for the sample (a) PP commercial and (b) PP graft MF1%.
Figure 10.
SEM micrograph (2,000×) and photograph of hydrophobicity test of: (a) untreated fibers and (b) copolymerized fibers.
Figure 10.
SEM micrograph (2,000×) and photograph of hydrophobicity test of: (a) untreated fibers and (b) copolymerized fibers.
The polymer fiber coverage after
in-situ polymerization and previous to composite preparation was qualitatively determined by a hydrophilic/hidrophobic test. The results are interpreted in terms of surface energy (or activity) change due to the attached material to GF. This technique includes the immersion of fibers in a mixture of two immiscible liquids with different polarity and density. A mixture of 50% v/v hexane/water was used in these tests. The densities at 20 °C were: hexane 0.675 g/cm
3 and water 1 g/cm
3. The GF surface behaves, initially, hydrophilicly since its superficial hydroxyl groups tend to form hydrogen bonds with water. However, such hydrophilicity should disappear as a consequence of the surface copolymerization and the resulting “PP coverage”. The different morphology exhibited by the fiber surface before and after copolymerization with 1% hydroxy-α-olefin is shown in
Figure 10. The complete PP coverage onto the GF is apparent from the comparison of
Figure 10a, b. This is consistent with possible surface activity variations that occurred with the polymerization onto the fibers. Untreated GF, which have surface hydroxy groups [
1], are hydrophilic, hence they sunk in water, as their density is 2.5 times greater. However, propylene copolymerized fibers remained at the water-hexane interphase. Since the “coating” is non-polar, the fibers became more compatible with the organic phase. Then, as
Figure 10 clearly shows, the “attraction forces” of the organic phase are greater than the gravity effect, showing that the GF surface energy radically changed. This simple test gives a qualitative but strong indication of the surface change that occurred to the fibers with the copolymerization treatment.
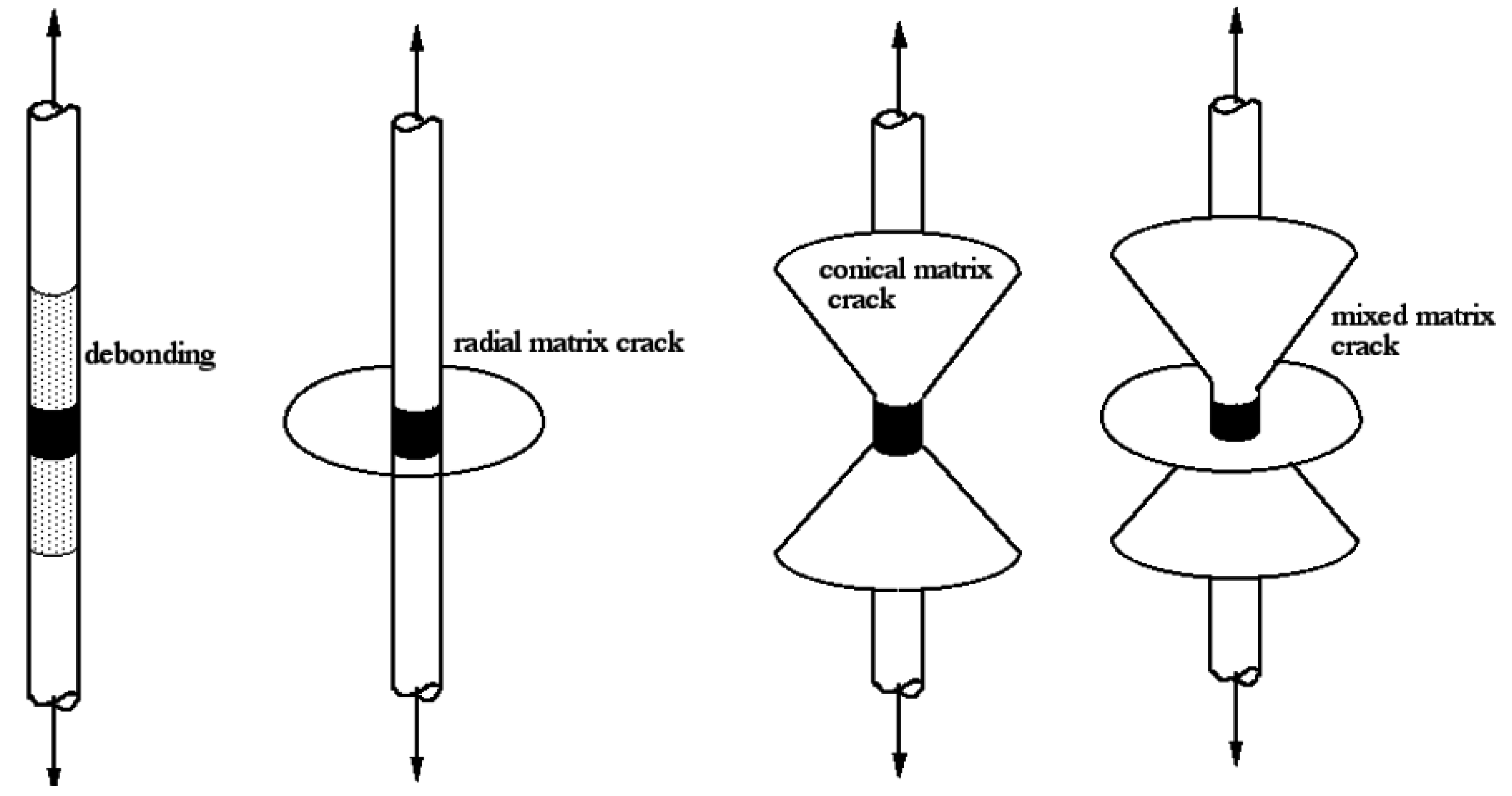
6. Mechanical Properties of PP/GF/PP Composites
Tensile mechanical behavior of laminated PP/GF/PP was evaluated using unidirectional arrays. The mechanical properties obtained were interpreted according to the adhesion results and phenomenological evidence.
The tensile behavior of composite materials is difficult to predict, as the fracture mechanics are so complex. Is known that the composite shows a marked anisotropy, that is to say, its properties vary significantly when measured in different directions. This usually arises because the harder constituent is in a fibrous form with the fiber axes preferentially aligned in particular directions [
3]. Hence, samples were prepared with uni-axially oriented continuous fibers, where the rule of mixtures can be used to predict modulus and strength. According to this rule, the mechanical strength for continuous, uni-axially oriented fiber composites is just based on the properties of each component and the relative concentration and orientation of the fibers. These approaches assume that the charge transfer is complete and the variations in adhesion between the fiber and the matrix are not taken into account. For this reason, they are, in principle, more suitable to predict the modulus value than the mechanical strength. The elastic modulus is measured as the slope of the stress-strain curve at low deformation, as the charge transfer is less critical in this property determination. Very low deformation materials react to an atomic-molecular level separately. However, the mechanical strength is the maximum tension that the material can resist, and it usually occurs at higher deformation, where charge transfer is a decisive factor, and consequently, adhesion influences its value. It is remarkable that the mechanical strength values, calculated from rule of mixture, assume full adhesion and thus overestimate the actual value. This problem is most evident in the case of transverse mechanical resistance, where the matrix “strip” of the fibers goes through the interface. For this reason the equation arises only as an approximation. Tensile tests were carried out on PP, GF and composite samples.
Table 5 summarizes the mechanical properties of the PP matrix and the fibers used in the composites. Note that these values are used as parameters, because they were measured under similar conditions with samples of equal size.
Table 5.
Mechanical properties of PP and GFs.
Table 5.
Mechanical properties of PP and GFs.
| Property | PP | GF |
|---|
| E [MPa] | 685 ± 69.6 | 47,600 ± 2,015 |
| σu [MPa] | 25.7 ± 1.7 | 234 ± 31.3 |
| εy [%] | 10.0 ± 1.8 | ---- |
| εb [%] | 412 ± 92 | 2.6 ± 0.9 |
The fiber concentration of the tensile specimens was measured by weight difference before and after washing them. The achieved weight and volumetric fractions are summarized in
Table 6 [
41].
Table 6.
Average concentration of fiber composites.
Table 6.
Average concentration of fiber composites.
| Fraction [%] | PP/GF/PP | PP/MAO-GF/PP | PP/COP-GF/PP |
|---|
| Volume | 53.8 ± 5.9 | 50.8 ± 4.8 | 46.6 ± 5.3 |
| Weight | 76.4 ± 4.35 | 74.2 ± 3.66 | 70.8 ± 4.58 |
Continuous parallel fiber PP/GF composites, containing about 1800 aligned GF, were prepared by compression molding in a hydraulic press. The fibers were carefully aligned and compounded by sandwiching them within two PP sheets. Before compounding, the GF received different surface conditioning: no pretreatment, in situ polymerization and just MAO treated surface. These composites were named PP/GF/PP, PP/COP-GF/PP and PP/MAO-GF/PP, respectively. Rectangular specimens for tensile tests were obtained by careful cutting with a sharp blade in order to have net edges and minimize errors during experimental tensile measurements.
The fracture shape, as well as the morphological features of fiber surface and microcomposites subjected to mechanical tests were studied by SEM.
Figure 19 shows tensile stress-strain curves for all the composites tested, PP/GF/PP, PP/MAO-GF/PP and PP/COP-GF/PP. The corresponding properties are summarized in
Table 7.
Table 7.
Mechanical properties for the composites PP/GF/PP.
Table 7.
Mechanical properties for the composites PP/GF/PP.
| Property | PP/GF/PP | PP/MAO-GF/PP | PP/COP-GF/PP |
|---|
| E [MPa] | 8,125 ± 1,060 | 8,491±1,029 | 8,852 ± 1,017 |
| σu [MPa] | 101.6 ± 14.1 | 103.4 ± 12.3 | 104.7 ± 11.1 |
| ε y [%] | 2.5 ± 0.8 | 1.6 ± 0.4 | 6.8 ± 0.9 |
| ε b [%] | 14.1 ± 8.3 | 16.56 ± 6.4 | 35 ± 7.2 |
| Toughness [J] | 12,654 ± 1,100 | 12,054 ± 512 | 38,579 ± 1,600 |
The possible occurrence of fiber damage caused by the MAO attack was studied in a previous study [
50], in which tensile stress-strain tests on both untreated and MAO treated single GFs were done. The strength data, statistically treated according to the Weibull distribution, showed that MAO induced only a slight decrease in the fiber tensile strength, indicating that a very small fiber damage could occur in the presence of MAO. This trend is also confirmed for composite samples, since only a small variation in strength and elongation properties was observed for composites having either untreated and MAO treated fibers (
Figure 19).
Figure 19.
Stress-strain curves of PP/GF/PP composites.
Figure 19.
Stress-strain curves of PP/GF/PP composites.
From the strain-dependent tensile behavior, it is observed that the mechanical strength of PP/GF/PP and PP/MAO-GF/PP composites is reached at 1 to 3% deformation; while for PP/COP-GF/PP, the deformation is close to 7%,
i.e., three times larger. This is related to the composite fracture mechanism. In the first two, the whole composite fracture occurs by fiber breakage, at ε < 3%, without any matrix contribution to resistance due to the low matrix-fiber adhesion. On the other hand, for copolymerized fiber composites, the fibers start breaking at about 2% strain (
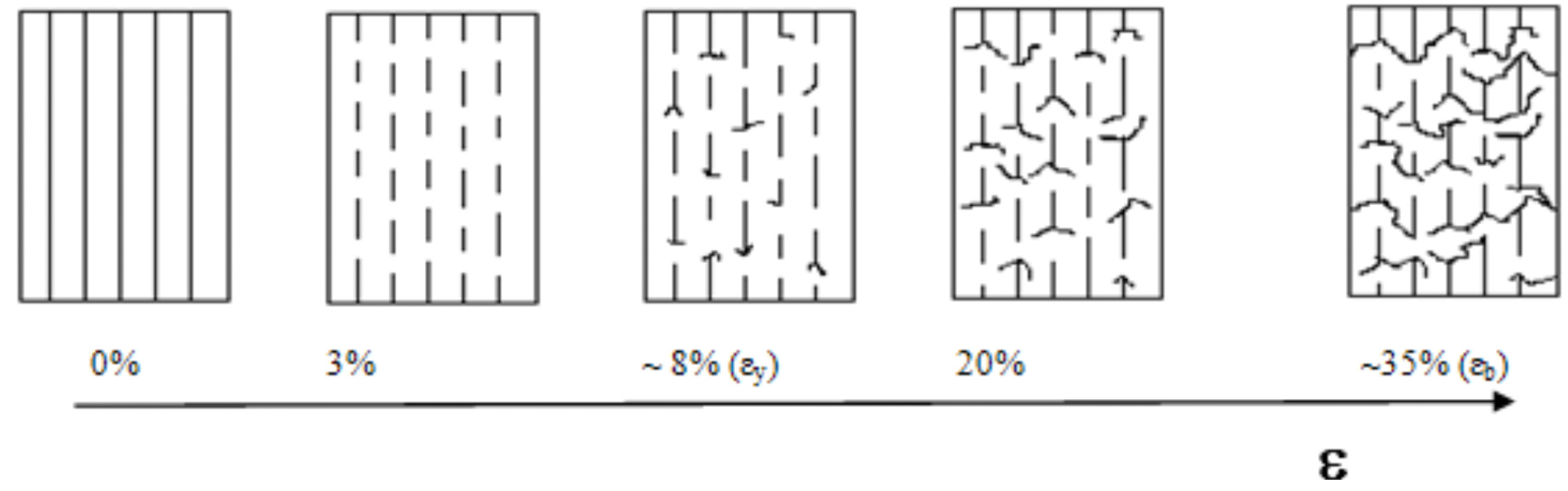
Figure 19), according to the main break strain of GF. However, the strain (at which strength is achieved) increases up to 7%, and can be attributed to a greater matrix/fiber adhesion. As was demonstrated above, the copolymerized fiber breaks into much smaller segments than samples containing untreated fibers. Then, although the first fracture occurs at the same strain for both treated and untreated fibers, the treated ones keep breaking as the deformation increases over this strain. This is due to the more effective matrix/fiber load transfer that requires greater energy consumption to fracture. In composites with polymerized fibers, GF/PP interface essentially resists by chemical anchorage. Once the fibers fracture, composites do not break immediately. This fact can be assigned to the greater stability of the glass-PP interface as compared to PP-PP entanglement resistance.
The composite initially contains continuous and aligned fibers, but once the first fiber fracture occurs, it gradually converts into a short fiber composite. Here the fiber fragments remain aligned with the load direction and well attached to the matrix. As the strain increases, each fiber fragment bears a growing stress up to its rupture limit. Significantly, the rate at which the load is transferred to the fibers decreases as voids begin to form and grow at the fiber tips.
Figure 20 outlines this mechanism.
It is clear that fibers always break at their fracture limit, however the mechanism for crack propagation, and consequently the composite mechanical strength, will be quite dependent on the fiber/matrix adhesion. When adhesion is low (composites with untreated fiber) the crack propagates mainly by fiber debonding. It results in fewer and relatively longer fiber segments. Instead, when polymerized (treated) fibers are used, cracks spread radially, preventing the fiber pullout. In this case, the propagating crack finds a greater amount of bonded fiber fragments in its way; so more energy is needed to break (or evade) them and allow the crack to grow. Therefore, when a large amount of short and well-adhered fiber segments are present, a considerable barrier to crack propagation is set, essentially by increments of the fissure path tortuosity. The proposed mechanism justifies the increment in elongation at break (more than twice for polymerized fiber samples as compared to PP/GF/PP and PP/MAO-GF/PP), as is shown in
Figure 20. Also, the toughness, calculated as the composite energy to break from the area under the stress-deformation curve, resulted three times greater for the treated fiber samples (see
Table 7). Furthermore, the elongation at yield (related to the energy required to generate cracks) indicated that PP/COP-GF/PP needed 5% more elongation than composites with non-copolymerized fibers to initiate cracks.
This evidence further validates the proposed mechanism. Other authors found similar behaviors, but attributed it to the ability of coupling agents to change the matrix nature, which could act as impediment of crack propagation. These authors did not report an increase in the strain at which the maximum stress occurs, but proposed just an increase of matrix ductility by the coupling agent use as the main cause of improvement [
54,
55,
56].
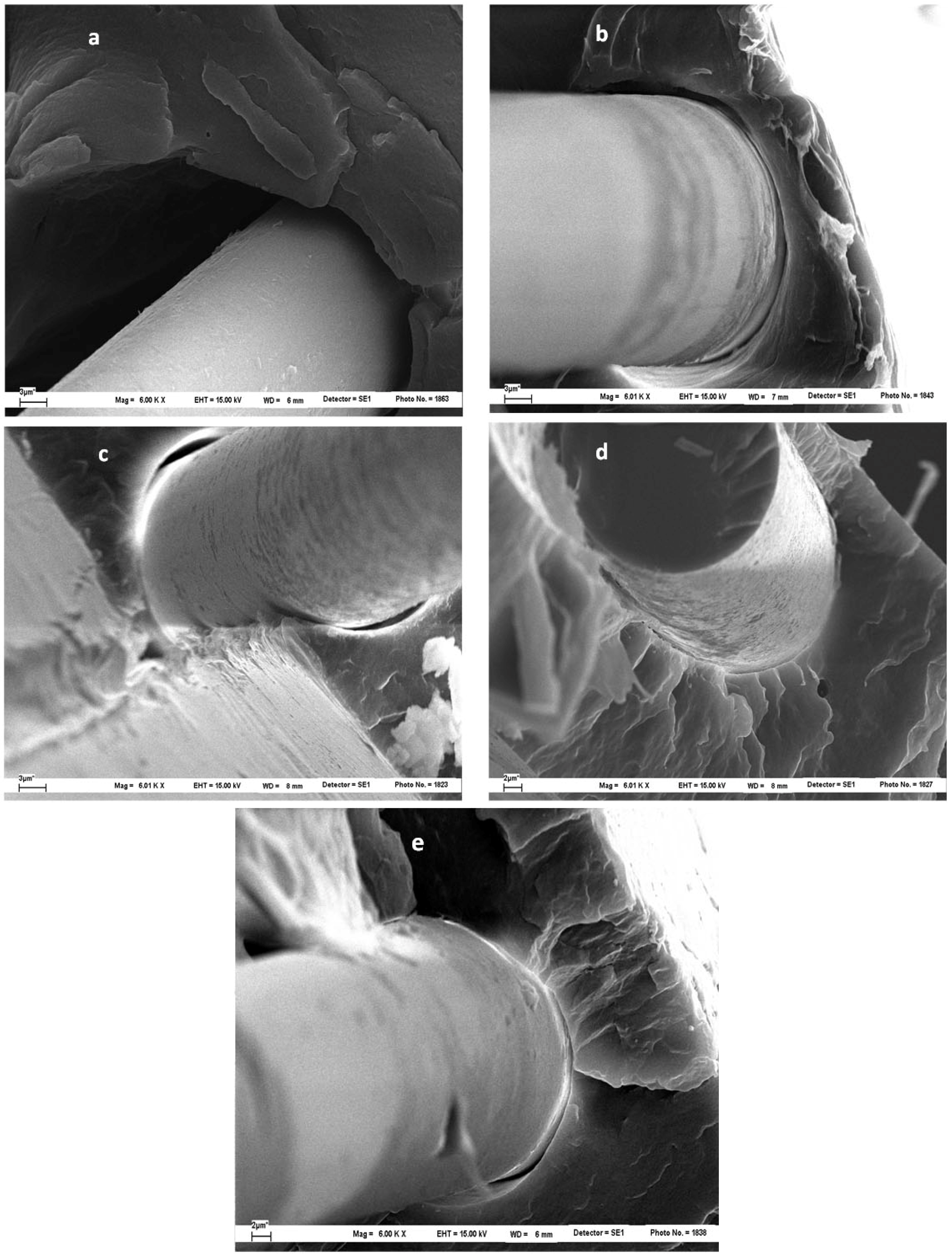
In order to corroborate the proposed fracture mechanism, and the adhesion enhancement, SEM characterization of specimens after tensile tests was carried out.
Figure 21 shows the fractured sample surfaces of PP/GF/PP, PP/MAO-GF/PP and PP/COP-GF/PP. It is clear from
Figure 21a, b that untreated and MAO treated composites breaks mainly by fiber debonding, hence PP remains unchanged. On the other hand, in PP/COP-GF/PP samples, the PP presents a lot of holes evenly distributed throughout the sample, indicating that fibers pull the matrix before composite fracture (
Figure 21c). This is consistent with the greater resistance of chemical anchorage relative to PP molecular entanglements. Moreover, the less regular overall fracture is consistent with a greater resistance to breakage. Also, PP patches remain adhered to the fiber surface after the composite fracture (see
Figure 22). The observed behavior agrees with the above-proposed model. Here the interphase presents a micro-damage mode of debonding accompanied by matrix cracks and promotes a stronger adhesion between matrix and fibers. The result is better resistance to development of damage and thus higher normal tensile stress. The interface debonding can reduce the effect of the matrix crack. The breakage (instead of debonding) of several small fiber bundles triggers larger matrix cracks, causing their propagation along the longitudinal direction.
Table 7 also shows the tensile modulus from non-polymerized and polymerized samples. As it is expected, no appreciable variation is observed since this is a zero strain property and neither fiber nor matrix modulus changed after treatment.
From these results it is clear that the in-situ polymerization technique improves the compatibility between phases allowing for more ductile composite materials without affecting their rigidity.
Figure 20.
Scheme proposed to explain the stress-strain behavior of PP/COP-GF/PP composites.
Figure 20.
Scheme proposed to explain the stress-strain behavior of PP/COP-GF/PP composites.
Figure 21.
SEM micrograph of composites after tensile break with different magnifications (a) PP/GF/PP at 53× and 270×; (b) PP/MAO-GF/PP at 53× and 270×, and (c) PP/COP-GF/PP at 53×.
Figure 21.
SEM micrograph of composites after tensile break with different magnifications (a) PP/GF/PP at 53× and 270×; (b) PP/MAO-GF/PP at 53× and 270×, and (c) PP/COP-GF/PP at 53×.
Figure 22.
SEM micrographs of PP/COP-GF/PP at: (a) 2,000× and (b) 4,000×.
Figure 22.
SEM micrographs of PP/COP-GF/PP at: (a) 2,000× and (b) 4,000×.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





















