

Corrosion Mitigation in Molten Salt Environments
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
- -
- The addition of a redox system;
- -
- The potentiostatic control of the material potential;
- -
- The addition of an amphoteric compound.
2. Experimental Methods
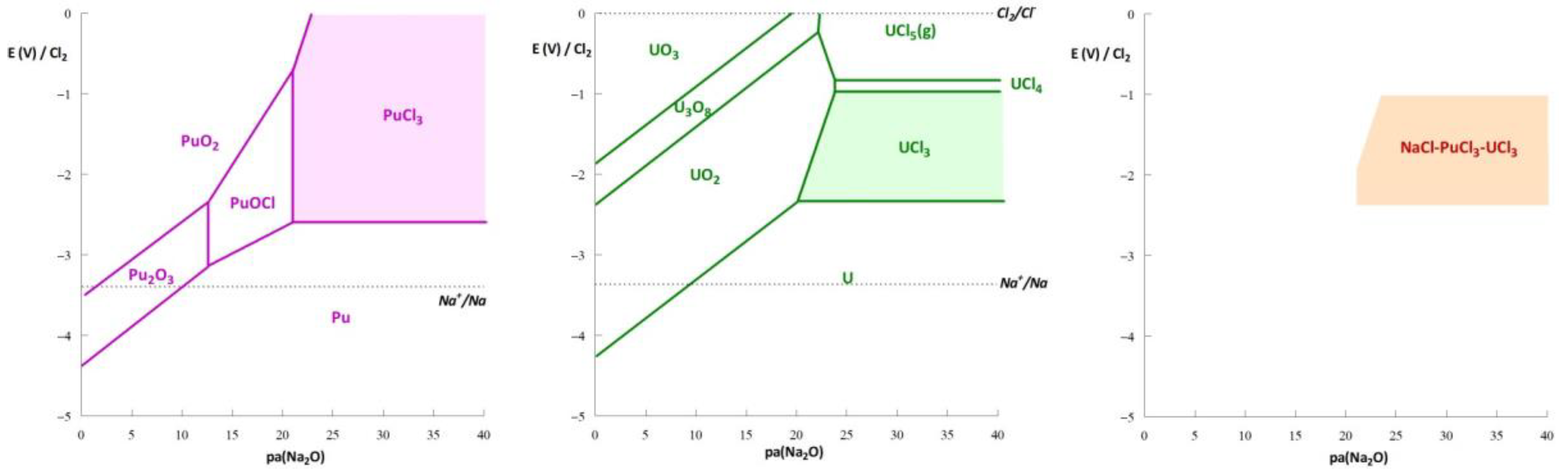
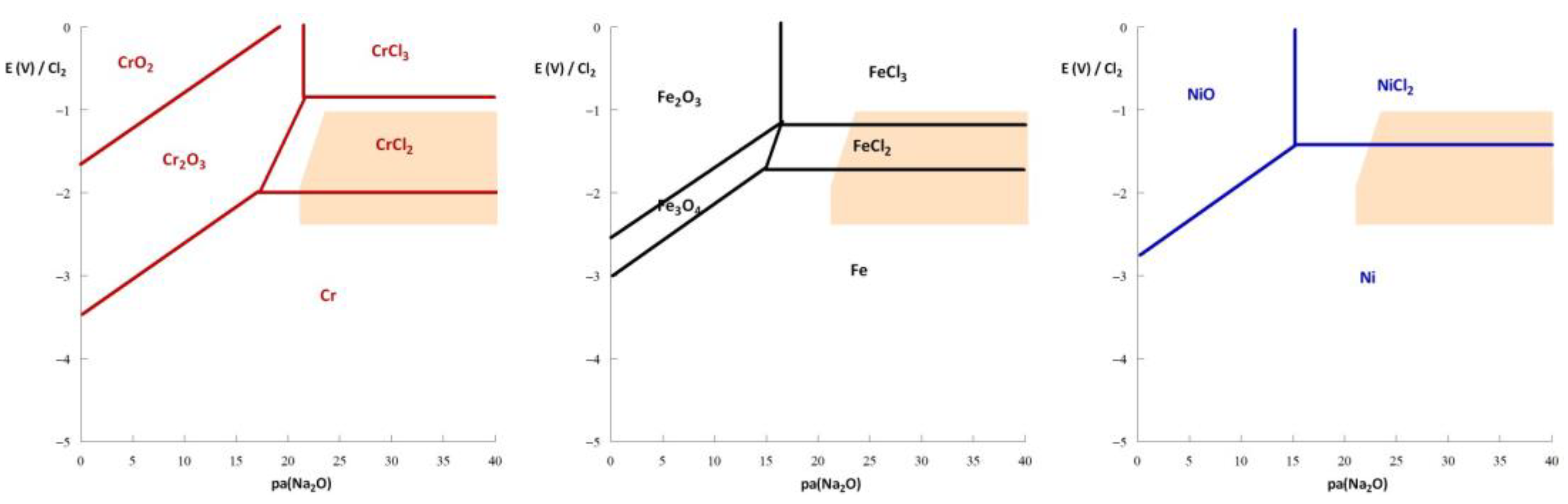
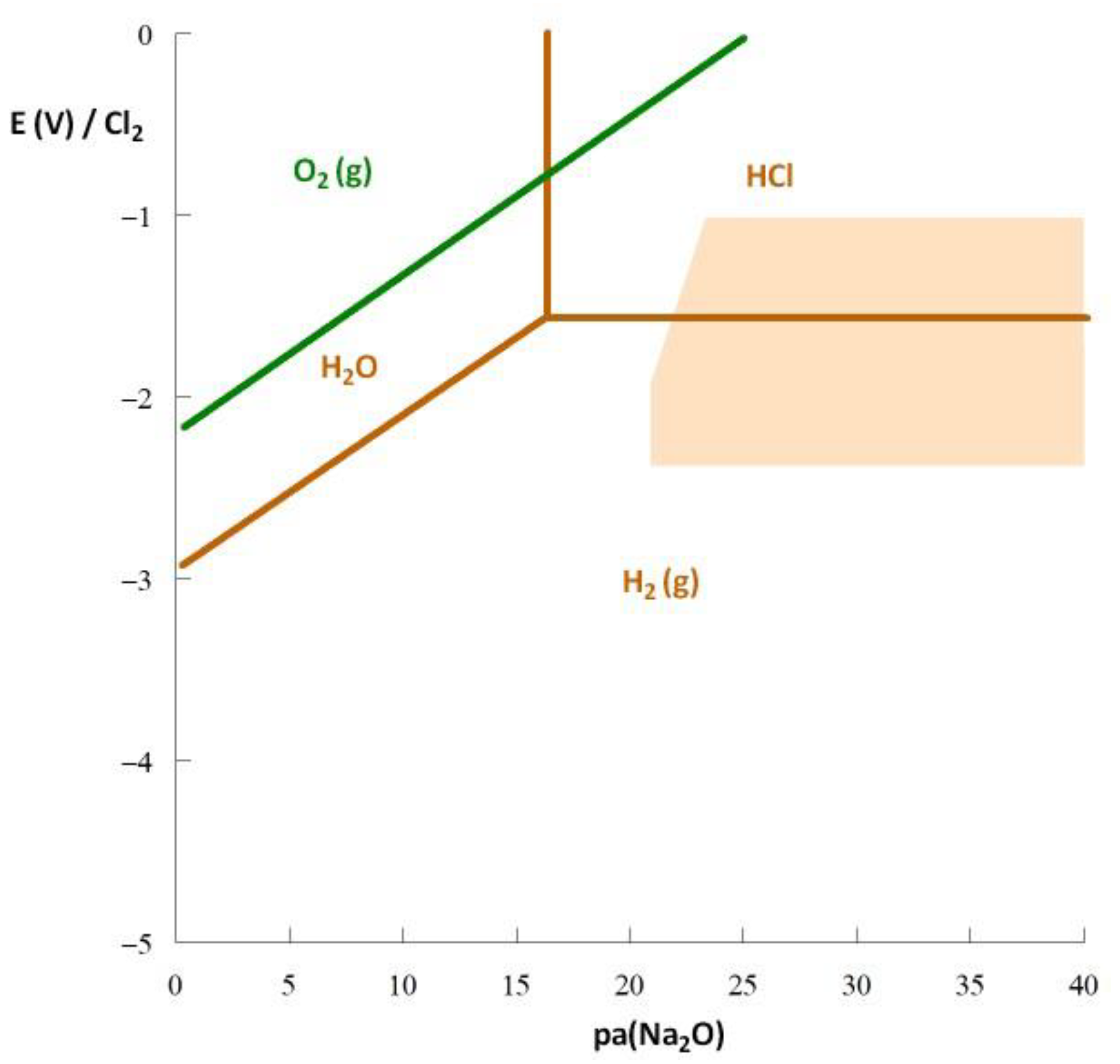
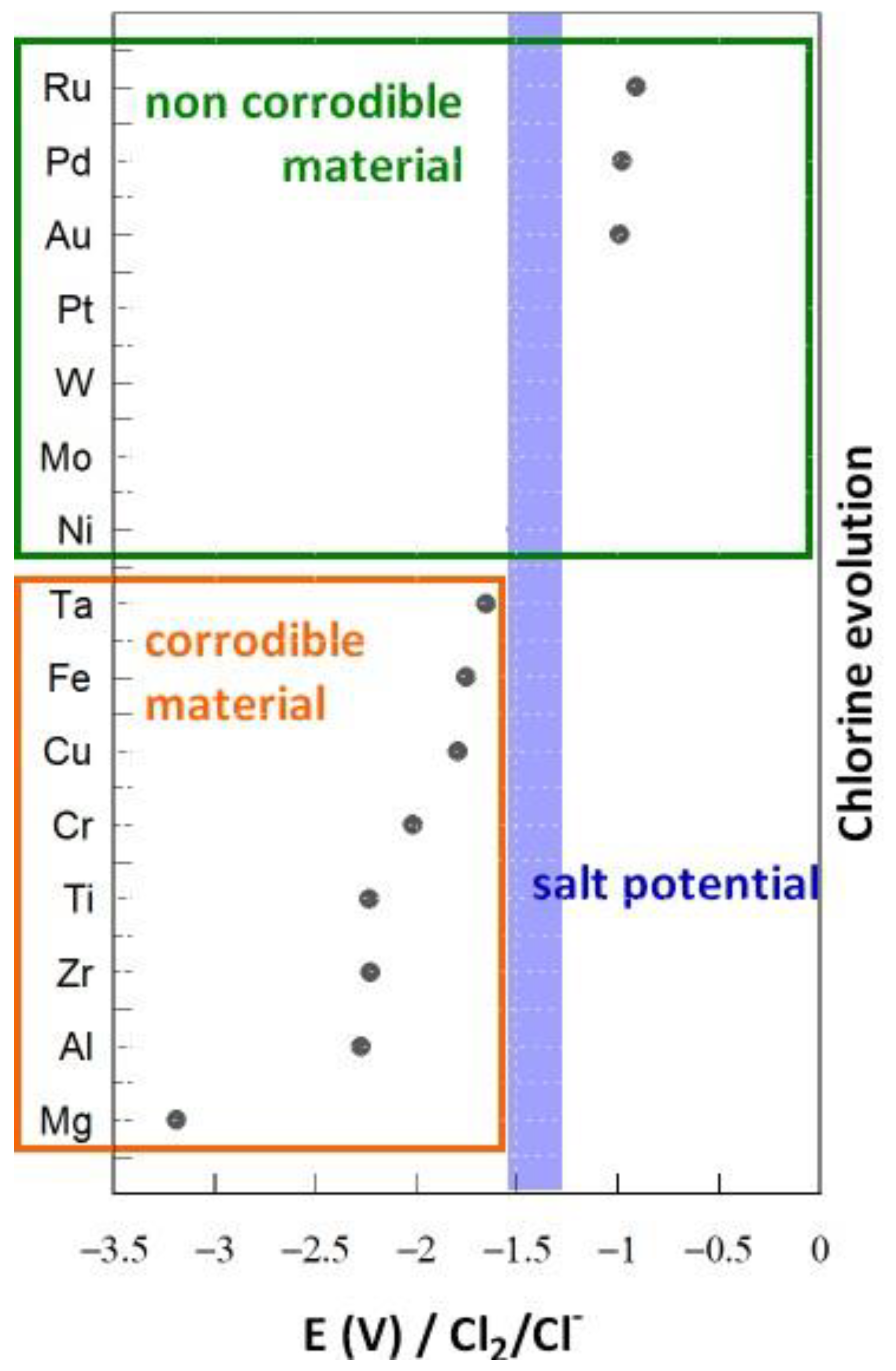
3. Thermodynamic Approach to Corrosion
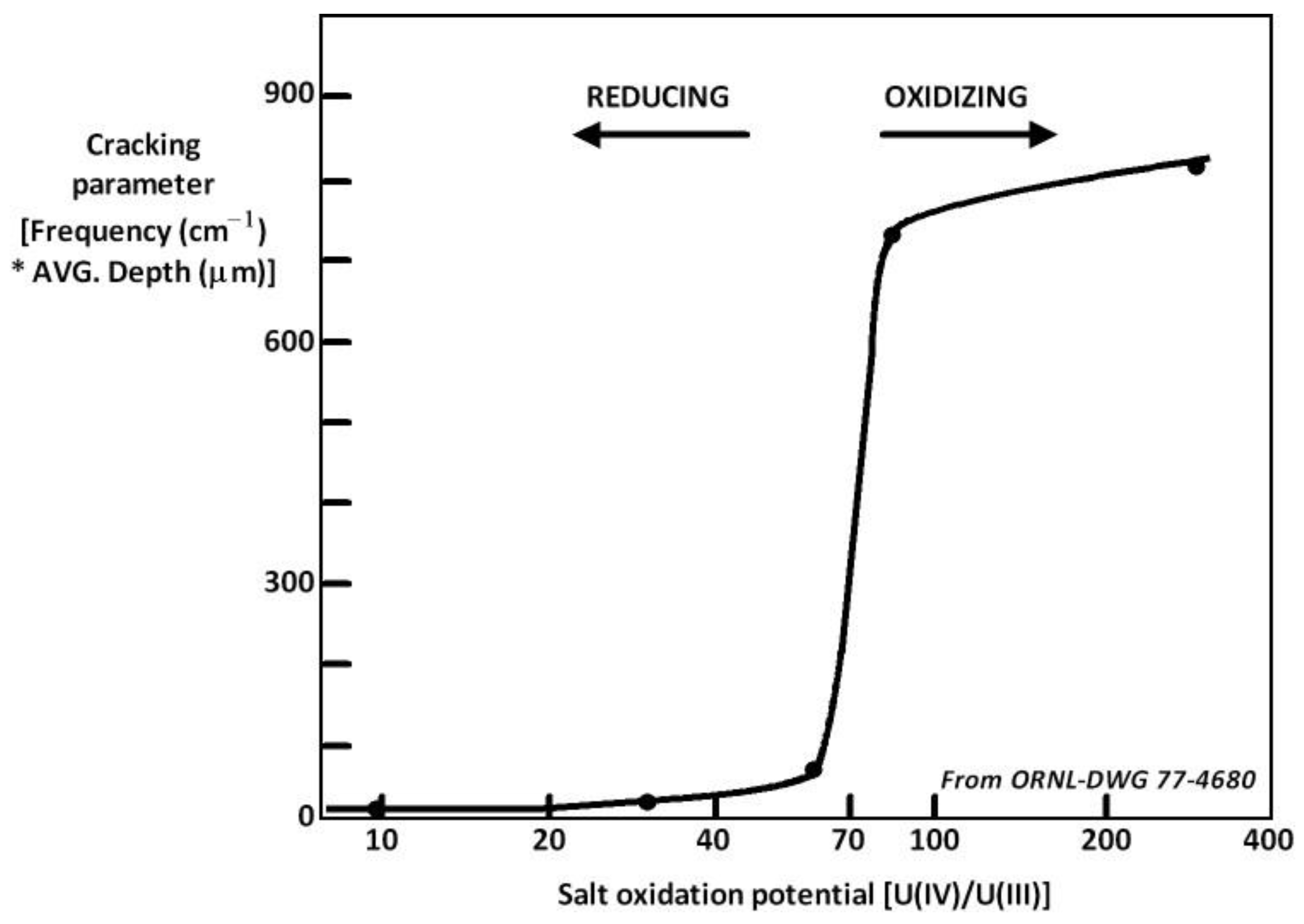
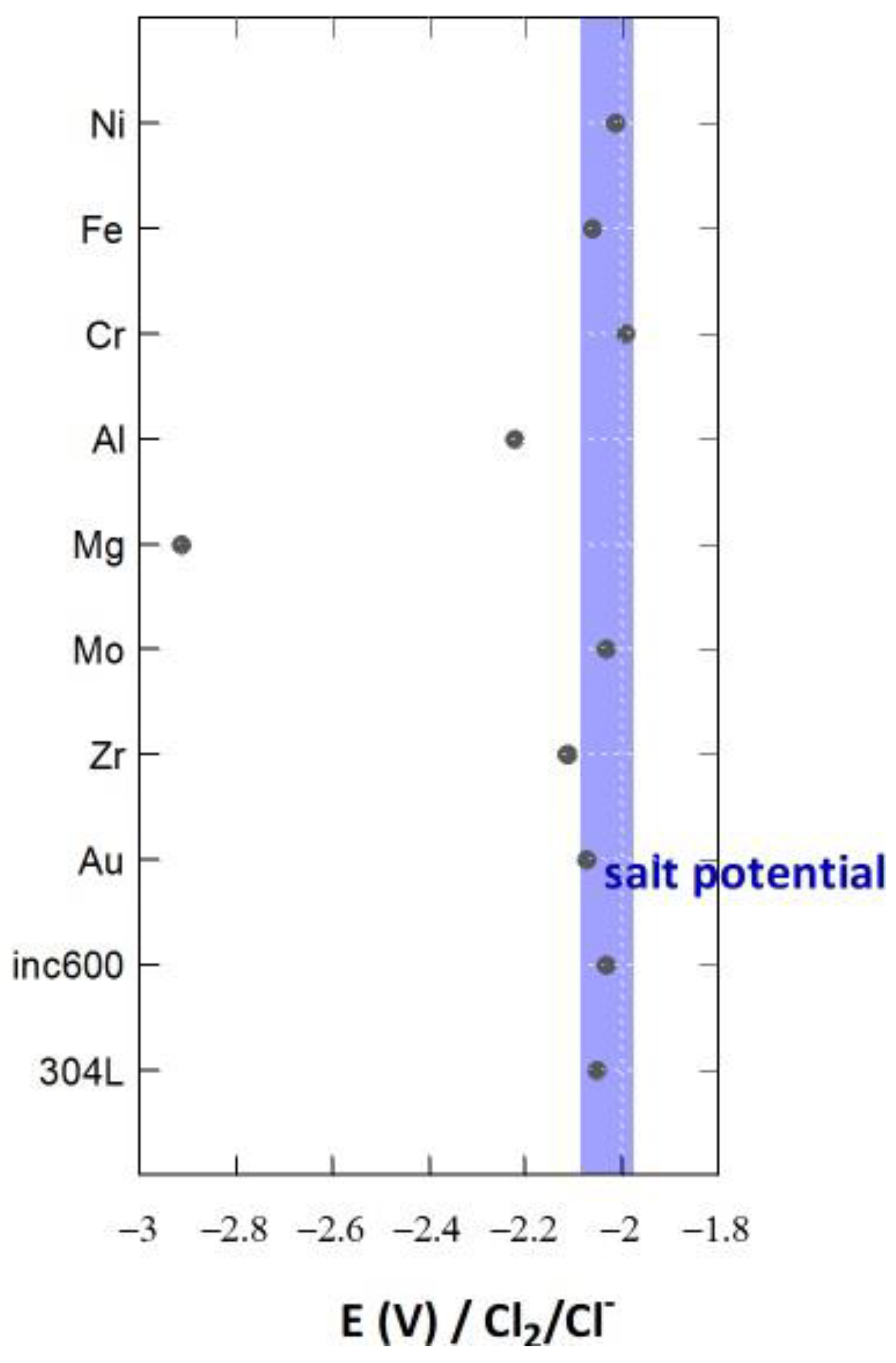
4. Salt Redox Potential Calculation and Measurement
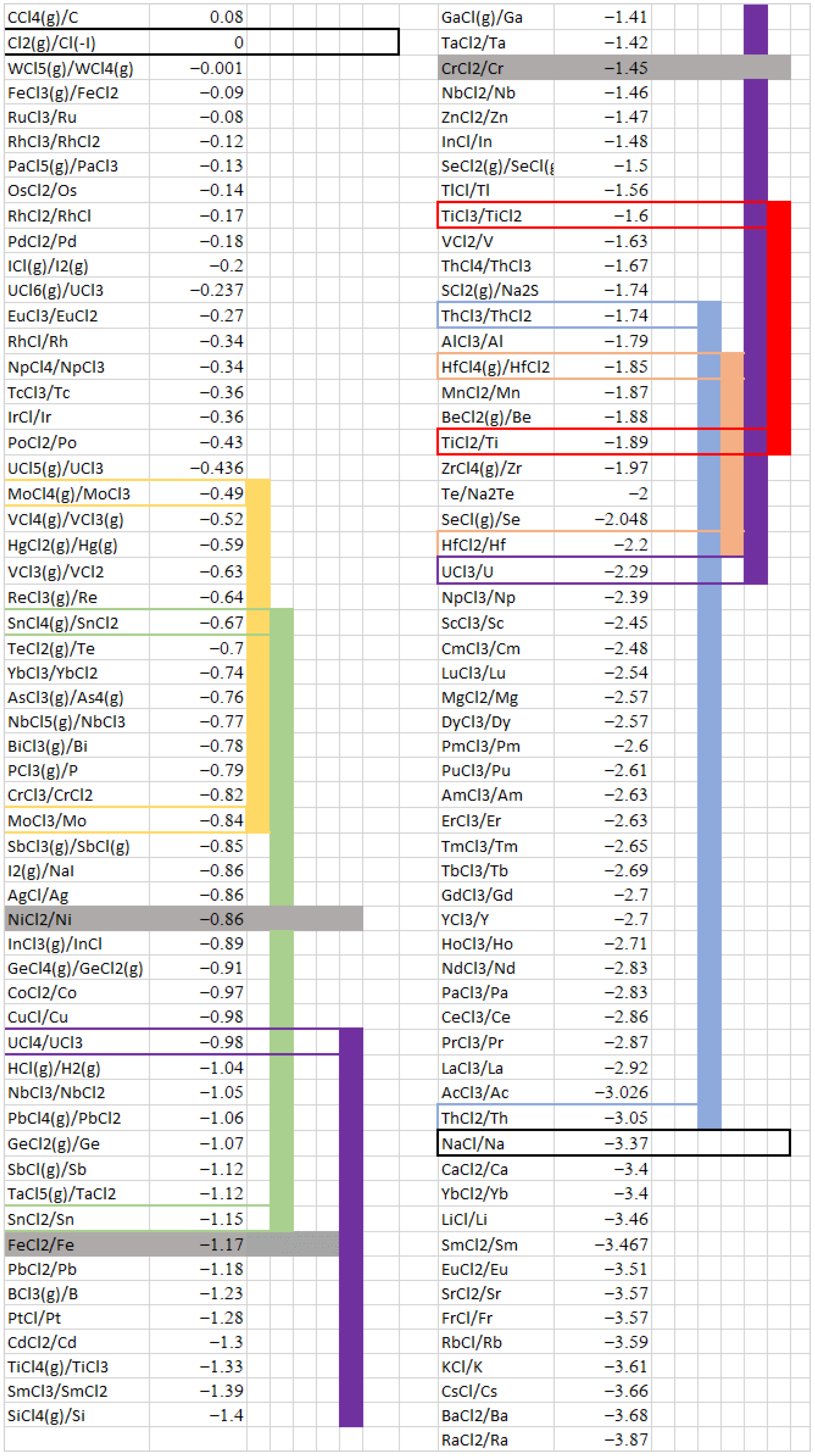
4.1. Theoretical Salt Redox Potential
- The case of LiCl-KCl (59–41 mol%) at 500 °C
- The case of NaCl-MgCl2 (55–45 mol%) at 500 °C
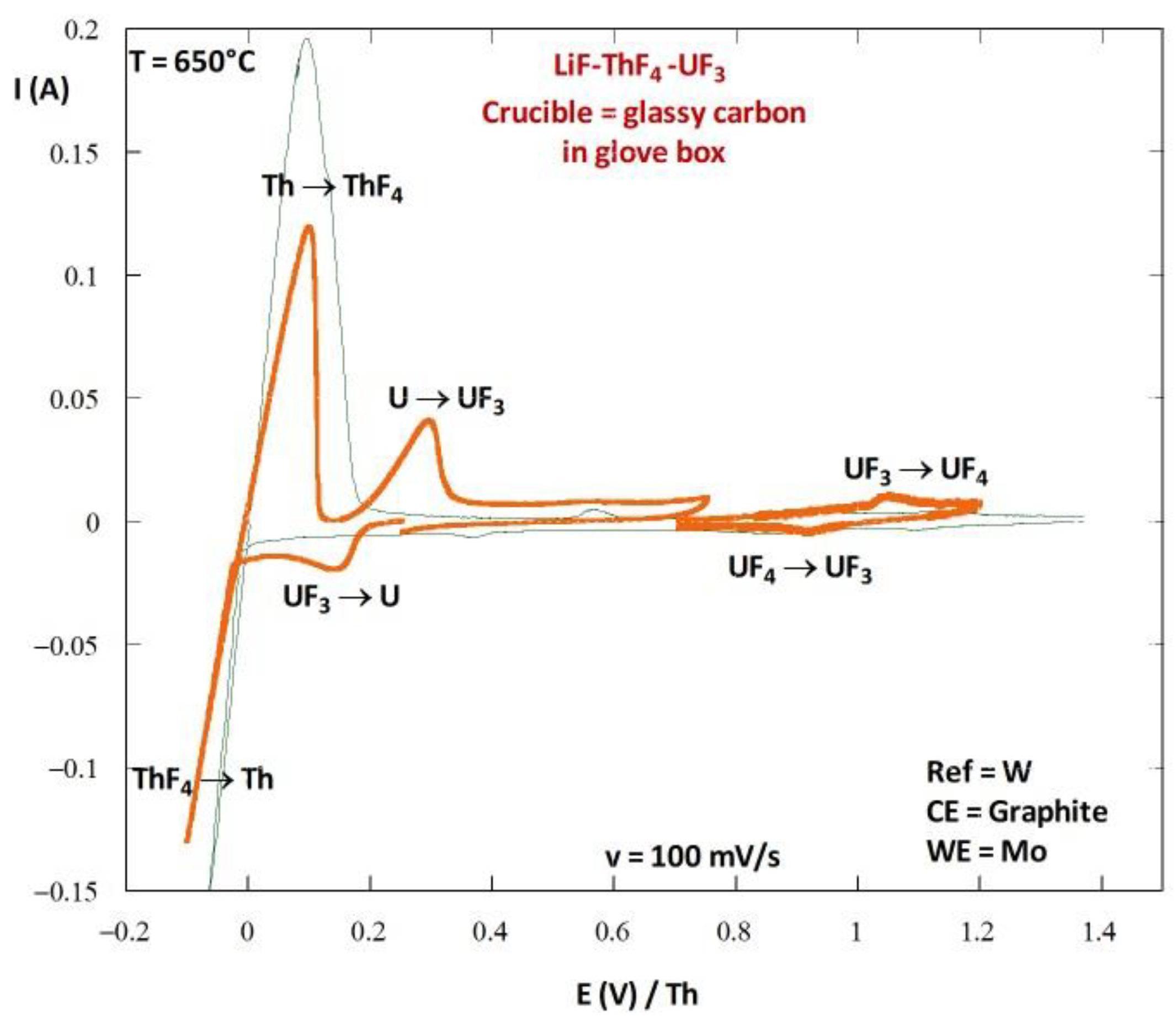
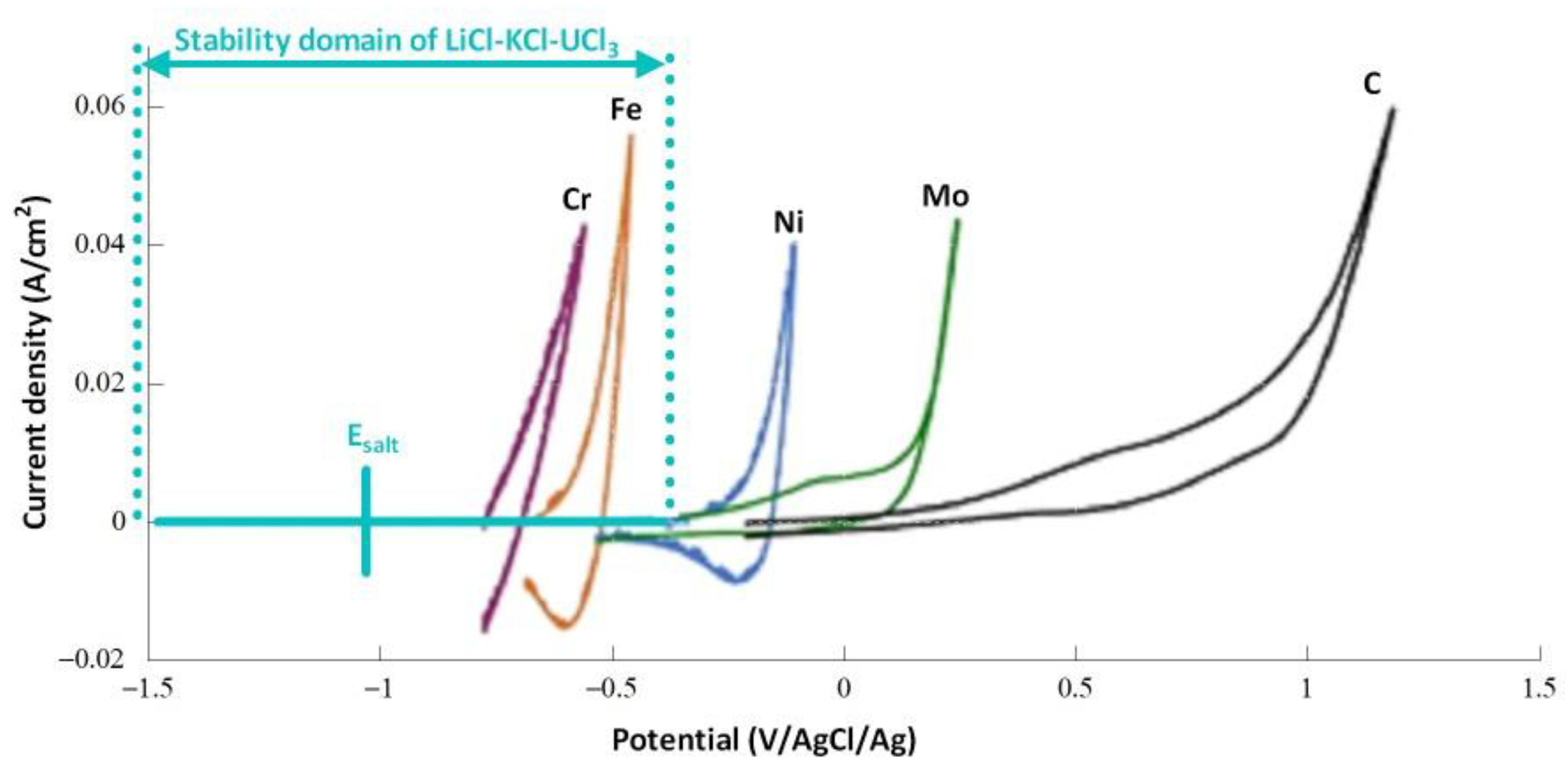
4.2. Experimental Salt Redox Potential Measurement
5. Corrosion Mitigation or Inhibition
5.1. Addition of a Redox System
5.2. Potentiostatic Control of the Material Potential
5.3. Addition of an Amphoteric Compound
6. Conclusions
Author Contributions
Funding

Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Roper, R.; Harkema, M.; Sabharwall, P.; Riddle, C.; Chisholm, B.; Day, B.; Marotta, P. Molten salt for advanced energy applications: A review. Ann. Nucl. Energy 2021, 169, 108924. [Google Scholar] [CrossRef]
- Leroux, C. Nitruration et Nitrocarburation—Procédés et Pratiques Industrielles. Les Techniques de l’Ingénieur. n°M1227 V2. 2012. Available online: https://www.techniques-ingenieur.fr/base-documentaire/materiaux-th11/traitements-thermiques-superficiels-et-thermochimiques-42501210/nitruration-et-nitrocarburation-m1227/ (accessed on 16 January 2024).
- Delpech, S. Molten salt for nuclear applications. In Molten Salt Chemistry: From Lab to Applications; Lantelme, F., Groult, H., Eds.; Elsevier: Amsterdam, The Netherlands, 2013; Chapter 24. [Google Scholar] [CrossRef]
- Grjotheim, K.; Kvande, H. Introduction to Aluminium Electrolysis. Understanding the Hall-Heroult Process; Aluminium Verlag GmbH: Düsseldorf, Germany, 1993; p. 260. [Google Scholar]
- Grinberg, I. Pombliere, Fabrique de Métaux Depuis 1898; Presse Universitaire de Grenoble: Fontaine, France, 1998. [Google Scholar]
- Hofmeister, M.; Klein, L.; Mira, H.; Rettig, R.; Virtanen, S.; Singer, R. Corrosion behaviour of stainless steels and a single crystal superalloy. Corros. Sci. 2015, 90, 46–53. [Google Scholar] [CrossRef]
- Raiman, S.; Lee, S. Aggregation and data analysis of corrosion studies in molten chloride and fluoride salts. J. Nucl. Mater. 2018, 511, 523–535. [Google Scholar] [CrossRef]
- Haubenreich, P.; Engel, J. Experience with the molten salt reactor experiment. Nucl. Appl. Technol. 1970, 8, 118–133. [Google Scholar] [CrossRef]
- Merle, E. Le Cycle Thorium en Réacteurs à Sels Fondus Peut-Il Être une Solution au Problème Énergétique du XXIème Siècle? Le Concept de TMSR-NM; Habilitation à Diriger les Recherches, Institut National Polytechnique de Grenoble: Saint-Martin-d’Hères, France, 2008. [Google Scholar]
- Allibert, M.; Delpech, S.; Gérardin, D.; Heuer, D.; Laureau, A.; Merle, E. Homogeneous Molten Salt Reactors: The molten Salt Fast Reactor Concept. In Handbook of Generation IV Nuclear Reactors; Woodhead Publishing Series in Energy; Woodhead Publishing: Montréal, QC, Canada, 2023. [Google Scholar]
- Plambeck, J. Volume X—Fused salt systems. In Encyclopedia of Electrochemistry of the Elements; Marcel Dekker, Inc.: New York, NY, USA; Basel, Switzerland, 1976. [Google Scholar]
- Pourbaix, M. Atlas D’équilibres Électrochimiques; Gauthier-Villars & Cie: Paris, France, 1963. [Google Scholar]
- Tremillon, B. La Chimie en Solvants Non-Aqueux; Presse Universitaires de France: Paris, France, 1971. [Google Scholar]
- Rouquette-Sanchez, S.; Picard, G.S. Chalcogenide chemistry in molten salts. I. Selenium(IV) acido-basic and redox properties in the LiCl–KCl eutectic melt at 450, 500, 550 and 600 °C. J. Electroanal. Chem. 2004, 572, 173–183. [Google Scholar] [CrossRef]
- Guo, S.; Zhang, J.; Wu, W.; Zhou, W. Corrosion in the molten fluoride and chloride salts and materials development for nuclear applications. Prog. Mater. Sci. 2018, 97, 448–487. [Google Scholar] [CrossRef]
- Tzvetkoff, T.; Gencheva, P. Mechanism of formation of corrosion layers on nickel and nickel-based alloys in melts containing oxyanions—A review. Mater. Chem. Phys. 2003, 82, 897. [Google Scholar] [CrossRef]
- Tzetkoff, T.; Kolchakov, J. Mechanism of growth, composition and structure of oxide films formed on ferrous alloys in molten salt electrolytes—A review. Mater. Chem. Phys. 2004, 87, 201–211. [Google Scholar] [CrossRef]
- Serp, J.; Allibert, M.; Benes, O.; Delpech, S.; Feynberg, O.; Ghetta, V.; Heuer, D.; Holcomb, D.; Ignatiev, V.; Kloosterman, J.L.; et al. The molten salt reactor (MSR) in generation IV: Overview and perspectives. Prog. Nucl. Energy 2014, 77, 308–319. [Google Scholar] [CrossRef]
- Liu, B.; Wei, X.; Wang, W.; Lu, J.; Ding, J. Corrosion behavior of Ni-based alloys in molten NaCl-CaCl2-MgCl2 eutectic salt for concentrating solar power. Sol. Energy Mater. Sol. Cells 2017, 170, 77–86. [Google Scholar] [CrossRef]
- Choi, S.; Orabona, N.; Dale, O.; Okabe, P.; Inman, C.; Simpson, M. Effect of Mg dissolution on cyclic voltammetry and open circuit potentiometry of molten MgCl2–KCl–NaCl candidate heat transfer fluid for concentrating solar power. Sol. Energy Mater. Sol. Cells 2019, 202, 110087. [Google Scholar] [CrossRef]
- Suna, H.; Wanga, J.; Tanga, Z.; Liua, Y.; Wanga, C. Assessment of effects of Mg treatment on corrosivity of molten NaCl-KCl-MgCl2 salt with Raman and Infrared spectra. Corros. Sci. 2020, 164, 108350. [Google Scholar] [CrossRef]
- Shimkevitch, A. Introduction on the band theory of molten salt. Phys. Lett. A 2019, 383, 1207–1213. [Google Scholar] [CrossRef]
- Alekseev, A.; Shimkevich, A. On voltage-sensitive managing the redox potential of MSR fuel composition. In Proceedings of the 16th International Conference on Nuclear Engineering ICONE16, Orlando, FL, USA, 11–15 May 2008. [Google Scholar]
- Lumdsen, J. Thermodynamics of Molten Salt Mixtures; Academic Press: New York, NY, USA, 1966. [Google Scholar]
- Castrillejo, Y.; Hernandez, P.; Rodrigues, J.; Vega, M.; Barrado, E. Electrochemistry of scandium in the eutectic LiCl–KCl. Electrochim. Acta 2012, 71, 166–172. [Google Scholar] [CrossRef]
- Swain, L.; Pakhui, G.; Jain, A.; Ghosh, S. Electrochemical behaviour of LiCl-KCl eutectic melts containing moisture as impurity. Part I: Inert tungsten electrode. J. Electroanal. Chem. 2022, 910, 116125. [Google Scholar] [CrossRef]
- Keiser, J. Status of Tellurium-Hastelloy N Studies in Molten Fluoride Salts. Report ORNL/TM-6002. 1977. Available online: https://www.osti.gov/biblio/7295251 (accessed on 16 January 2024).
- Keiser, J.; Manning, D.; Clausing, R. Corrosion resistance of some nicekl-base alloys to molten fluoride salts containing UF4 and tellurium. In Proceedings of the International Symposium on Molten Salts, Lexington, MA, USA; 1976. [Google Scholar]
- Ignatiev, V.; Surenkov, A. Voltammetric measurements on the [U(IV)]/[U(III)] couple and embrittlement of high nickel alloys in fuel LiF-BeF2-UF4 salt with tellurium addition in application to molten salt reactor. Corros. Sci. 2019, 160, 108164. [Google Scholar] [CrossRef]
- Doligez, X. Influence du Retraitement Physico-Chimique de sel Combustible sur le Comportement du MSFR et sur le Dimensionnement de Son Unité de Retraitement. Thèse de l’Université de Grenoble. 2010. Available online: https://theses.hal.science/tel-00553238/ (accessed on 16 January 2024).
- Delpech, S.; Cabet, C.; Slim, C.; Picard, G.S. Molten fluorides for nuclear applications. Mater. Today 2010, 13, 34. [Google Scholar] [CrossRef]
- Kudyakov, V.; Smirnov, M.; Chukreev, N.; Posokhin, Y. Formation of divalent thorium in a medium of fused potassium chloride. Transl. At. Energiya 1968, 24, 448–452. [Google Scholar] [CrossRef]
- Smirnov, M.; Kudakov, V. Complexing in molten mixtures of thorium and alkali halides. Electrochim. Acta 1983, 28, 1349–1359. [Google Scholar] [CrossRef]
- Chiotti, P.; Fuller, J.; Dock, C.; Jha, M. The binary systems ThCl4-Th and ThCl4-ThOCl2. J. Less-Common Met. 1973, 31, 365–376. [Google Scholar] [CrossRef]
- Tumidajs, P.; Flengas, S. Potential measurements of thorium tetrachloride in alkali halide solutions. Can. J. Chem. 1991, 69, 462–467. [Google Scholar] [CrossRef]
- Hoover, R.; Shaltry, M.; Martin, S.; Sridharan, K.; Phongikaroon, S. Electrochemical studies and analysis of 1–10 wt% UCl3 concentrations in molten LiCl–KCl eutectic. J. Nucl. Mater. 2014, 452, 389–396. [Google Scholar] [CrossRef]
- Polovov, I.; Abramov, A.; Dedov, K.; Karpov, V.; Zhiyakov, A.; Gibadullina, A.; Belikov, S.; Volkovich, V.; Rebrin, O. Corrosion of Austenitic Steels and Their Components in Uranium-Containing Chloride Melts. ECS Trans. 2017, 77, 847–855. [Google Scholar] [CrossRef]
- Kuznetsov, S. Thermodynamic Properties of Hafnium Chlorides Dissolved in the NaCl-KCl Melt Obtained by Electrochemical Transient Techniques. ECS Trans. 2018, 86, 193–201. [Google Scholar] [CrossRef]
- Yan, B.L.; Wang, J.; Yang, T.; Yan, Y.D.; Zhang, M.L.; Qiu, M.; Zhu, W.J. Synthesis of Ti powders with different morphologies via controlling the valence state of the titanium ion in KCl-NaCl molten salt. J. Electroanal. Chem. 2020, 876, 114496. [Google Scholar] [CrossRef]
- Martinez, A.; Castrillejo, Y.; Barrado, E.; Haarberg, G.; Picard, G.S. A chemical and electrochemical study of titanium ions in the molten equimolar CaCl2-NaCl mixture at 550 °C. J. Electroanal. Chem. 1998, 449, 67–80. [Google Scholar] [CrossRef]
- Ferry, D. Comportements Chimique et Electrochimique des Chlorures et Oxydes de Titane dans l’Eutectique LiCl-KCl à 743 K. Possibilité de Chloruration du Rutile et de L’ilmenite. These de Doctorat de l’Université Paris 6 1985. Available online: https://pascal-francis.inist.fr/vibad/index.php?action=getRecordDetail&idt=8743453 (accessed on 16 January 2024).
- Ferry, D.; Picard, G.; Tremillon, B. Low temperature molten salt process for extraction of titanium metal—Electrochemical study of chlorination and reduction stages. Trans. Inst. Min. Metall. C Min. Proc. Ext. Metall. 1988, 97, C21. [Google Scholar]
- Lambertin, D.; Lacquement, J.; Sanchez, S.; Picard, G.S. Americium chemical properties in molten LiCl–KCl eutectic at 743 K. Plasmas Ions 2000, 3, 65–72. [Google Scholar] [CrossRef]














Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Delpech, S.; Carrière, C.; Chmakoff, A.; Martinelli, L.; Rodrigues, D.; Cannes, C. Corrosion Mitigation in Molten Salt Environments. Materials 2024, 17, 581. https://doi.org/10.3390/ma17030581
Delpech S, Carrière C, Chmakoff A, Martinelli L, Rodrigues D, Cannes C. Corrosion Mitigation in Molten Salt Environments. Materials. 2024; 17(3):581. https://doi.org/10.3390/ma17030581
Chicago/Turabian StyleDelpech, Sylvie, Charly Carrière, Alexandre Chmakoff, Laure Martinelli, Davide Rodrigues, and Céline Cannes. 2024. "Corrosion Mitigation in Molten Salt Environments" Materials 17, no. 3: 581. https://doi.org/10.3390/ma17030581