
Impact of Nitroxyl Radicals on Photovoltaic Conversion Properties of Dye-Sensitized Solar Cells


Abstract
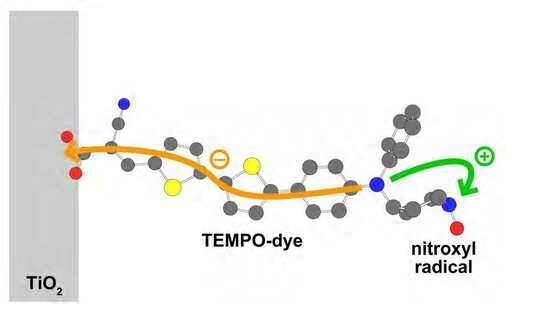
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Characterization
2.3. Fabrication of DSSCs and Photovoltaic Measurements
2.4. Synthesis
2.4.1. Compound 1: 1-Bromo-4-hexyloxybenzene
2.4.2. Compound 2: 4-(4-Hexyloxy-phenylamino)-2,2,6,6-tetramethyl-piperidine 1-oxyl
2.4.3. Compound 3: 5-Formyl-2,2′-bithiophene
2.4.4. Compound 4: 5-Formyl-5′-(4-bromophenyl)-2,2′-bithiophene
2.4.5. Compound 5: 2-[5′-(4-Bromophenyl)-2,2′-bithiophen-5-Yl]-1,3-dioxolane
2.4.6. Compound 6: 4-[N-(4-(5′-(1,3-Dioxolan-2-Yl)-2,2′-bithiophen-5-Yl)phenyl)-N-(4-hexyloxy-phenyl)amino]-tetramethylpiperidine 1-oxyl
2.4.7. Compound 7: 4-[N-(4-(5′-Formyl-2,2′-bithiophen-5-Yl)phenyl)-N-(4-hexyloxy-phenyl)amino]-tetramethylpiperidine 1-oxyl
2.4.8. Compound 8: 2-Cyano-3-(5′-{4-[N-(4-hexyloxy-phenyl)-N-(1-oxyl-2,2,6,6-tetramethyl-piperidin-4-Yl)amino]phenyl}-2,2′-bithiophen-5-Yl)acrylic Acid (TEMPO-Dye)
3. Results
3.1. Design and Synthesis of Dye Compound
3.2. Calculating Proportion of Residual Radicals and Controlling It via Reaction Time
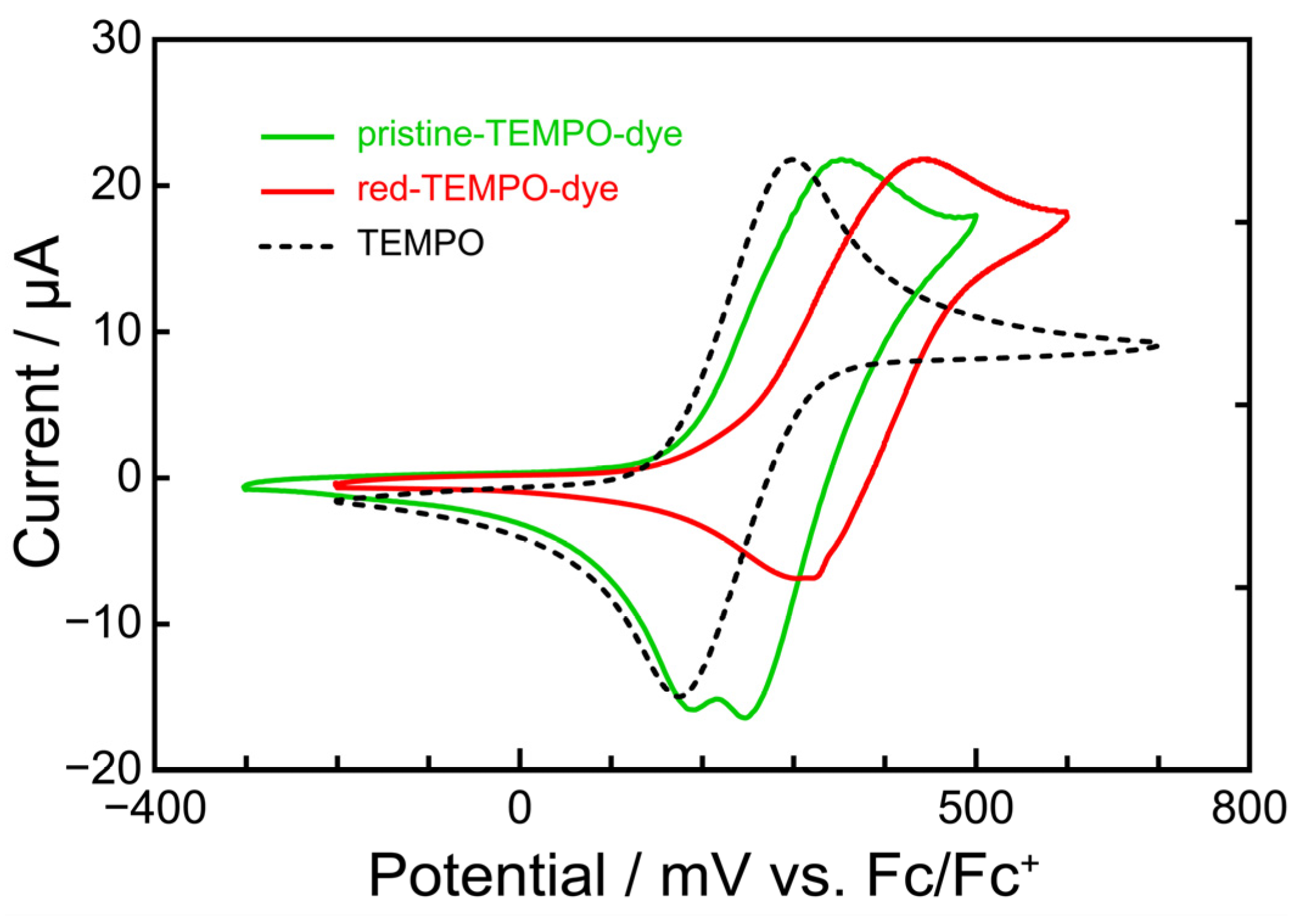
3.3. Optical and Electrochemical Properties of TEMPO-Dye
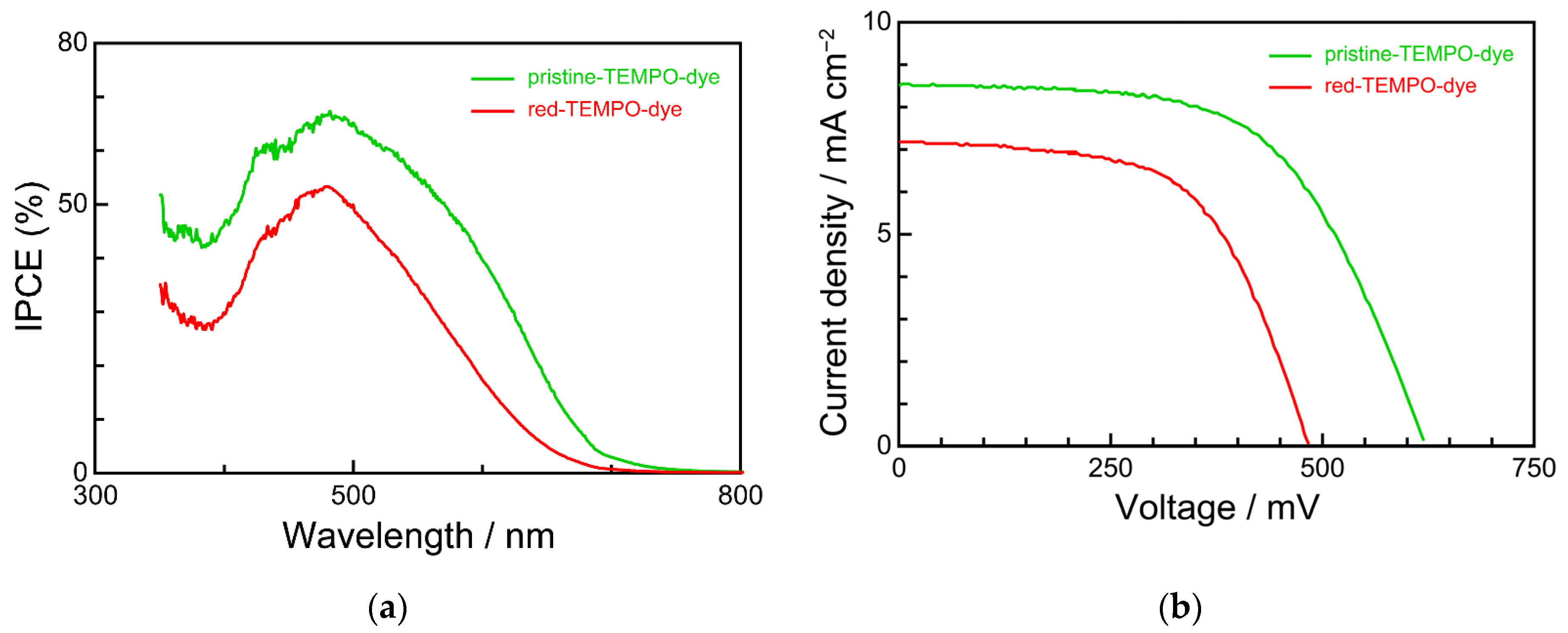
3.4. Device Characteristics
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- O’Regan, B.; Grätzel, M. A low-cost, high-efficiency solar cell based on dye-sensitized colloidal TiO2 films. Nature 1991, 353, 737–740. [Google Scholar] [CrossRef]
- Hagfeldt, A.; Boschloo, G.; Sun, L.; Kloo, L.; Pettersson, H. Dye-sensitized solar cells. Chem. Rev. 2010, 110, 6595–6663. [Google Scholar] [CrossRef] [PubMed]
- Yum, J.H.; Walter, P.; Huber, S.; Rentsch, D.; Geiger, T.; Nüesch, F.; De Angelis, F.; Grätzel, M.; Nazeeruddin, M.K. Efficient far red sensitization of nanocrystalline TiO2 films by an unsymmetrical squaraine dye. J. Am. Chem. Soc. 2007, 129, 10320–10321. [Google Scholar] [CrossRef] [PubMed]
- Imae, I.; Koshima, T.; Korai, K.; Ooyama, Y.; Komaguchi, K.; Harima, Y. Enhanced photovoltaic performances of panchromatic EDOT-containing dye by introducing bulky dialkylfluorene units in donor moiety. Dyes Pigment. 2016, 132, 262–269. [Google Scholar] [CrossRef]
- Ren, Y.; Zhang, D.; Suo, J.; Cao, Y.; Eickemeyer, F.T.; Vlachopoulos, N.; Zakeeruddin, S.M.; Hagfeldt, A.; Grätzel, M. Hydroxamic acid pre-adsorption raises the efficiency of cosensitized solar cells. Nature 2023, 613, 60–65. [Google Scholar] [CrossRef] [PubMed]
- Kano, Y.; Kushimoto, K.; Komaguchi, K.; Ooyama, Y.; Imae, I.; Ohshita, J.; Harima, Y. Intermolecular distances of carboxylated TEMPO derivatives on TiO2 evaluated by spin-probe ESR. Phys. Chem. Chem. Phys. 2012, 14, 15988–15990. [Google Scholar] [CrossRef] [PubMed]
- Nishide, H.; Oyaizu, K. Toward flexible batteries. Science 2008, 319, 737–738. [Google Scholar] [CrossRef] [PubMed]
- Pavlishchuk, V.V.; Addison, A.W. Conversion constants for redox potentials measured versus different reference electrodes in acetonitrile solutions at 25 °C. Inorg. Chim. Acta 2000, 298, 97–102. [Google Scholar] [CrossRef]
- Peng, H.; Chen, Y.; Chen, L.; He, X.; Li, F. Luminescent mesogen jacketed poly(1-alkyne) bearing lateral terphenyl with hexyloxy tail. J. Polym. Sci. A Polym. Chem. 2010, 48, 5679–5692. [Google Scholar] [CrossRef]
- Vilsmeier, A.; Haack, A. Über die Einwirkung von Halogenphosphor auf Alkyl-formanilide. Eine neue Methode zur Darstellung sekundärer und tertiärer p-Alkylamino-benzaldehyde. Chem. Ber. 1927, 60, 119–122. [Google Scholar] [CrossRef]
- Paul, F.; Patt, J.; Hartwig, J.F. Palladium-catalyzed formation of carbon-nitrogen bonds. Reaction intermediates and catalyst improvements in the hetero cross-coupling of aryl halides and tin amides. J. Am. Chem. Soc. 1994, 116, 5969–5970. [Google Scholar] [CrossRef]
- Gozzi, C.; Lavenot, L.; Llg, K.; Penalva, V.; Lemaire, M. Direct thiophene arylation catalysed by palladium. Tetrahedron Lett. 1997, 38, 8867–8870. [Google Scholar] [CrossRef]
- Steinberger, S.; Mishra, A.; Reinold, E.; Mena-Osteritz, E.; Müller, H.; Uhrich, C.; Pfeiffer, M.; Bäuerle, P. Synthesis and characterizations of red/near-IR absorbing A–D–A–D–A-type oligothiophenes containing thienothiadiazole and thienopyrazine central units. J. Mater. Chem. 2012, 22, 2701–2712. [Google Scholar] [CrossRef]
- Knoevenagel, E. Condensation von Malonsäure mit aromatischen Aldehyden durch Ammoniak und Amine. Ber. Dtsch. Chem. Ges. 1898, 31, 2596–2619. [Google Scholar] [CrossRef]
- Hagfeldt, A.; Grätzel, M. Light-induced redox reactions in nanocrystalline systems. Chem. Rev. 1995, 95, 49–68. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| λmax of Absorption /nm | λmax of Emission /nm | Band Gap (E0–0) /V | HOMO/V vs. NHE | LUMO/V vs. NHE |
|---|---|---|---|---|
| 461 | 624 | 2.30 | 0.93 | –1.37 |
| Dye | Jsc/mA cm−2 | Voc/mV | ff/- | η (%) |
|---|---|---|---|---|
| pristine-TEMPO-dye | 8.69 | 618 | 0.59 | 3.16 |
| red-TEMPO-dye | 7.22 | 485 | 0.59 | 2.06 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Imae, I.; Akazawa, R.; Harima, Y. Impact of Nitroxyl Radicals on Photovoltaic Conversion Properties of Dye-Sensitized Solar Cells. Materials 2024, 17, 77. https://doi.org/10.3390/ma17010077
Imae I, Akazawa R, Harima Y. Impact of Nitroxyl Radicals on Photovoltaic Conversion Properties of Dye-Sensitized Solar Cells. Materials. 2024; 17(1):77. https://doi.org/10.3390/ma17010077
Chicago/Turabian StyleImae, Ichiro, Ryosuke Akazawa, and Yutaka Harima. 2024. "Impact of Nitroxyl Radicals on Photovoltaic Conversion Properties of Dye-Sensitized Solar Cells" Materials 17, no. 1: 77. https://doi.org/10.3390/ma17010077