
Coarse-Grained Monte Carlo Simulations with Octree Cells for Geopolymer Nucleation at Different pH Values


Abstract
:1. Introduction
2. Simulation Model and Method
2.1. Specification of Silicate Particles in Each System
2.2. Monte Carlo Approach: Implementation in MATLAB Code
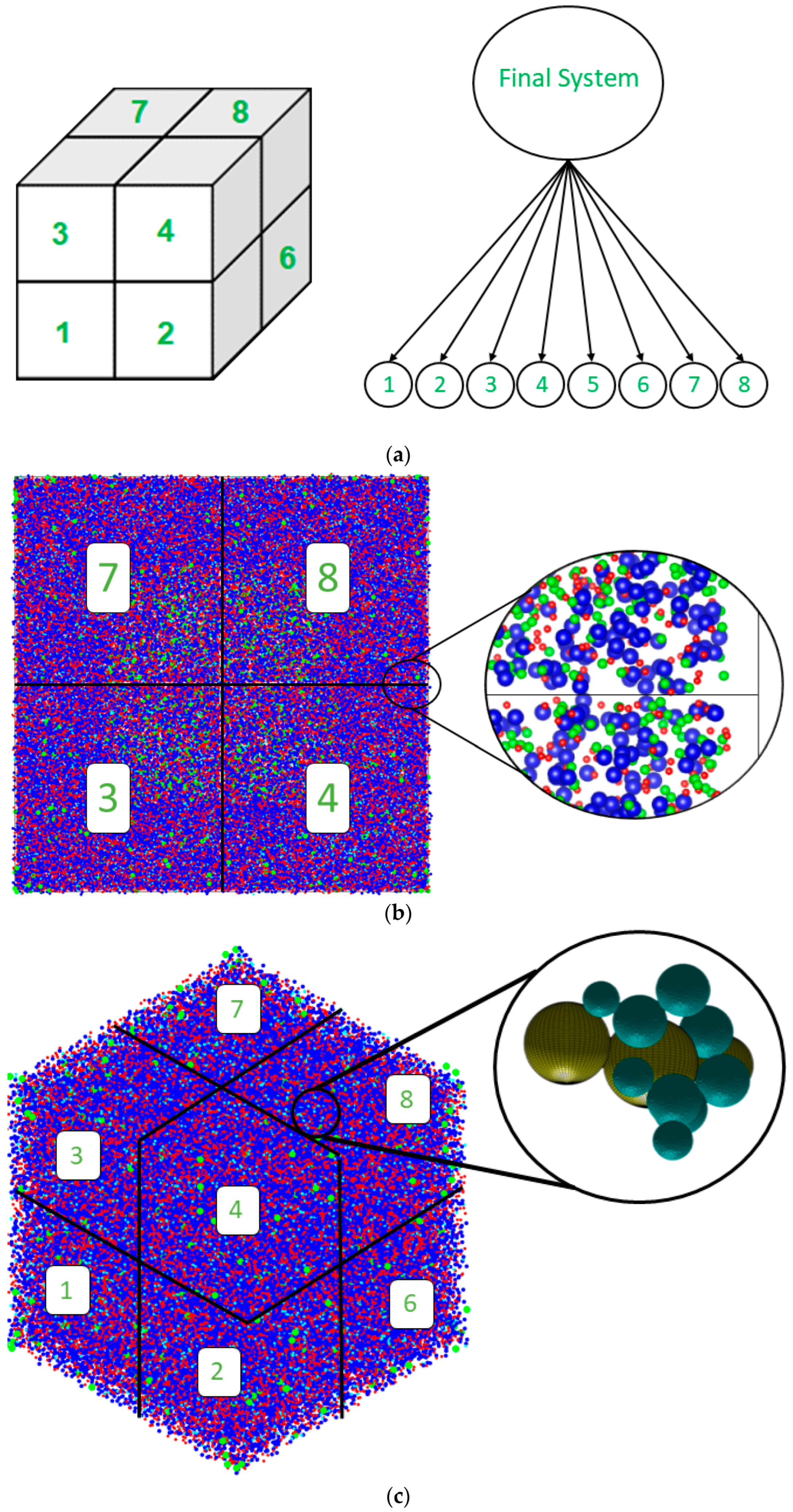
2.3. Octree Cell Approach: Development of the MATLAB Program
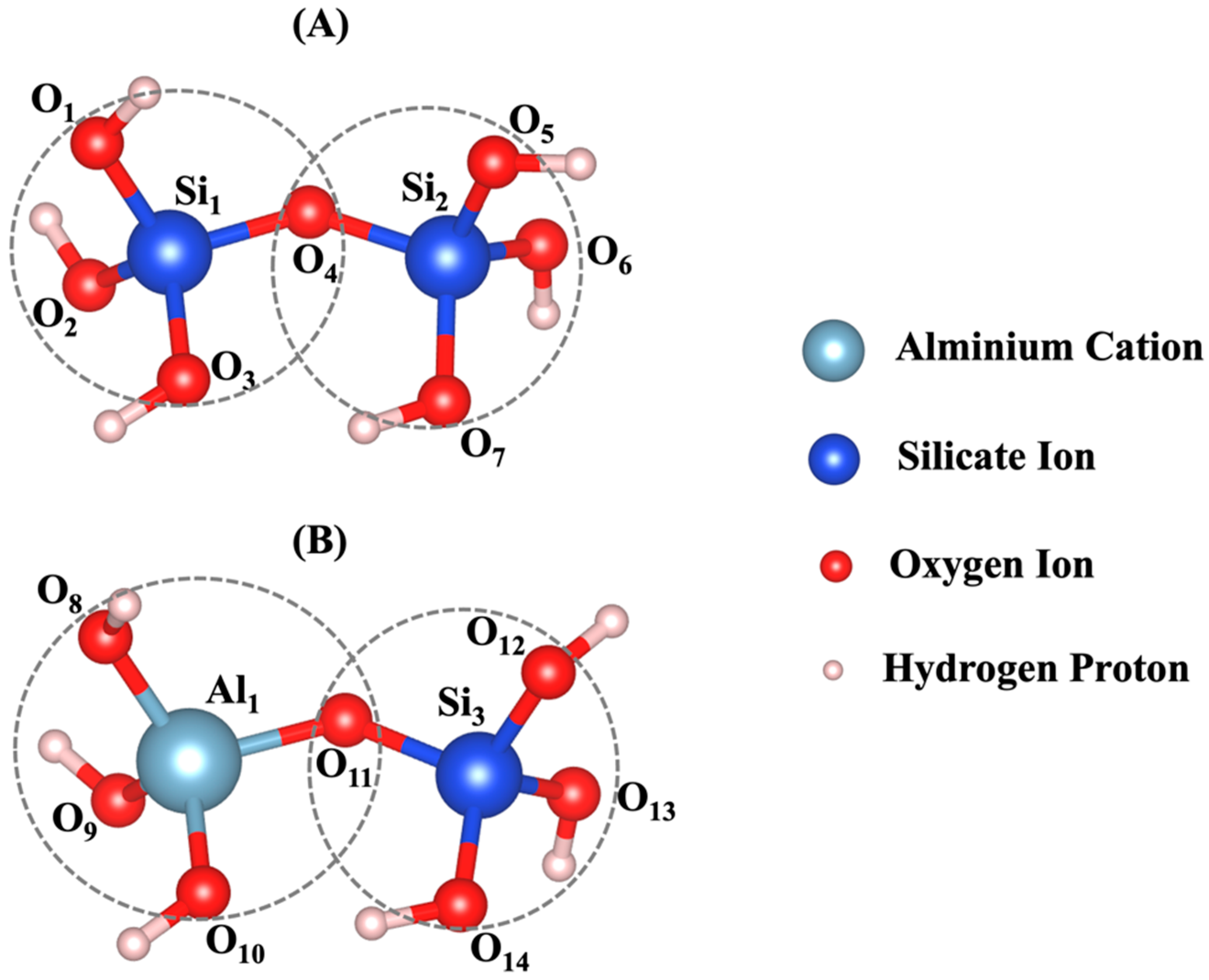
2.4. Density Functional Theory (DFT) Calculation
3. Results and Discussion
4. Conclusions
- A methodology for efficient expansion of CGMC system sizes with increased particle counts using the Octree Cell method, reduces waiting times, and harnesses the benefits of parallel high-performance computers.
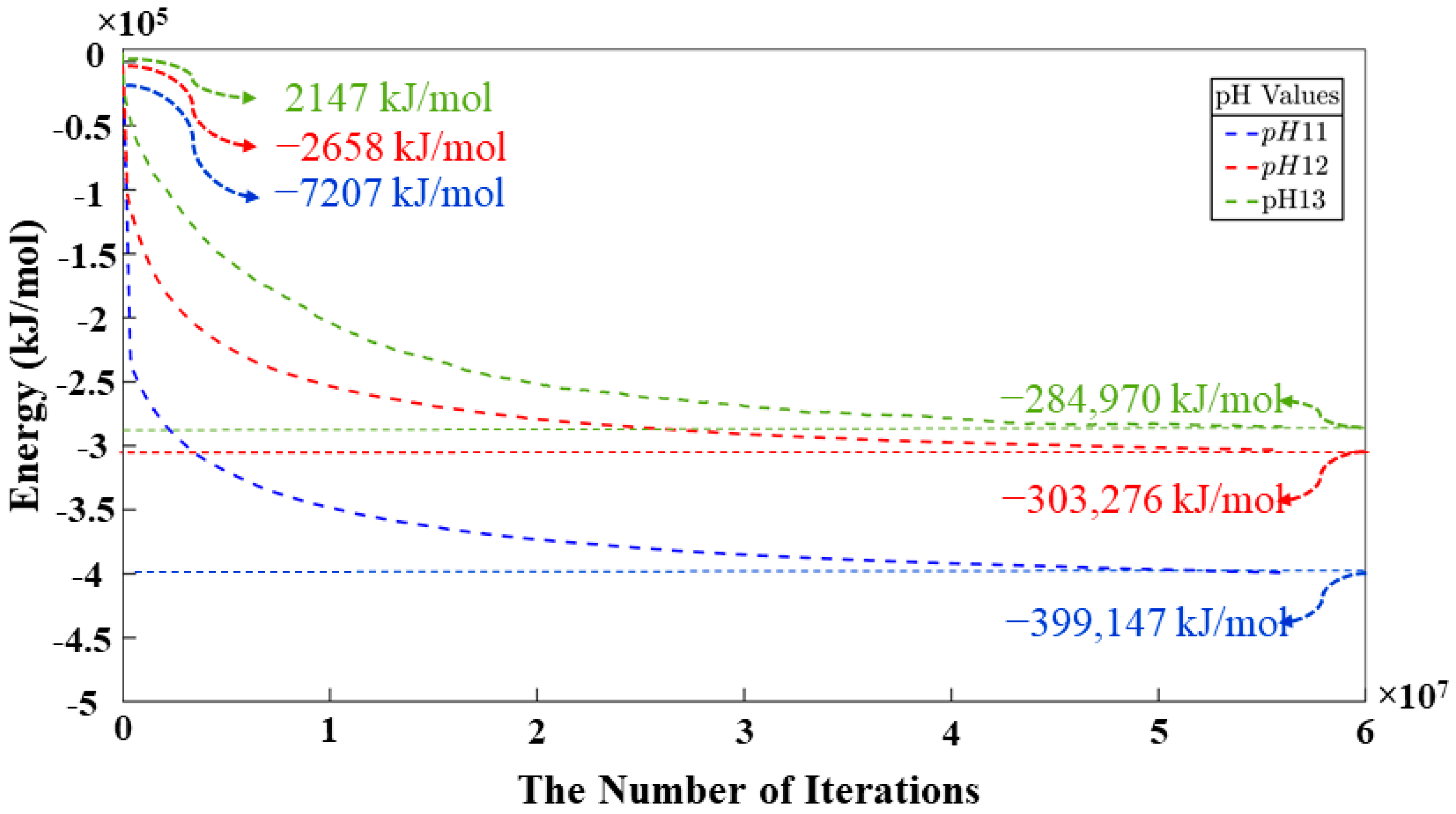
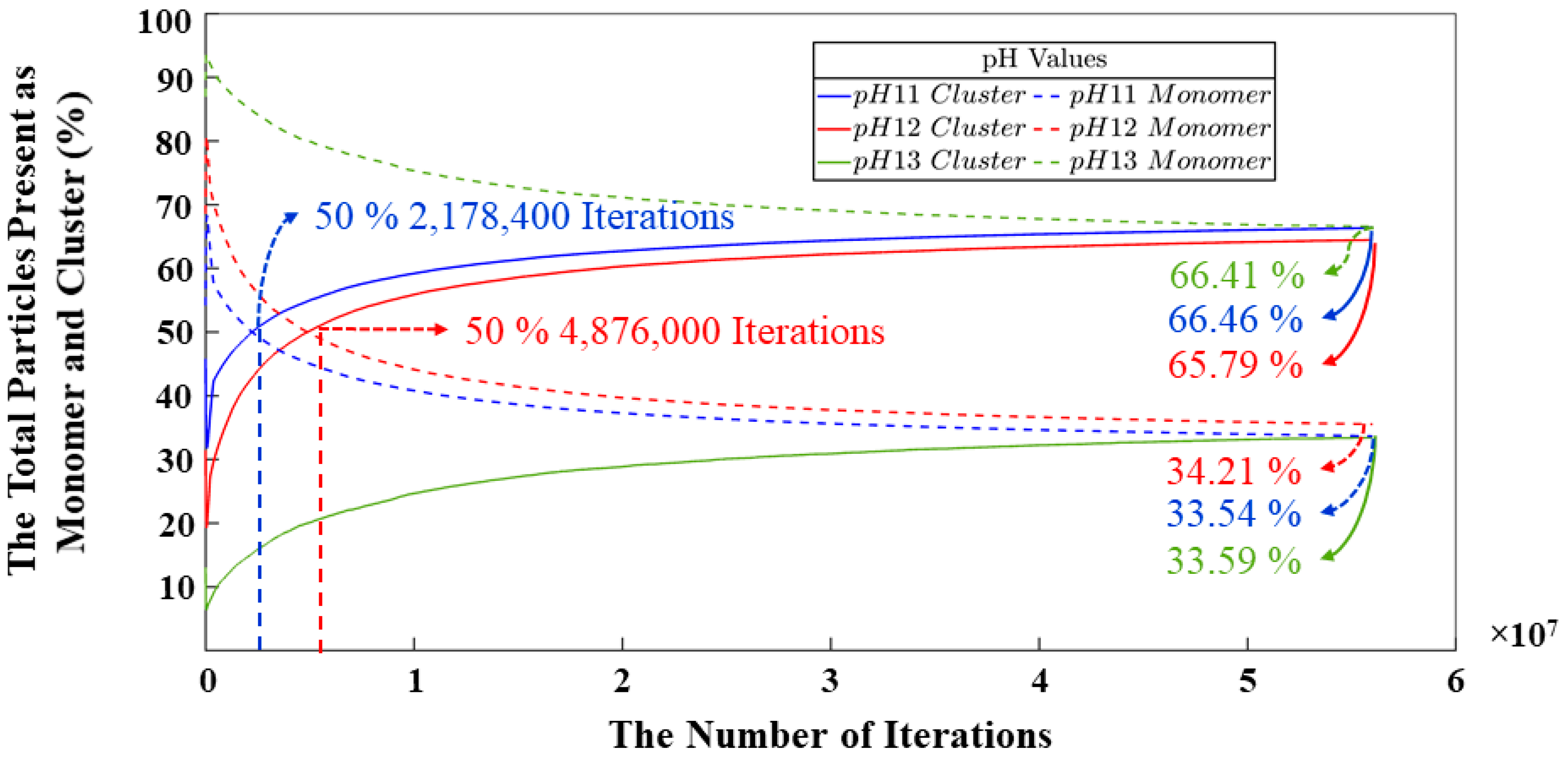
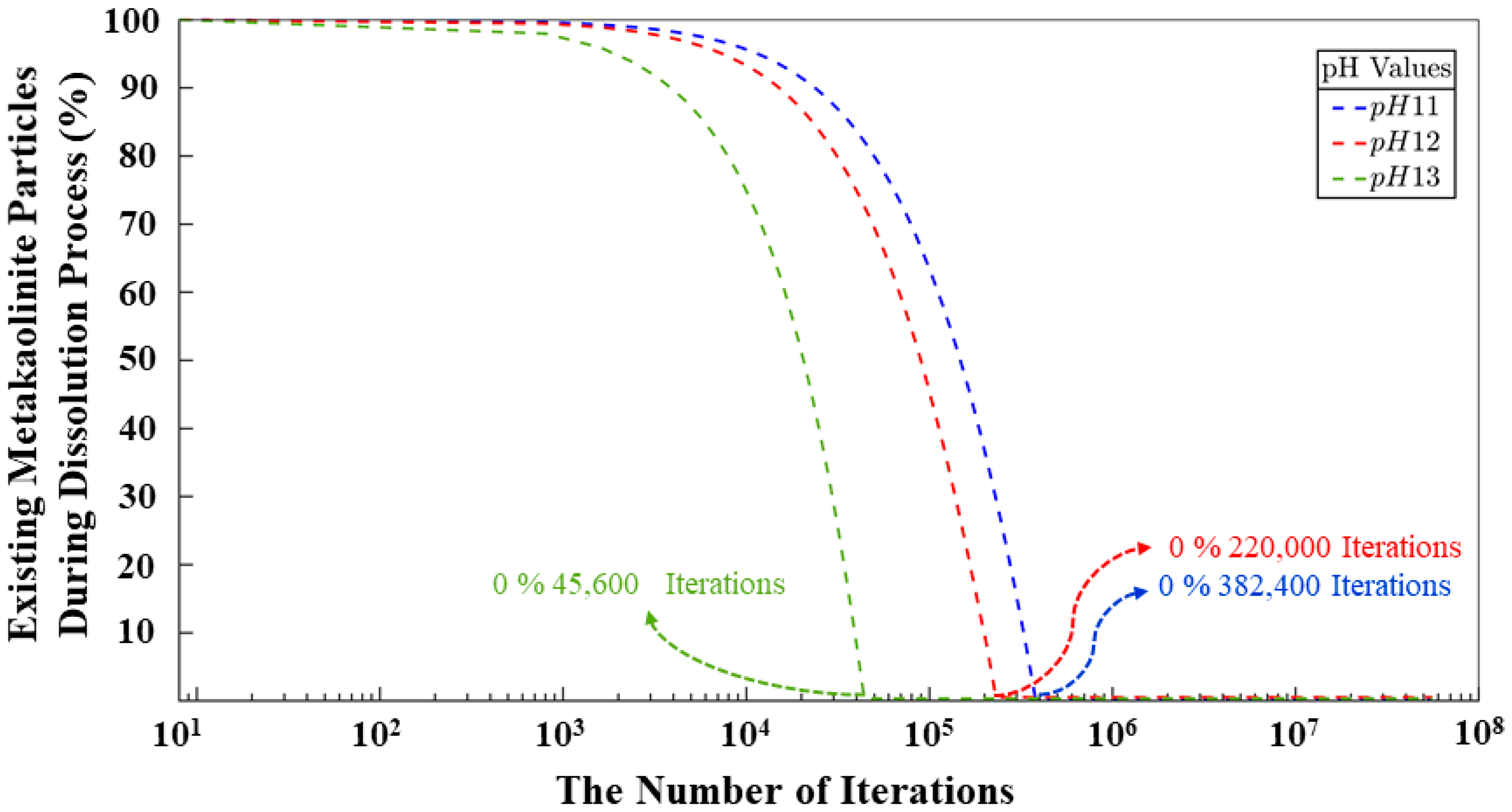
- A CGMC methodology to computationally study the critical role of pH in governing dissolution, polycondensation, and reorganization mechanisms in geopolymer materials. As a result, systems with pH levels of 11 and 12 exhibit notably similar behavior regarding the smaller quantity of remaining monomers at the end of simulations, displaying a marginal difference of merely 0.67%. Conversely, the system with pH 13 showcases a higher monomer content by the simulation’s conclusion, depicting a 32.87% increase compared to the pH 11 system and a similar 32.20% increase in contrast to the pH 12 system. The same values are represented for the behavior of the monomers in the polycondensation process; nevertheless, a difference of 2,697,600 (out of total 56 millions iterations) iterations is identified between the systems with pH 11 and 12 to reach the equilibrium between particles contributing to cluster formation and those persisting as monomers throughout the simulation. It is evident that the pH 11 system, whith the largest number of particles among pH 11 to 13), achieved equilibrium in the shortest duration.
- Digital image analysis of porosity revealed a direct link between higher pH levels, increased porosity, and the prevalence of monomer species. Similarities in the polycondensation processes of systems at pH 11 and 12 tend to coarsen the final pore structure with increasing pH. The comparative analysis between systems 11 and 12 shows an observed increase of 10.31% in pore dimensions, while the comparison between systems 11 and 13 shows a discernible increase of 32.53% in pore dimensions. pH 13 contains pores with larger diameters, in agreement with the higher percentage of monomers at the end of the simulation.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Atoms | Bond Length (Angstrom) |
|---|---|
| O1-Si1 | 1.64 |
| O2-Si1 | 1.64 |
| O3-Si1 | 1.66 |
| O4-Si1 | 1.63 |
| O4-Si2 | 1.64 |
| O5-Si2 | 1.64 |
| O6-Si2 | 1.64 |
| O7-Si2 | 1.64 |
| O8-Al1 | 1.76 |
| O9-Al1 | 1.76 |
| O10-Al1 | 1.77 |
| O11-Al1 | 1.76 |
| O11-Si3 | 1.63 |
| O12-Si3 | 1.65 |
| O13-Si3 | 1.65 |
| O14-Si3 | 1.64 |
| Atoms | Angle (Degree) |
|---|---|
| O1-Si1-O2 | 106.74° |
| O2-Si1-O3 | 106.97° |
| O3-Si1-O4 | 104.25° |
| O4-Si1-O1 | 110.56° |
| Si1-O4-Si2 | 138.28° |
| O4-Si2-O5 | 104.21° |
| O5-Si2-O6 | 107.15° |
| O6-Si2-O7 | 108.41° |
| O7-Si2-O4 | 109.46° |
| O8-Al1-O9 | 107.72° |
| O9-Al1-O10 | 110.84° |
| O10-Al1-O11 | 102.20° |
| O11-Al1-O8 | 111.33° |
| Al1-O11-Si3 | 135.12° |
| O11-Si3-O12 | 109.02° |
| O12-Si3-O13 | 104.84° |
| O13-Si3-O14 | 111.35° |
| O14-Si3-O11 | 110.30° |
References
- Davidovits, J. Geopolymers: Inorganic polymeric new materials. J. Therm. Anal. 1991, 37, 1633–1656. [Google Scholar] [CrossRef]
- Chitsaz, S.; Tarighat, A. Molecular dynamics simulation of N-A-S-H geopolymer macro molecule model for prediction of its modulus of elasticity. Constr. Build. Mater. 2020, 243, 118176. [Google Scholar] [CrossRef]
- Duxson, P.; Fernández-Jiménez, A.; Provis, J.L.; Lukey, G.C.; Palomo, A.; Van Deventer, J.S.J. Geopolymer technology: The current state of the art. J. Mater. Sci. 2007, 42, 2917–2933. [Google Scholar] [CrossRef]
- Vogt, O.; Ballschmiede, C.; Ukrainczyk, N.; Koenders, E. Evaluation of Sulfuric Acid-Induced Degradation of Potassium Silicate Activated Metakaolin Geopolymers by Semi-Quantitative SEM-EDX Analysis. Materials 2020, 13, 4522. [Google Scholar] [CrossRef] [PubMed]
- Izadifar, M.; Ukrainczyk, N.; Koenders, E. Silicate Dissolution Mechanism from Metakaolinite Using Density Functional Theory. Nanomaterials 2023, 13, 1196. [Google Scholar] [CrossRef]
- Buchwald, A.; Zellmann, H.-D.; Kaps, C. Condensation of aluminosilicate gels—Model system for geopolymer binders. J. Non-Cryst. Solids 2011, 357, 1376–1382. [Google Scholar] [CrossRef]
- White, C.E.; Provis, J.L.; Proffen, T.; Van Deventer, J.S.J. Quantitative Mechanistic Modeling of Silica Solubility and Precipitation during the Initial Period of Zeolite Synthesis. J. Phys. Chem. C 2011, 115, 9879–9888. [Google Scholar] [CrossRef]
- Zhang, L.; Lüttge, A. Aluminosilicate Dissolution Kinetics: A General Stochastic Model. J. Phys. Chem. B 2008, 112, 1736–1742. [Google Scholar] [CrossRef]
- Lloyd, R.R.; Provis, J.L.; Van Deventer, J.S.J. Pore solution composition and alkali diffusion in inorganic polymer cement. Cem. Concr. Res. 2010, 40, 1386–1392. [Google Scholar] [CrossRef]
- White, C.E.; Provis, J.L.; Proffen, T.; Van Deventer, J.S.J. Molecular mechanisms responsible for the structural changes occurring during geopolymerization: Multiscale simulation. AIChE J. 2012, 58, 2241–2253. [Google Scholar] [CrossRef]
- Bakharev, T. Resistance of geopolymer materials to acid attack. Cem. Concr. Res. 2005, 35, 658–670. [Google Scholar] [CrossRef]
- Ukrainczyk, N.; Muthu, M.; Vogt, O.; Koenders, E. Geopolymer, Calcium Aluminate, and Portland Cement-Based Mortars: Comparing Degradation Using Acetic Acid. Materials 2019, 12, 3115. [Google Scholar] [CrossRef] [PubMed]
- Koenig, A.; Herrmann, A.; Overmann, S.; Dehn, F. Resistance of alkali-activated binders to organic acid attack: Assessment of evaluation criteria and damage mechanisms. Constr. Build. Mater. 2017, 151, 405–413. [Google Scholar] [CrossRef]
- Drugă, B.; Ukrainczyk, N.; Weise, K.; Koenders, E.; Lackner, S. Interaction between wastewater microorganisms and geopolymer or cementitious materials: Biofilm characterization and deterioration characteristics of mortars. Int. Biodeterior. Biodegrad. 2018, 134, 58–67. [Google Scholar] [CrossRef]
- Izadifar, M.; Valencia, N.C.; Xiao, P.; Ukrainczyk, N.; Koenders, E. 3D Off-Lattice Coarse-Grained Monte Carlo Simulations for Nucleation of Alkaline Aluminosilicate Gels. Materials 2023, 16, 1863. [Google Scholar] [CrossRef] [PubMed]
- White, C.E.; Provis, J.L.; Kearley, G.J.; Riley, D.P.; Van Deventer, J.S.J. Density functional modelling of silicate and aluminosilicate dimerisation solution chemistry. Dalton Trans. 2011, 40, 1348–1355. [Google Scholar] [CrossRef]
- Izadifar, M.; Thissen, P.; Steudel, A.; Kleeberg, R.; Kaufhold, S.; Kaltenbach, J.; Schuhmann, R.; Dehn, F.; Emmerich, K. Comprehensive examination of dehydroxylation of kaolinite, disordered kaolinite, and dickite: Experimental studies and density functional theory. Clays Clay Miner. 2020, 68, 319–333. [Google Scholar] [CrossRef]
- Izadifar, M.; Dolado, J.S.; Thissen, P.; Ukrainczyk, N.; Koenders, E.; Ayuela, A. Theoretical Elastic Constants of Tobermorite Enhanced with Reduced Graphene Oxide through Hydroxyl vs Epoxy Functionalization: A First-Principles Study. J. Phys. Chem. C 2023, 127, 18117–18126. [Google Scholar] [CrossRef]
- Mortazavi, B. Structural, electronic, thermal and mechanical properties of C60-based fullerene two-dimensional networks explored by first-principles and machine learning. Carbon 2023, 213, 118293. [Google Scholar] [CrossRef]
- Mortazavi, B. Machine Learning Interatomic Potentials: Keys to First-Principles Multiscale Modeling. In Machine Learning in Modeling and Simulation; Rabczuk, T., Bathe, K.-J., Eds.; Computational Methods in Engineering & the Sciences; Springer International Publishing: Cham, Switzerland, 2023; pp. 427–451. ISBN 978-3-031-36643-7. [Google Scholar] [CrossRef]
- Franchini, C.; Bayer, V.; Podloucky, R.; Paier, J.; Kresse, G. Density functional theory study of MnO by a hybrid functional approach. Phys. Rev. B 2005, 72, 045132. [Google Scholar] [CrossRef]
- Duxson, P.; Provis, J.L.; Lukey, G.C.; Mallicoat, S.W.; Kriven, W.M.; Van Deventer, J.S.J. Understanding the relationship between geopolymer composition, microstructure and mechanical properties. Colloids Surf. Physicochem. Eng. Asp. 2005, 269, 47–58. [Google Scholar] [CrossRef]
- Duxson, P.; Provis, J.L.; Lukey, G.C.; Van Deventer, J.S.J. The role of inorganic polymer technology in the development of ‘green concrete’. Cem. Concr. Res. 2007, 37, 1590–1597. [Google Scholar] [CrossRef]
- Lee, W.K.W.; Van Deventer, J.S.J. Structural reorganisation of class F fly ash in alkaline silicate solutions. Colloids Surf. Physicochem. Eng. Asp. 2002, 211, 49–66. [Google Scholar] [CrossRef]
- Šefčík, J.; McCormick, A.V. Thermochemistry of aqueous silicate solution precursors to ceramics. AIChE J. 1997, 43, 2773–2784. [Google Scholar] [CrossRef]
- Salah Uddin, K.M.; Izadifar, M.; Ukrainczyk, N.; Koenders, E.; Middendorf, B. Dissolution of Portlandite in Pure Water: Part 1 Molecular Dynamics (MD) Approach. Materials 2022, 15, 1404. [Google Scholar] [CrossRef]
- Salah Uddin, K.M.; Izadifar, M.; Ukrainczyk, N.; Koenders, E.; Middendorf, B. Dissolution of β-C2S Cement Clinker: Part 1 Molecular Dynamics (MD) Approach for Different Crystal Facets. Materials 2022, 15, 6388. [Google Scholar] [CrossRef]
- Izadifar, M.; Ukrainczyk, N.; Salah Uddin, K.; Middendorf, B.; Koenders, E. Dissolution of Portlandite in Pure Water: Part 2 Atomistic Kinetic Monte Carlo (KMC) Approach. Materials 2022, 15, 1442. [Google Scholar] [CrossRef]
- Izadifar, M.; Ukrainczyk, N.; Salah Uddin, K.M.; Middendorf, B.; Koenders, E. Dissolution of β-C2S Cement Clinker: Part 2 Atomistic Kinetic Monte Carlo (KMC) Upscaling Approach. Materials 2022, 15, 6716. [Google Scholar] [CrossRef]
- Jorge, M.; Auerbach, S.M.; Monson, P.A. Modeling Spontaneous Formation of Precursor Nanoparticles in Clear-Solution Zeolite Synthesis. J. Am. Chem. Soc. 2005, 127, 14388–14400. [Google Scholar] [CrossRef]
- Kohn, W.; Sham, L.J. Self-Consistent Equations Including Exchange and Correlation Effects. Phys. Rev. 1965, 140, A1133–A1138. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B 1993, 47, 558–561. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Hafner, J. Ab-initio simulations of materials using VASP: Density-functional theory and beyond. J. Comput. Chem. 2008, 29, 2044–2078. [Google Scholar] [CrossRef] [PubMed]
- Sun, G.; Kürti, J.; Rajczy, P.; Kertesz, M.; Hafner, J.; Kresse, G. Performance of the Vienna ab initio simulation package (VASP) in chemical applications. J. Mol. Struct. Theochem. 2003, 624, 37–45. [Google Scholar] [CrossRef]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Momma, K.; Izumi, F. ıt VESTA: A three-dimensional visualization system for electronic and structural analysis. J. Appl. Crystallogr. 2008, 41, 653–658. [Google Scholar] [CrossRef]
- Mortazavi, B.; Cuniberti, G. Atomistic modeling of mechanical properties of polycrystalline graphene. Nanotechnology 2014, 25, 215704. [Google Scholar] [CrossRef]
- Mortazavi, B.; Silani, M.; Podryabinkin, E.V.; Rabczuk, T.; Zhuang, X.; Shapeev, A.V. First-Principles Multiscale Modeling of Mechanical Properties in Graphene/Borophene Heterostructures Empowered by Machine-Learning Interatomic Potentials. Adv. Mater. 2021, 33, 2102807. [Google Scholar] [CrossRef] [PubMed]
- Izadifar, M.; Sekkal, W.; Dubyey, L.; Ukrainczyk, N.; Zaoui, A.; Koenders, E. Theoretical Studies of Adsorption Reactions of Aluminosilicate Aqueous Species on Graphene-Based Nanomaterials: Implications for Geopolymer Binders. ACS Appl. Nano Mater. 2023, 6, 16318–16331. [Google Scholar] [CrossRef]














| Percentage (%) | |||
|---|---|---|---|
| pH System | Water + Na | Silicate in Solution | Metakaolinite |
| 11 12 13 | 68.4% | 10.6 | 21% |
| Number of Particles | ||
|---|---|---|
| pH | Silicate in Solution | Metakaolinite |
| 11 | 17,560 | 97,336 |
| 12 | 10,640 | 64,000 |
| 13 | 5824 | 17,576 |
| pH Systems | |||
|---|---|---|---|
| Silicate Monomer Species | 11 | 12 | 13 |
| M | 872 | 0 | 0 |
| M−.Na+.3H2O | 15,816 | 8512 | 1744 |
| M2−.2Na+.6H2O | 872 | 2128 | 4080 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Valencia, N.C.; Izadifar, M.; Ukrainczyk, N.; Koenders, E. Coarse-Grained Monte Carlo Simulations with Octree Cells for Geopolymer Nucleation at Different pH Values. Materials 2024, 17, 95. https://doi.org/10.3390/ma17010095
Valencia NC, Izadifar M, Ukrainczyk N, Koenders E. Coarse-Grained Monte Carlo Simulations with Octree Cells for Geopolymer Nucleation at Different pH Values. Materials. 2024; 17(1):95. https://doi.org/10.3390/ma17010095
Chicago/Turabian StyleValencia, Nicolas Castrillon, Mohammadreza Izadifar, Neven Ukrainczyk, and Eduardus Koenders. 2024. "Coarse-Grained Monte Carlo Simulations with Octree Cells for Geopolymer Nucleation at Different pH Values" Materials 17, no. 1: 95. https://doi.org/10.3390/ma17010095