
The Effect of Colloidal Nanoparticles on Phase Separation of Block and Heteroarm Star Copolymers Confined between Polymer Brushes

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
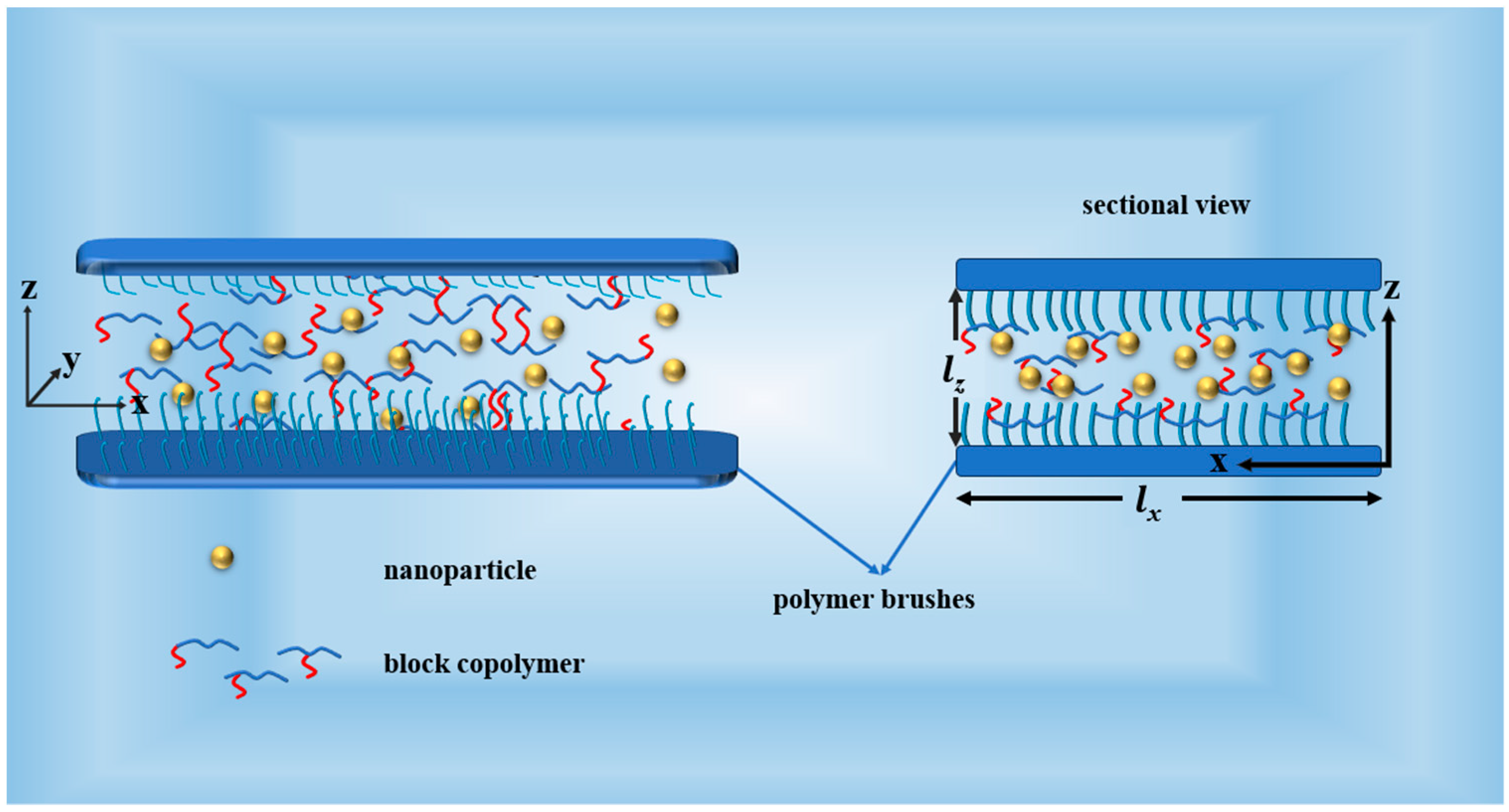
2. Model and Method
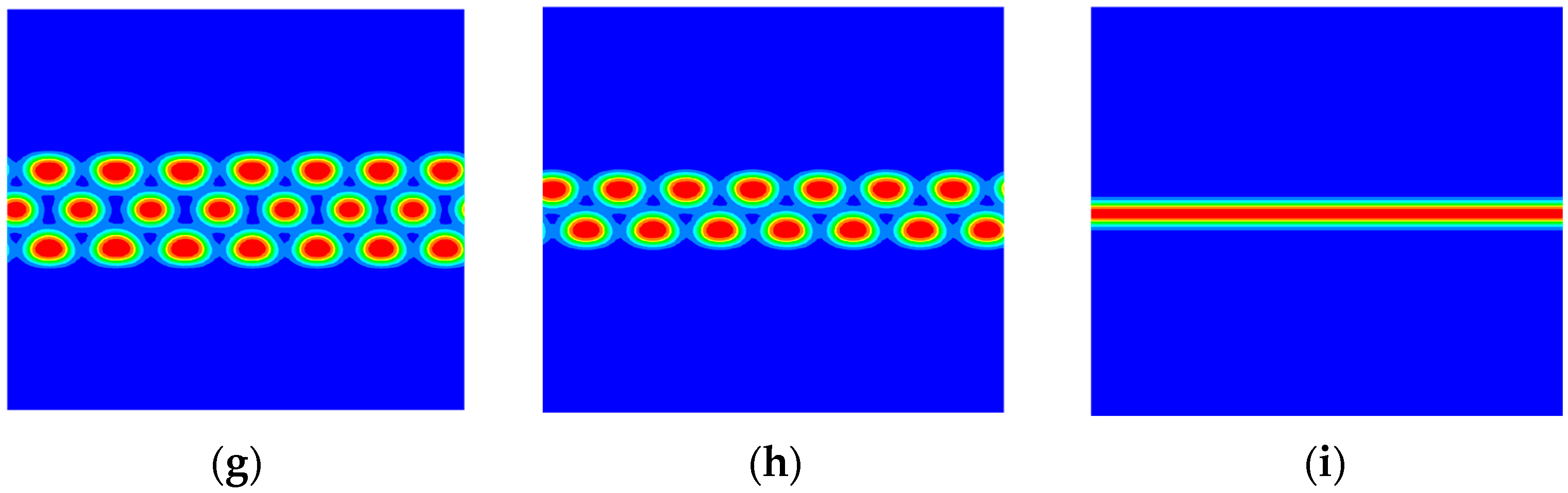
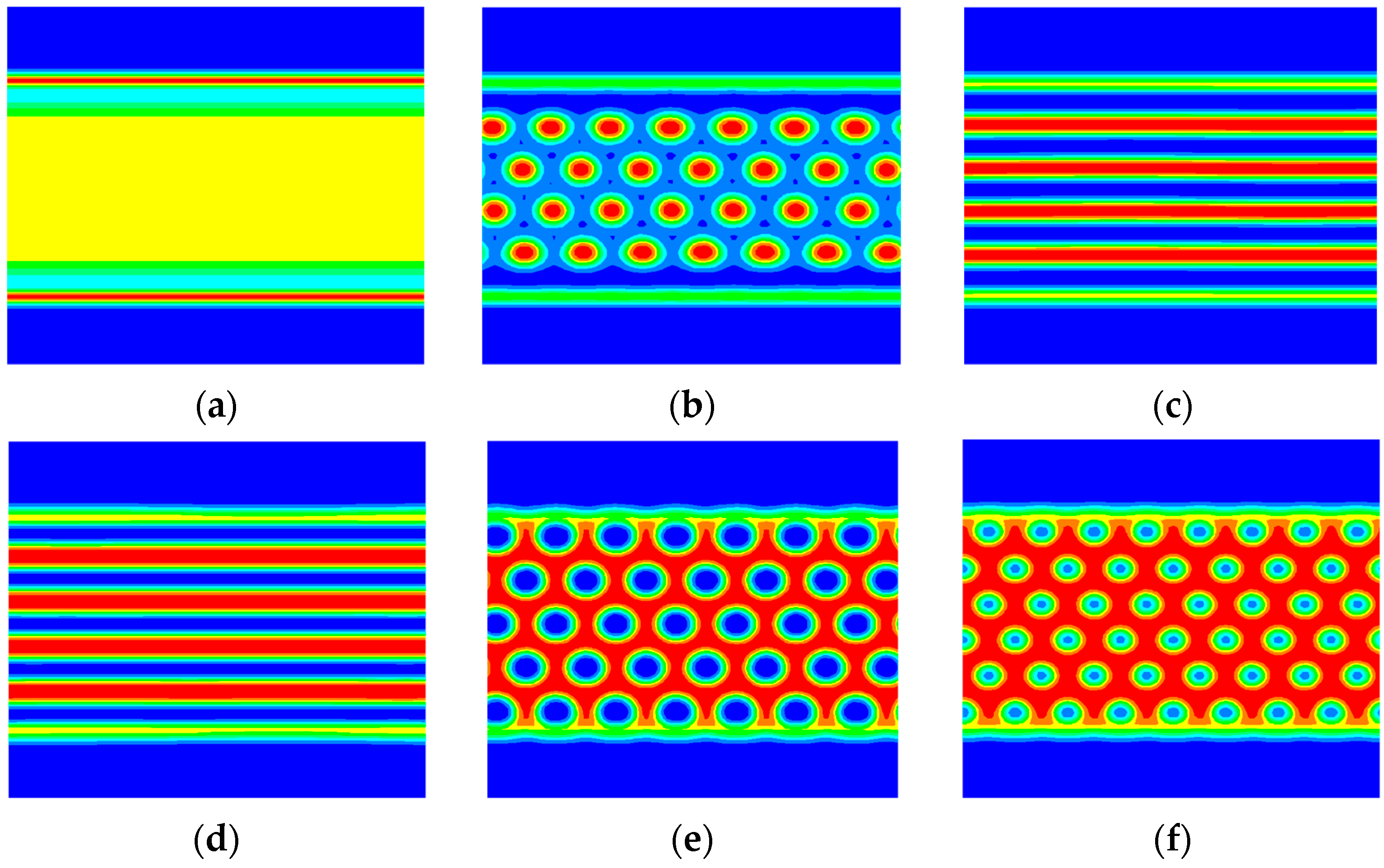
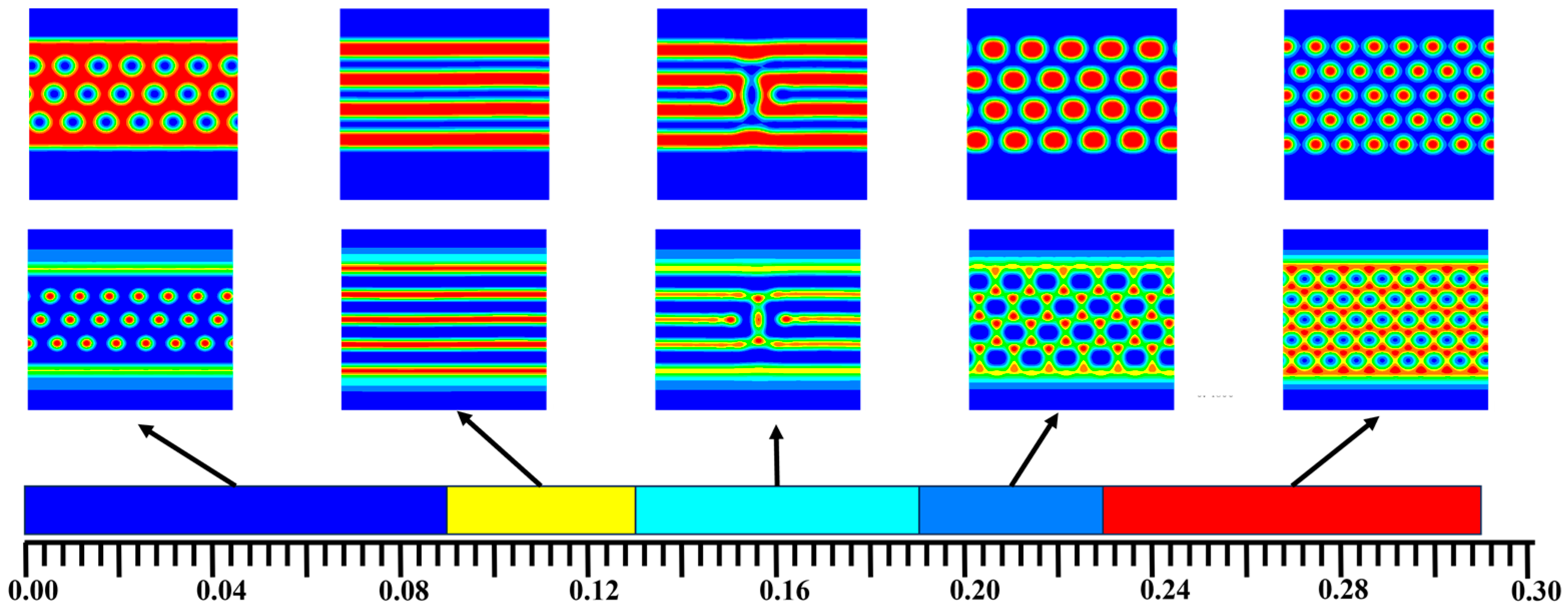
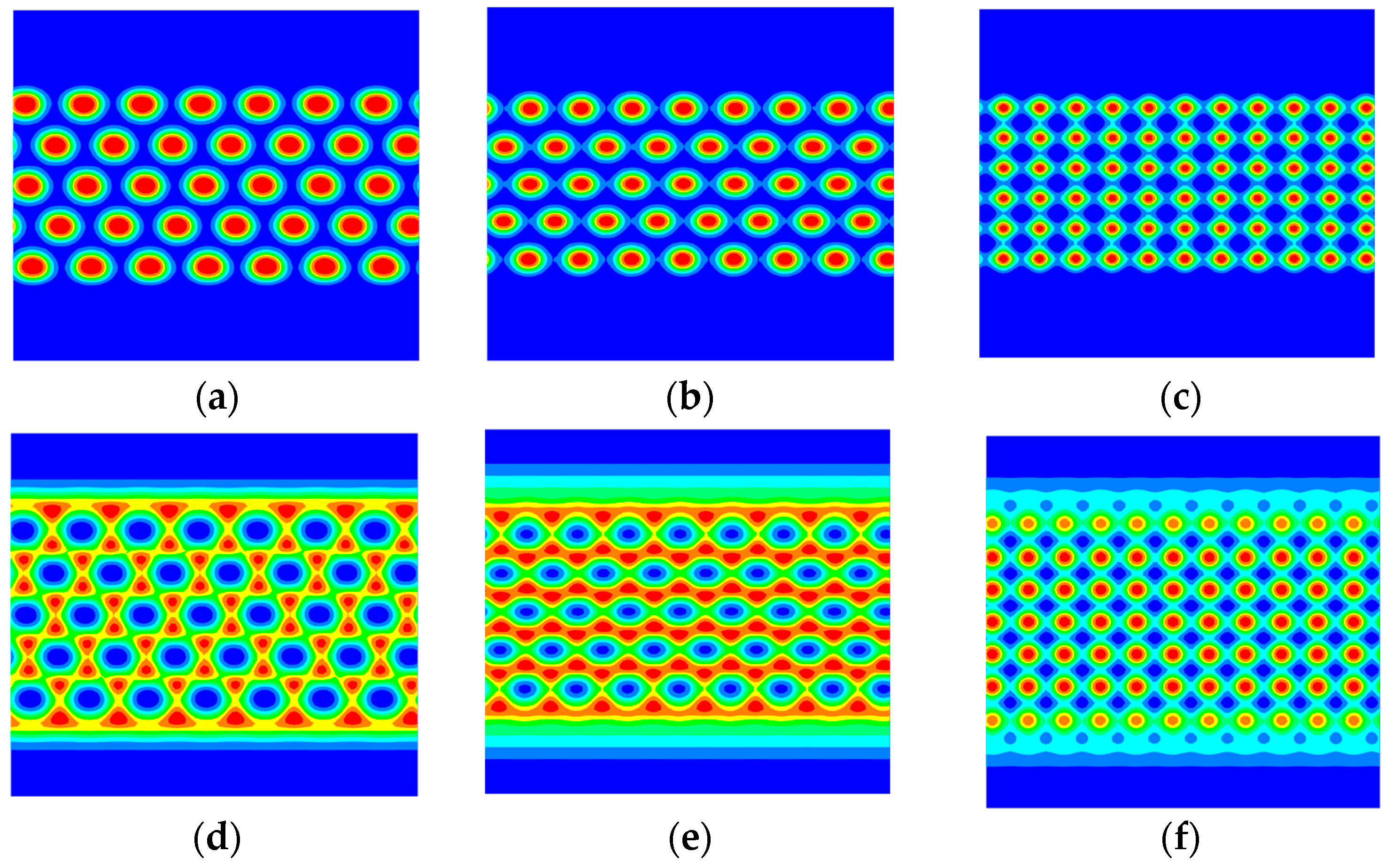
3. Numerical Results and Discussions
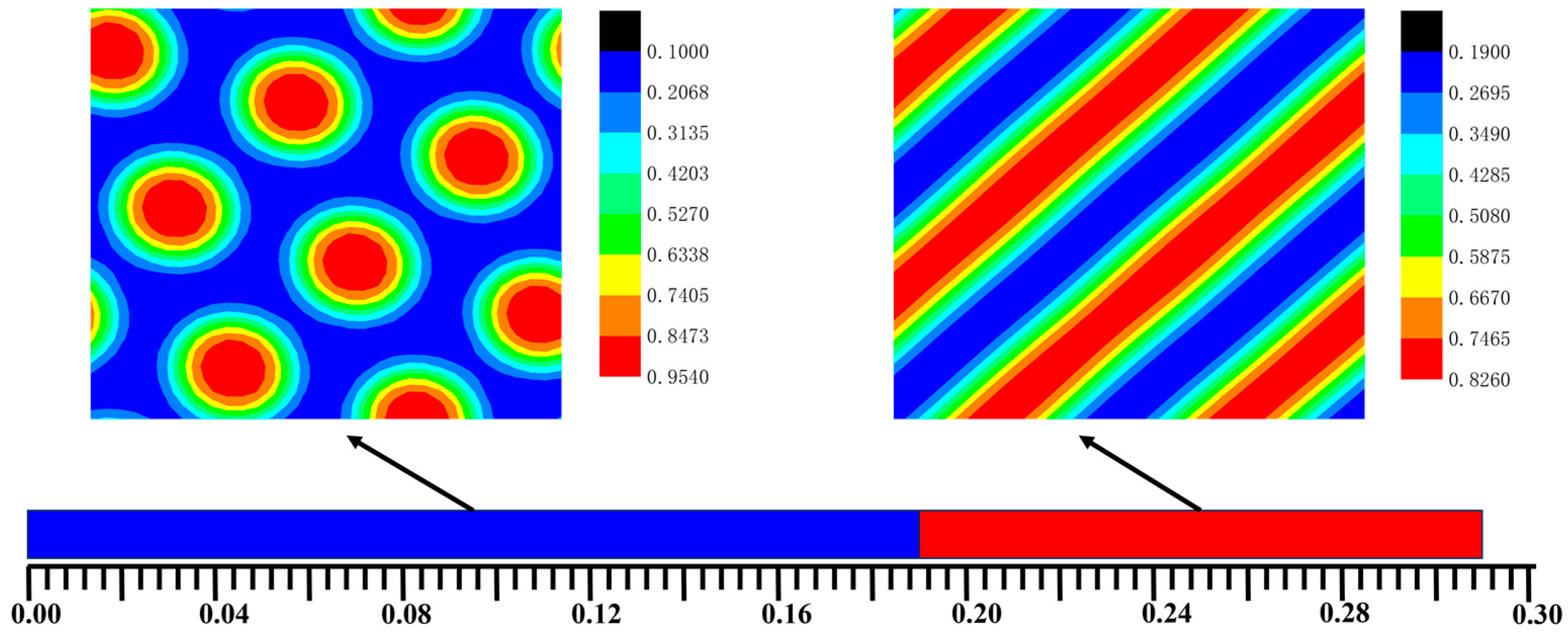
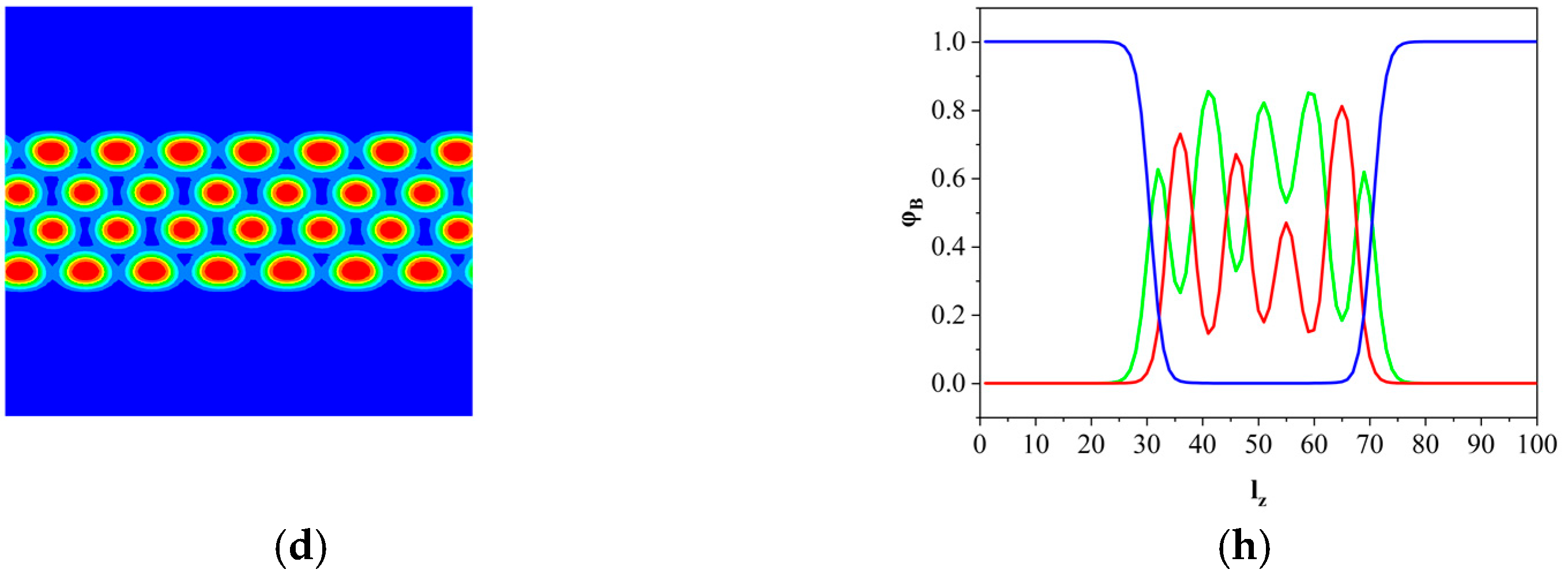
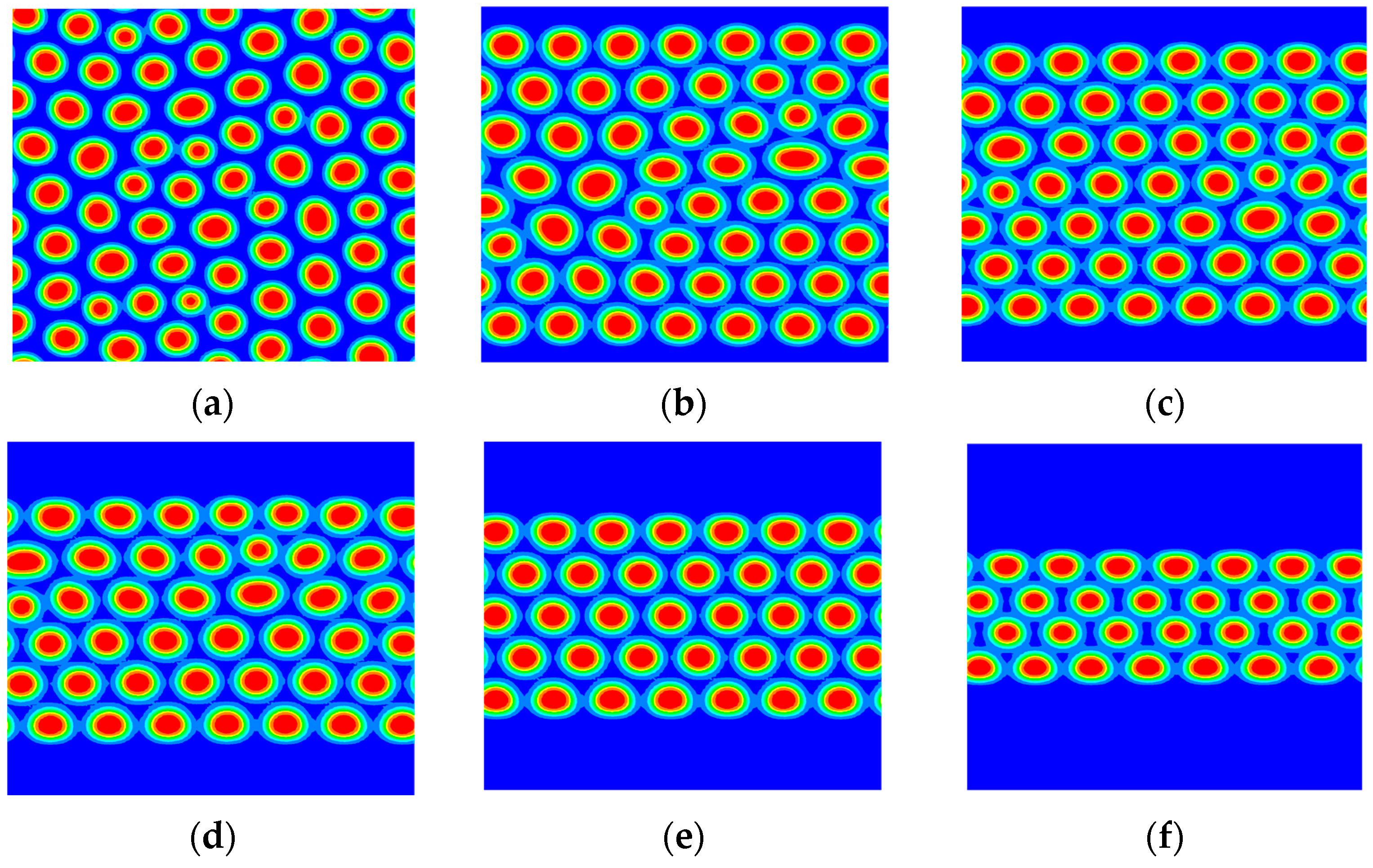
3.1. The Phase Separation of the AB Diblock Copolymers, A1A2B, and A2B Heteroarm Star Copolymers
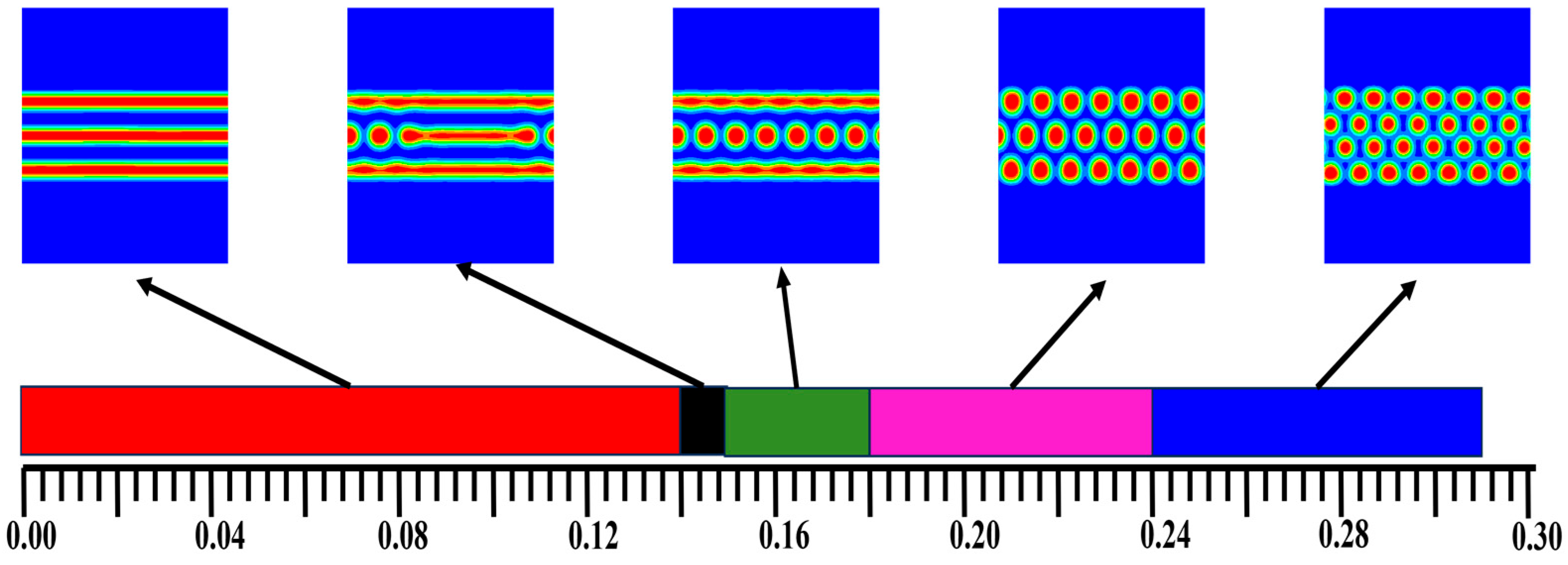
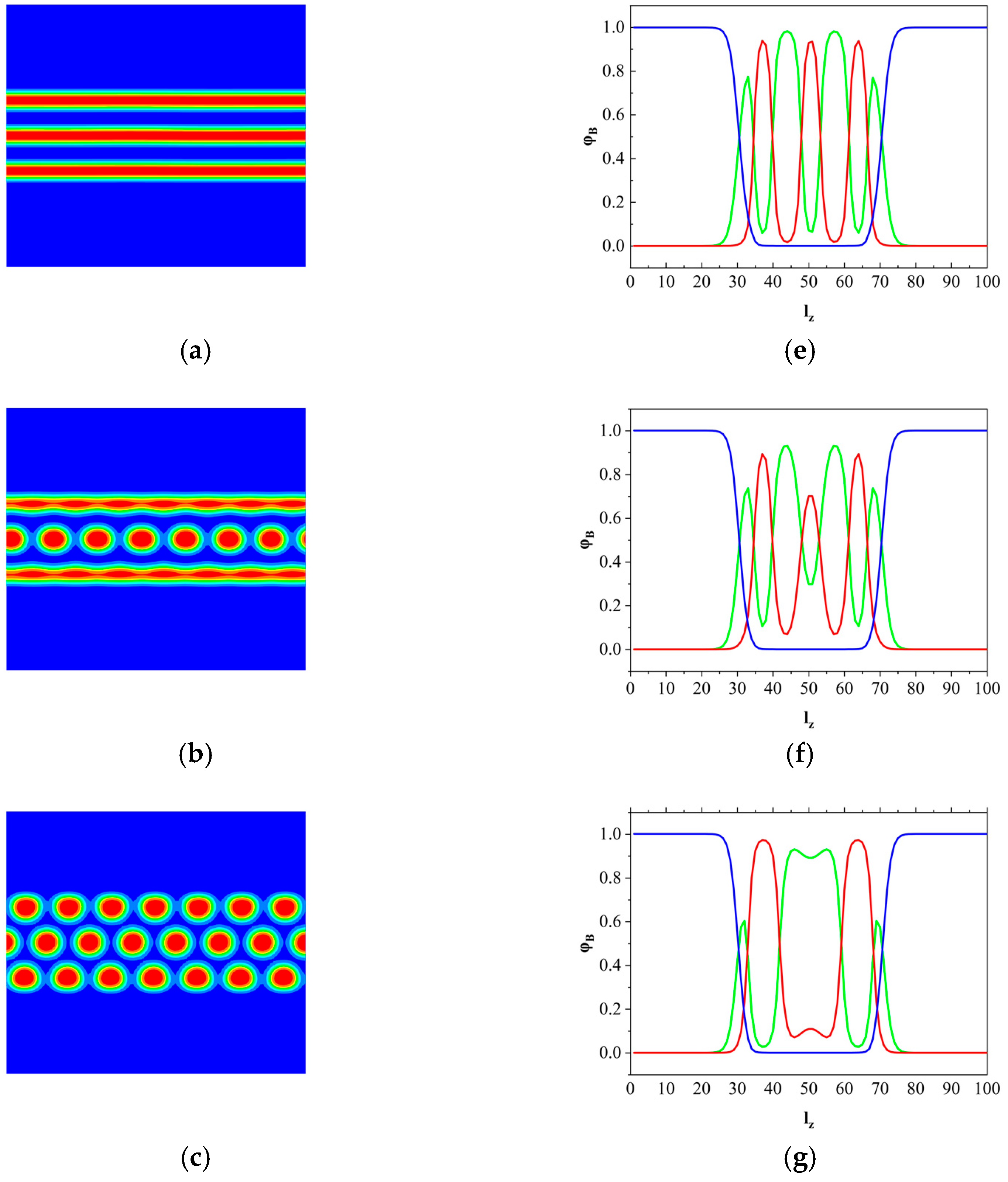
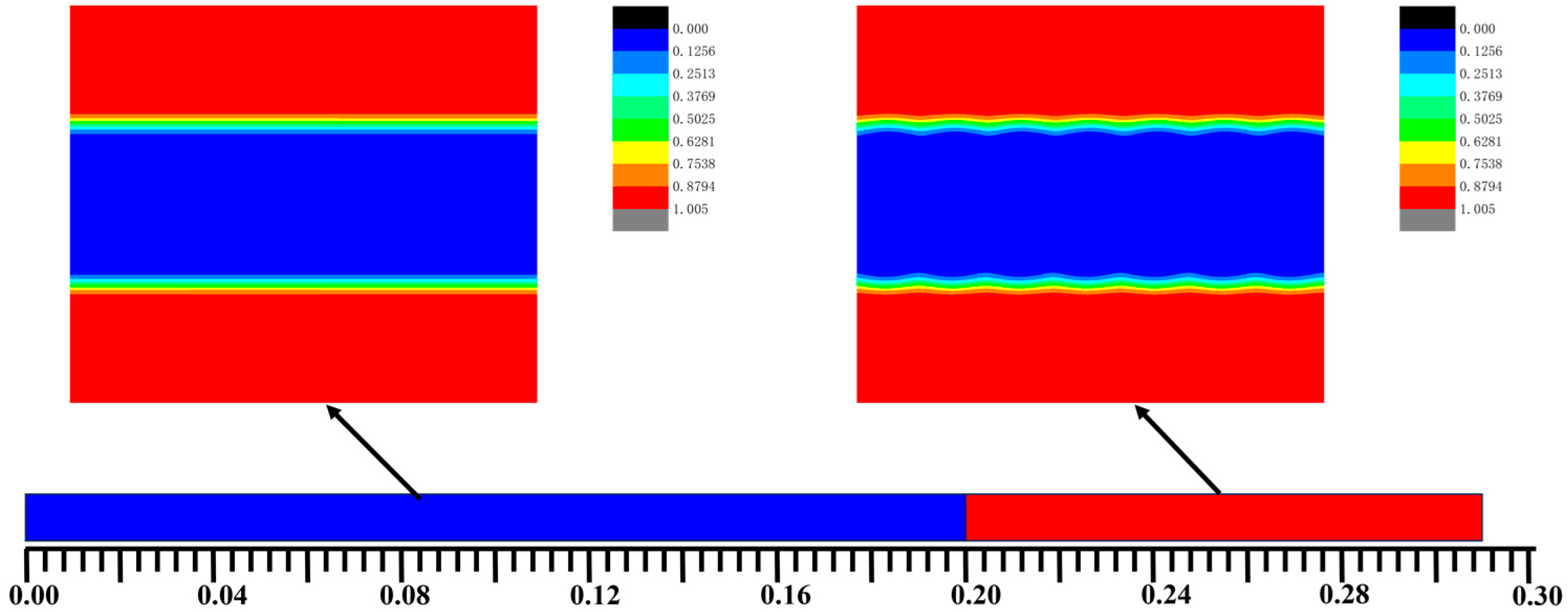
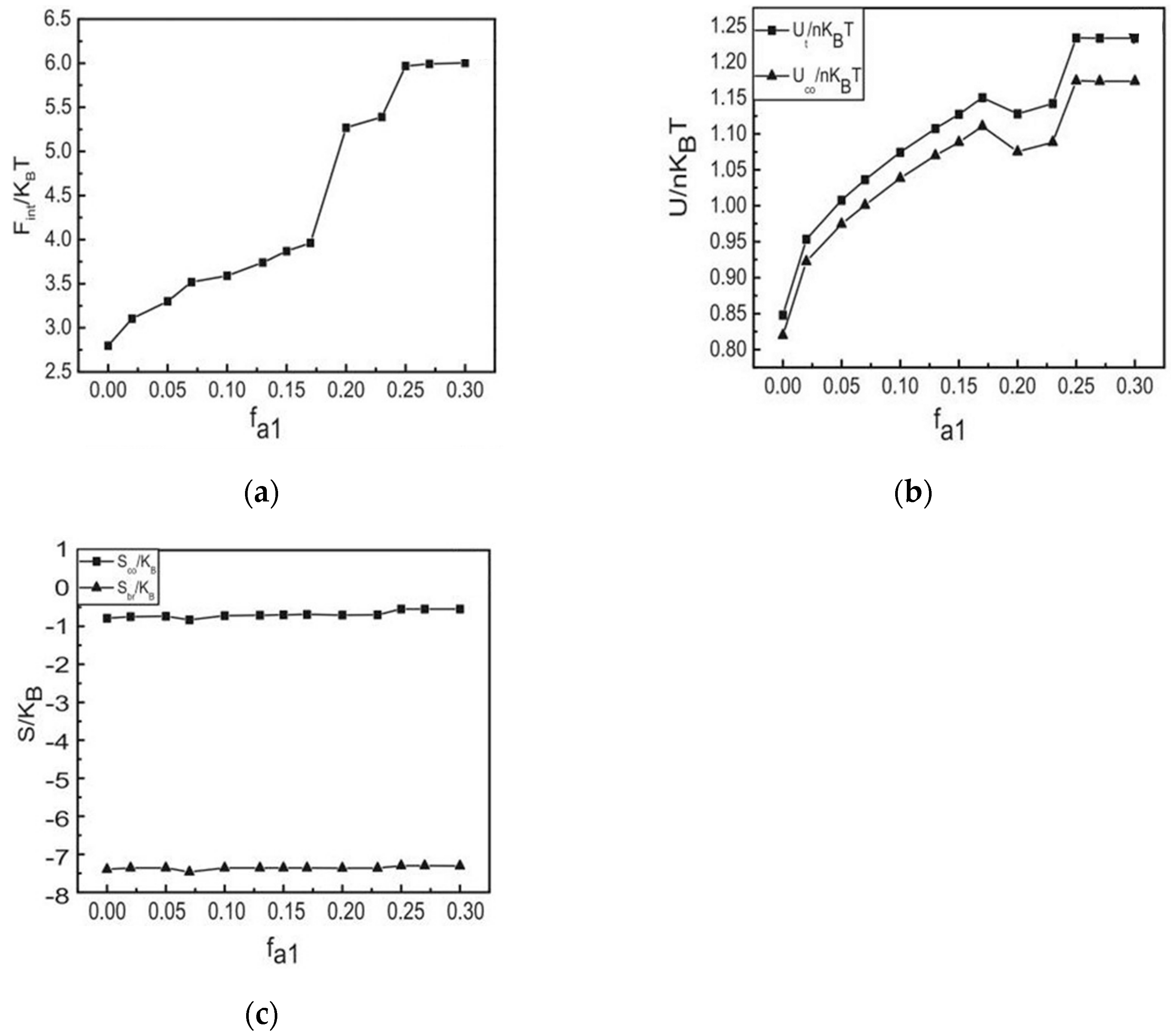
3.2. The Effect of Colloidal Nanoparticle Dispersion on Phase Separation of Copolymers
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Caseri, W. Nanocomposites of polymers and inorganic particles: Preparation, structure and properties. Mater. Sci. Technol. 2006, 22, 807–817. [Google Scholar] [CrossRef]
- Yoo, M.; Kim, S.; Jang, S.G.; Choi, S.-H.; Yang, H.; Kramer, E.J.; Lee, W.B.; Kim, B.J.; Bang, J. Controlling the orientation of block copolymer thin films using thermally-stable gold nanoparticles with tuned surface chemistry. Macromolecules 2011, 44, 9356–9365. [Google Scholar] [CrossRef]
- Horechyy, A.; Nandan, B.; Zafeiropoulos, N.E.; Jehnichen, D.; Göbel, M.; Stamm, M.; Pospiech, D. Nanoparticle directed domain orientation in thin films of asymmetric block copolymers. Colloid Polym. Sci. 2014, 292, 2249–2260. [Google Scholar] [CrossRef]
- Paras; Yadav, K.; Kumar, P.; Teja, D.R.; Chakraborty, S.; Chakraborty, M.; Mohapatra, S.S.; Sahoo, A.; Chou, M.M.C.; Liang, C.-T.; et al. A Review on Low-Dimensional Nanomaterials: Nanofabrication, Characterization and Applications. Nanomaterials 2022, 13, 160. [Google Scholar] [CrossRef] [PubMed]
- Hoheisel, T.N.; Hur, K.; Wiesner, U.B. Block copolymer-nanoparticle hybrid self-assembly. Prog. Polym. Sci. 2015, 40, 3–32. [Google Scholar] [CrossRef]
- Horvat, A.; Lyakhova, K.; Sevink, G.; Zvelindovsky, A.; Magerle, R. Phase behavior in thin films of cylinder-forming ABA block copolymers: Mesoscale modeling. J. Chem. Phys. 2004, 120, 1117–1126. [Google Scholar] [CrossRef] [PubMed]
- Shin, K.; Xiang, H.; Moon, S.I.; Kim, T.; McCarthy, T.J.; Russell, T.P. Curving and frustrating flatland. Science 2004, 306, 76. [Google Scholar] [CrossRef]
- He, X.; Liang, H.; Song, M.; Pan, C. Possibility of Design of Nanodevices by Confined Macromolecular Self-Assembly. Macromol. Theory Simul. 2002, 11, 379–382. [Google Scholar] [CrossRef]
- Feng, J.; Liu, H.; Hu, Y. Mesophase separation of diblock copolymer confined in a cylindrical tube studied by dissipative particle dynamics. Macromol. Theory Simul. 2006, 15, 674–685. [Google Scholar] [CrossRef]
- Chen, D.; Zhao, W.; Wei, D.; Russell, T.P. Dewetting on curved interfaces: A simple route to polymer nanostructures. Macromolecules 2011, 44, 8020–8027. [Google Scholar] [CrossRef]
- Yu, B.; Jin, Q.; Ding, D.; Li, B.; Shi, A.-C. Confinement-induced morphologies of cylinder-forming asymmetric diblock copolymers. Macromolecules 2008, 41, 4042–4054. [Google Scholar] [CrossRef]
- Chen, P.; He, X.; Liang, H. Effect of surface field on the morphology of a symmetric diblock copolymer under cylindrical confinement. J. Chem. Phys. 2006, 124, 104906. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Zhang, L.; Zhao, X. Creasing to cratering instability in polymers under ultrahigh electric fields. Phys. Rev. Lett. 2011, 106, 118301. [Google Scholar] [CrossRef] [PubMed]
- Morozov, A.; Zvelindovsky, A.; Fraaije, J. Orientational phase transitions in the hexagonal phase of a diblock copolymer melt under shear flow. Phys. Rev. E 2000, 61, 4125. [Google Scholar] [CrossRef] [PubMed]
- Travasso, R.D.; Kuksenok, O.; Balazs, A.C. Exploiting photoinduced reactions in polymer blends to create hierarchically ordered, defect-free materials. Langmuir 2006, 22, 2620–2628. [Google Scholar] [CrossRef] [PubMed]
- Tsori, Y. Colloquium: Phase transitions in polymers and liquids in electric fields. Rev. Mod. Phys. 2009, 81, 1471. [Google Scholar] [CrossRef]
- Sun, M.; Zhang, J.-j.; Wang, B.; Wu, H.-s.; Pan, J. Domain patterns in a diblock copolymer-diblock copolymer mixture with oscillatory particles. Phys. Rev. E 2011, 84, 011802. [Google Scholar] [CrossRef] [PubMed]
- Xiang, H.; Shin, K.; Kim, T.; Moon, S.I.; McCarthy, T.J.; Russell, T.P. Block copolymers under cylindrical confinement. Macromolecules 2004, 37, 5660–5664. [Google Scholar] [CrossRef]
- Zhao, Y.; Fang, J. Positioning and alignment of lipid tubules on patterned Au substrates. Langmuir 2006, 22, 1891–1895. [Google Scholar] [CrossRef]
- Chen, H.; Chen, X.; Ye, Z.; Liu, H.; Hu, Y. Competitive adsorption and assembly of block copolymer blends on nanopatterned surfaces. Langmuir 2010, 26, 6663–6668. [Google Scholar] [CrossRef]
- Ye, X.; Edwards, B.J.; Khomami, B. Elucidating the formation of block copolymer nanostructures on patterned surfaces: A self-consistent field theory study. Macromolecules 2010, 43, 9594–9597. [Google Scholar] [CrossRef]
- Zhang, C.; Chen, Y.; Deng, Z.; Shi, M. Role of rough surface topography on gas slip flow in microchannels. Phys. Rev. E 2012, 86, 016319. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.A.; McCarthy, T.J. Polymer surface modification: Topography effects leading to extreme wettability behavior. Macromolecules 2007, 40, 3965–3969. [Google Scholar] [CrossRef]
- Mazloomi, A.; Moosavi, A. Thin liquid film flow over substrates with two topographical features. Phys. Rev. E 2013, 87, 022409. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.-X.; Zhang, J.-J.; Wang, B.-F.; Wu, H.-S.; Sun, M.-N. A Diblock-Diblock Copolymer Mixture under Parallel Wall Confinement. Chin. Phys. Lett. 2013, 30, 046401. [Google Scholar] [CrossRef]
- Hu, T.; Ren, Y.; Zhang, L.; Li, W. Impact of Architecture of Symmetric Block Copolymers on the Stability of a Dislocation Defect. Macromolecules 2021, 54, 773–782. [Google Scholar] [CrossRef]
- Platonova, O.; Bronstein, L.; Solodovnikov, S.; Yanovskaya, I.; Obolonkova, E.; Valetsky, P.; Wenz, E.; Antonietti, M. Cobalt nanoparticles in block copolymer micelles: Preparation and properties. Colloid Polym. Sci. 1997, 275, 426–431. [Google Scholar] [CrossRef]
- Ren, C.-l.; Ma, Y.-q. Reentrant ordering transition of asymmetric copolymer solution film confined between polymer-grafted surfaces. Phys. Rev. E 2005, 72, 051804. [Google Scholar] [CrossRef]
- Kim, Y.; Pyun, J.; Fréchet, J.M.; Hawker, C.J.; Frank, C.W. The dramatic effect of architecture on the self-assembly of block copolymers at interfaces. Langmuir 2005, 21, 10444–10458. [Google Scholar] [CrossRef]
- Li, W.; Müller, M. Directed self-assembly of block copolymers by chemical or topographical guiding patterns: Optimizing molecular architecture, thin-film properties, and kinetics. Prog. Polym. Sci. 2016, 54, 47–75. [Google Scholar] [CrossRef]
- Matsen, M.W. Effect of architecture on the phase behavior of AB-type block copolymer melts. Macromolecules 2012, 45, 2161–2165. [Google Scholar] [CrossRef]
- Yun, J.; Faust, R.; Szilágyi, L.S.; Kéki, S.; Zsuga, M. Effect of Architecture on the Micellar Properties of Amphiphilic Block Copolymers: Comparison of AB Linear Diblock, AAB, and A2B Heteroarm Star Block Copolymers. Macromolecules 2003, 36, 1717–1723. [Google Scholar] [CrossRef]
- Drolet, F.; Fredrickson, G.H. Combinatorial screening of complex block copolymer assembly with self-consistent field theory. Phys. Rev. Lett. 1999, 83, 4317. [Google Scholar] [CrossRef]
- Drolet, F.; Fredrickson, G.H. Optimizing chain bridging in complex block copolymers. Macromolecules 2001, 34, 5317–5324. [Google Scholar] [CrossRef]
- Barbon, S.M.; Song, J.-A.; Chen, D.; Zhang, C.; Lequieu, J.; Delaney, K.T.; Anastasaki, A.; Rolland, M.; Fredrickson, G.H.; Bates, M.W. Architecture effects in complex spherical assemblies of (AB) n-type block copolymers. ACS Macro Lett. 2020, 9, 1745–1752. [Google Scholar] [CrossRef] [PubMed]
- Constantinou, A.P.; Wang, L.; Wang, S.; Georgiou, T.K. Thermoresponsive block copolymers of increasing architecture complexity: A review on structure–property relationships. Polym. Chem. 2023, 14, 223–247. [Google Scholar] [CrossRef]
- Lee, C.; Gido, S.P.; Pitsikalis, M.; Mays, J.W.; Tan, N.B.; Trevino, S.F.; Hadjichristidis, N. Asymmetric single graft block copolymers: Effect of molecular architecture on morphology. Macromolecules 1997, 30, 3732–3738. [Google Scholar] [CrossRef]
- Modica, K.J.; Martin, T.B.; Jayaraman, A. Effect of polymer architecture on the structure and interactions of polymer grafted particles: Theory and simulations. Macromolecules 2017, 50, 4854–4866. [Google Scholar] [CrossRef]
- Moinuddin, M.; Tripathy, M. Effect of Architecture and Topology on the Self-Assembly of Polymer-Grafted Nanoparticles. Macromolecules 2022, 55, 9312–9323. [Google Scholar] [CrossRef]
- Xu, M.; Ku, K.H.; Lee, Y.J.; Kim, T.; Shin, J.J.; Kim, E.J.; Choi, S.-H.; Yun, H.; Kim, B.J. Effect of polymer ligand conformation on the self-assembly of block copolymers and polymer-grafted nanoparticles within an evaporative emulsion. Macromolecules 2021, 54, 3084–3092. [Google Scholar] [CrossRef]
- Choi, J.; Hore, M.J.; Clarke, N.; Winey, K.I.; Composto, R.J. Nanoparticle brush architecture controls polymer diffusion in nanocomposites. Macromolecules 2014, 47, 2404–2410. [Google Scholar] [CrossRef]
- Che, J.; Park, K.; Grabowski, C.A.; Jawaid, A.; Kelley, J.; Koerner, H.; Vaia, R.A. Preparation of ordered monolayers of polymer grafted nanoparticles: Impact of architecture, concentration, and substrate surface energy. Macromolecules 2016, 49, 1834–1847. [Google Scholar] [CrossRef]
- Goel, V.; Pietrasik, J.; Poling-Skutvik, R.; Jackson, A.; Matyjaszewski, K.; Krishnamoorti, R. Structure of block copolymer grafted silica nanoparticles. Polymer 2018, 159, 138–145. [Google Scholar] [CrossRef]
- Sunday, D.F.; Dolejsi, M.; Chang, A.B.; Richter, L.J.; Li, R.; Kline, R.J.; Nealey, P.F.; Grubbs, R.H. Confinement and Processing Can Alter the Morphology and Periodicity of Bottlebrush Block Copolymers in Thin Films. ACS Nano 2020, 14, 17476–17486. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Yun, H.; Liao, K.; Kim, B. Entropy-Driven Assembly Behavior of Polymer-Grafted Nanoparticles in 3D Confined Block Copolymers. In Proceedings of the APS March Meeting Abstracts, Virtual, 15–19 March 2021; p. 63. [Google Scholar]
- Ali, I.; Kareem, F.; Rahim, S.; Perveen, S.; Ahmed, S.; Shah, M.R.; Malik, M.I. Architecture based selectivity of Amphiphilic block copolymers of poly (ethylene oxide) and poly (ε-caprolactone) for drug delivery. React. Funct. Polym. 2020, 150, 104553. [Google Scholar] [CrossRef]
- Chen, K.; Chen, C.-Y.; Chen, H.-L.; Komaki, R.; Kawakami, N.; Isono, T.; Satoh, T.; Hung, D.-Y.; Liu, Y.-L. Self-Assembly Behavior of Sugar-Based Block Copolymers in the Complex Phase Window Modulated by Molecular Architecture and Configuration. Macromolecules 2022, 56, 28–39. [Google Scholar] [CrossRef]
- Wang, M.; Han, L.; Zhu, Y.; Qi, R.; Tian, L.; He, F. Formation of Hierarchical Architectures with Dimensional and Morphological Control in the Self-Assembly of Conjugated Block Copolymers. Small Methods 2020, 4, 1900470. [Google Scholar] [CrossRef]
- Algarni, F.; Musteata, V.E.; Falca, G.; Chisca, S.; Hadjichristidis, N.; Nunes, S.P. Thermo-responsive membranes from blends of PVDF and PNIPAM-b-PVDF block copolymers with linear and star architectures. Macromolecules 2021, 54, 10235–10250. [Google Scholar] [CrossRef]
- Gupta, S.; Chokshi, P. Self-assembly of polymer grafted nanoparticles within spherically confined diblock copolymers. J. Phys. Chem. B 2020, 124, 11738–11749. [Google Scholar] [CrossRef]
- Willinger, M.; Reimhult, E. Thermoresponsive nanoparticles with cyclic-polymer-grafted shells are more stable than with linear-polymer-grafted shells: Effect of polymer topology, molecular weight, and core size. J. Phys. Chem. B 2021, 125, 7009–7023. [Google Scholar] [CrossRef]
- Schmidt, B.V.K.J.; Wang, C.X.; Kraemer, S.; Connal, L.A.; Klinger, D. Highly functional ellipsoidal block copolymer nanoparticles: A generalized approach to nanostructured chemical ordering in phase separated colloidal particles. Polym. Chem. 2018, 9, 1638–1649. [Google Scholar] [CrossRef]
- Alexandridis, P.; Tsianou, M. Block copolymer-directed metal nanoparticle morphogenesis and organization. Eur. Polym. J. 2011, 47, 569–583. [Google Scholar] [CrossRef]
- Guo, Y.; Harirchian-Saei, S.; Izumi, C.M.S.; Moffitt, M.G. Block Copolymer Mimetic Self-Assembly of Inorganic Nanoparticles. ACS Nano 2011, 5, 3309–3318. [Google Scholar] [CrossRef]












Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, M.; Chen, W.; Qin, L.; Xie, X.-M. The Effect of Colloidal Nanoparticles on Phase Separation of Block and Heteroarm Star Copolymers Confined between Polymer Brushes. Materials 2024, 17, 804. https://doi.org/10.3390/ma17040804
Sun M, Chen W, Qin L, Xie X-M. The Effect of Colloidal Nanoparticles on Phase Separation of Block and Heteroarm Star Copolymers Confined between Polymer Brushes. Materials. 2024; 17(4):804. https://doi.org/10.3390/ma17040804
Chicago/Turabian StyleSun, Minna, Wenyu Chen, Lei Qin, and Xu-Ming Xie. 2024. "The Effect of Colloidal Nanoparticles on Phase Separation of Block and Heteroarm Star Copolymers Confined between Polymer Brushes" Materials 17, no. 4: 804. https://doi.org/10.3390/ma17040804