
Clarithromycin-Loaded Submicron-Sized Carriers: Pharmacokinetics and Pharmacodynamic Evaluation

Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
2.2.1. Preparation of CLA-DLCs
2.2.2. Characterization of CLA-DLCs
Size, Polydispersity Index, and Zeta Potential
Entrapment Efficiency
Solid State Characterization
2.2.3. Drug Release Studies
2.2.4. Stability Studies
2.2.5. In Vitro Antimicrobial Activity
Minimum Inhibitory Concentration (MIC) Determination
Minimum Biofilm Inhibitory Concentration (MBIC) Evaluation
2.2.6. In Vivo Studies
2.2.7. Pharmacokinetic Evaluation
2.2.8. Pharmacodynamic Studies
2.2.9. Histopathological Evaluation
2.2.10. Statistical Analysis
3. Results and Discussion
3.1. Formulation Optimization
3.2. Solid State Characterization
3.3. Dissolution Study
3.4. Stability Studies
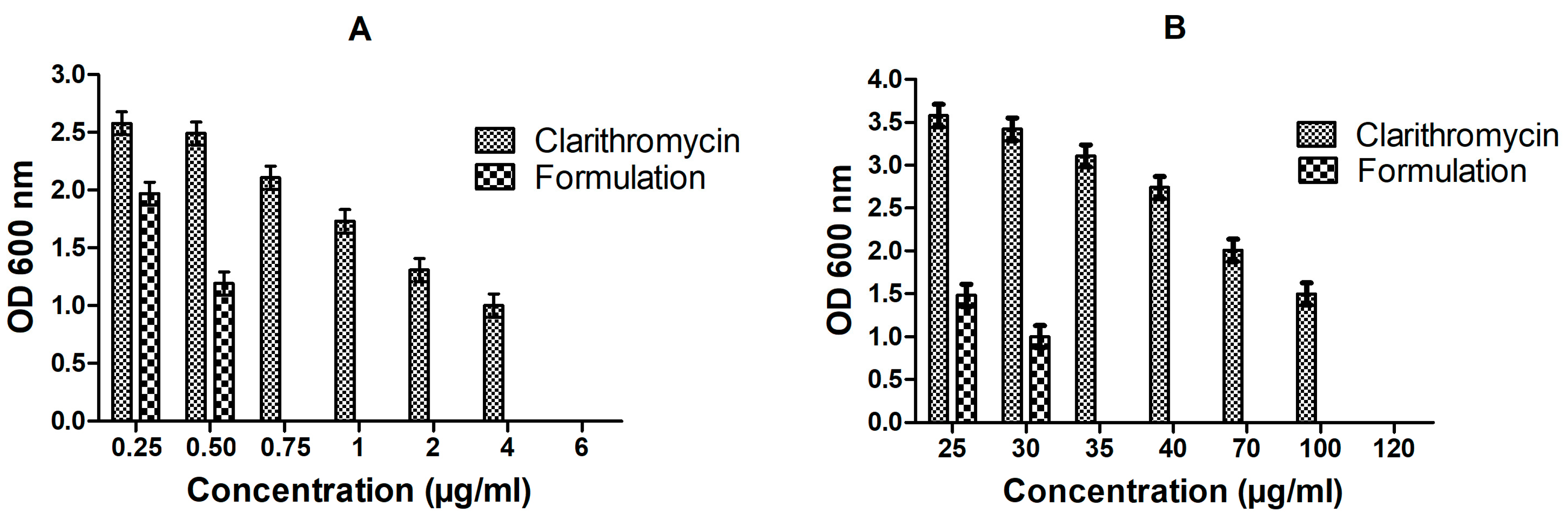
3.5. In Vitro Anti-Bacterial Studies
3.6. Pharmacokinetic Profile
3.7. Pharmacodynamic Study
Effect of CLA-DLCs on Oxidative Stress Biomarkers
3.8. Histology
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Qi, L.; Li, H.; Zhang, C.; Liang, B.; Li, J.; Wang, L.; Du, X.; Liu, X.; Qiu, S.; Song, H. Relationship between antibiotic resistance, biofilm formation, and biofilm-specific resistance in Acinetobacter baumannii. Front. Microbiol. 2016, 7, 483. [Google Scholar] [CrossRef] [PubMed]
- Cecconi, M.; Evans, L.; Levy, M.; Rhodes, A. Sepsis and septic shock. Lancet 2018, 392, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Bahr, G.; González, L.J.; Vila, A.J. Metallo-β-lactamases in the age of multidrug resistance: From structure and mechanism to evolution, dissemination, and inhibitor design. Chem. Rev. 2021, 121, 7957–8094. [Google Scholar] [CrossRef] [PubMed]
- Launey, Y.; Duteurtre, B.; Larmet, R.; Nesseler, N.; Tawa, A.; Mallédant, Y.; Seguin, P. Risk factors for mortality in postoperative peritonitis in critically ill patients. World J. Crit. Care Med. 2017, 6, 48–55. [Google Scholar] [CrossRef]
- Elkhatib, W.; Noreddin, A. Efficacy of ciprofloxacin-clarithromycin combination against drug-resistant Pseudomonas aeruginosa mature biofilm using in vitro experimental model. Microb. Drug Resist. 2014, 20, 575–582. [Google Scholar] [CrossRef]
- Domon, H.; Maekawa, T.; Yonezawa, D.; Nagai, K.; Oda, M.; Yanagihara, K.; Terao, Y. Mechanism of macrolide-induced inhibition of pneumolysin release involves impairment of autolysin release in macrolide-resistant Streptococcus pneumoniae. Antimicrob. Agents Chemother. 2018, 62, e00161-18. [Google Scholar] [CrossRef]
- Iskandar, I.; Walters, J.D. Clarithromycin accumulation by phagocytes and its effect on killing of Aggregatibacter actinomycetemcomitans. J. Periodontol. 2011, 82, 497–504. [Google Scholar] [CrossRef]
- Cepas, V.; López, Y.; Muñoz, E.; Rolo, D.; Ardanuy, C.; Martí, S.; Xercavins, M.; Horcajada, J.P.; Bosch, J.; Soto, S.M. Relationship between biofilm formation and antimicrobial resistance in gram-negative bacteria. Microb. Drug Resist. 2019, 25, 72–79. [Google Scholar] [CrossRef]
- Sharma, M.; Gupta, N.; Gupta, S. Implications of designing clarithromycin loaded solid lipid nanoparticles on their pharmacokinetics, antibacterial activity and safety. RSC Adv. 2016, 6, 76621–76631. [Google Scholar] [CrossRef]
- Patra, J.K.; Das, G.; Fraceto, L.F.; Campos, E.V.R.; Torres, M.P.R.; Torres, L.S.A.; Torres, L.A.D.; Grillo, R.; Swamy, M.K.; Sharma, S.; et al. Nano based drug delivery systems: Recent developments and future prospects. J. Nanobiotechnol. 2018, 16, 71. [Google Scholar] [CrossRef]
- Lu, Y.; Zhang, Y.; Yang, Z.; Tang, X. Formulation of an intravenous emulsion loaded with a clarithromycin–phospholipid complex and its pharmacokinetics in rats. Int. J. Pharm. 2009, 366, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Nie, S.; Yang, X.; Wang, C.; Cui, S.; Pan, W. Optimization of tocol emulsions for the intravenous delivery of clarithromycin. Int. J. Pharm. 2008, 356, 282–290. [Google Scholar] [CrossRef] [PubMed]
- Alhajlan, M.; Alhariri, M.; Omri, A. Efficacy and safety of liposomal clarithromycin and its effect on pseudomonas aeruginosa virulence factors. Antimicrob. Agents Chemother. 2013, 57, 2694–2704. [Google Scholar] [CrossRef] [PubMed]
- Ye, T.; Yu, J.; Luo, Q.; Wang, S.; Chan, H.K. Inhalable clarithromycin liposomal dry powders using ultrasonic spray freeze drying. Powder Technol. 2017, 305, 63–70. [Google Scholar] [CrossRef]
- Rajarajan, S.; Rao, B.P.; Selvamuthu, K.S. Design and development of chitosan beta-cyclodextrin based nasal mucoadhesive microspheres of clarithromycin. Int. J. Pharm. Investig. 2020, 10, 279–285. [Google Scholar] [CrossRef]
- Dimer, F.; Wodarz, D.S.C.; Haupenthal, C.; Hartmann, J.; Lehr, R.; Michael, C. Inhalable clarithromycin microparticles for treatment of respiratory infections. Pharm. Res. 2015, 32, 3850–3861. [Google Scholar] [CrossRef] [PubMed]
- Gong, H.; Geng, S.; Zheng, Q.; Wang, P.; Luo, L.; Wang, X.; Zhang, Y.; Zhang, Y.; He, H.; Tang, X. An intravenous clarithromycin lipid emulsion with a high drug loading, H-bonding and a hydrogen-bonded ion pair complex exhibiting excellent antibacterial activity. Asian J. Pharm.Sci. 2016, 11, 618–630. [Google Scholar] [CrossRef]
- Chen, Z.; Cai, Z.; Zhu, C.; Song, X.; Qin, Y.; Zhu, M.; Zhang, T.; Cui, W.; Tang, H.; Zheng, H. Injectable and self-healing hydrogel with anti-bacterial and anti-inflammatory properties for acute bacterial rhinosinusitis with micro invasive treatment. Adv. Healthc. Mater. 2020, 12, 2001032. [Google Scholar] [CrossRef]
- Panahi, Y.; Gharekhani, A.; Hamishekhar, H.; Milani, P.Z.; Gharekhani, H. Stomach-specific drug delivery of clarithromycin using a semi interpenetrating polymeric network hydrogel made of montmorillonite and chitosan: Synthesis, characterization and in vitro drug release study. Adv. Pharm. Bull. 2019, 9, 159–173. [Google Scholar] [CrossRef]
- Cong, Y.; Geng, J.; Wang, H.; Su, J.; Arif, M.; Dong, Q.; Chi, Z.; Liu, C. Ureido-modified carboxymethyl chitosan-graft-stearic acid polymeric nano-micelles as a targeted delivering carrier of clarithromycin for Helicobacter pylori: Preparation and in vitro evaluation. Int. J. Biol. Macromol. 2019, 129, 686–692. [Google Scholar] [CrossRef]
- Jain, S.K.; Haider, T.; Kumar, A.; Jain, A. Lectin-conjugated clarithromycin and acetohydroxamic acid –loaded PLGA nanoparticles: A novel approach for effective treatment of H. pyroli. AAPS PharmSciTech 2016, 17, 1131–1140. [Google Scholar] [CrossRef] [PubMed]
- Bin-Jumah, M.; Gilani, S.J.; Jahangir, M.A.; Alshehri, S.; Yasir, M.; Kala, C.; Taleuzzaman, M.; Imam, S.S. Clarithromycin-loaded ocular chitosan nanoparticle: Formulation, optimization, characterization, ocular irritation, and antimicrobial activity. Int. J. Nanomed. 2020, 15, 7861–7875. [Google Scholar] [CrossRef] [PubMed]
- Öztürk, A.A.; Aygül, A.; Şenel, B. Influence of glyceryl behenate, tripalmitin and stearic acid on the properties of clarithromycin incorporated solid lipid nanoparticles (SLNs): Formulation, characterization, antibacterial activity and cytotoxicity. J. Drug Deliv. Sci. Technol. 2019, 54, 101240. [Google Scholar] [CrossRef]
- Zaid Alkilani, A.; Musleh, B.; Hamed, R.; Swellmeen, L.; Basheer, H.A. Preparation and characterization of patch loaded with clarithromycin nanovesicles for transdermal drug delivery. J. Funct. Biomater. 2023, 14, 57. [Google Scholar] [CrossRef] [PubMed]
- Ramalingam, P.; Ko, Y.T. Enhanced oral delivery of curcumin from N-trimethyl chitosan surface-modified solid lipid nanoparticles: Pharmacokinetic and brain distribution evaluations. Pharm. Res. 2015, 32, 389–402. [Google Scholar] [CrossRef] [PubMed]
- Beloqui, A.; Solinis, M.A.; Gascon, A.R.; Almeida, A.J.; Preat, V. Nanostructured lipid carriers: Promising drug delivery systems for future clinics. Nanomedicine 2016, 12, 143–161. [Google Scholar] [CrossRef]
- Sharma, M.; Sharma, S.; Wadhwa, J. Improved uptake and therapeutic intervention of curcumin via designing binary lipid nanoparticulate formulation for oral delivery in inflammatory bowel disorder. Artif. Cells Nanomed. Biotechnol. 2019, 47, 45–55. [Google Scholar] [CrossRef]
- Jaiswal, P.; Gidwani, B.; Vyas, A. Nanostructured lipid carriers and their current application in targeted drug delivery. Artif. Cells Nanomed. Biotechnol. 2016, 44, 27–40. [Google Scholar] [CrossRef]
- Baek, J.S.; Cho, C.W. Surface modification of solid lipid nanoparticles for oral delivery of curcumin: Improvement of bioavailability through enhanced cellular uptake, and lymphatic uptake. Eur. J. Pharm. Biopharm. 2017, 117, 132–140. [Google Scholar] [CrossRef]
- Dos Santos Ramos, M.A.; Da Silva, P.B.; Spósito, L.; De Toledo, L.G.; Bonifácio, B.; Rodero, C.F.; Dos Santos, K.C.; Chorilli, M.; Bauab, T.M. Nanotechnology-based drug delivery systems for control of microbial biofilms: A review. Int. J. Nanomed. 2018, 13, 1179–1213. [Google Scholar] [CrossRef]
- Hu, C.; Qian, A.; Wang, Q.; Xu, F.; He, Y.; Xu, J.; Xia, Y.; Xia, Q. Industrialization of lipid nanoparticles: From laboratory-scale to large-scale production line. Eur. J. Pharm. Biopharm. 2016, 109, 206–213. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.K.; Mukerjee, A.; Pandey, H.; Mishra, S.B. Fabrication of solid lipid nanoparticles by hot high shear homogenization and optimization by Box—Behnken design: An accelerated stability assessment. J. Appl. Pharm. Sci. 2021, 11, 035–047. [Google Scholar]
- Sharma, M.; Gupta, N. Mucoadhesive cationic bromelain laden nanocarriers restore patency of airway hyperresponsive remodeling via nasal route. Adv. Ther. 2023, 2200302. [Google Scholar] [CrossRef]
- Sharma, M.; Sharma, R. Implications of designing a bromelain loaded enteric nanoformulation on its stability and anti-inflammatory potential upon oral administration. RSC Adv. 2018, 8, 2541–2551. [Google Scholar] [CrossRef] [PubMed]
- Sharma, M.; Chaudhary, D. In vitro and in vivo implications of rationally designed bromelain laden core-shell hybrid solid lipid nanoparticles for oral administration in thrombosis management. Nanomed. Nanotechnol. Biol. Med. 2022, 42, 102543. [Google Scholar] [CrossRef] [PubMed]
- Aburahma, M.H.; Badr-Eldin, S.M. Compritol 888 ATO: A multifunctional lipid excipient in drug delivery systems and nanopharmaceuticals. Expert Opin. Drug Deliv. 2014, 11, 1865–1883. [Google Scholar] [CrossRef]
- Nair, A.B.; Shah, J.; Al-Dhubiab, B.E.; Jacob, S.; Patel, S.S.; Venugopala, K.N.; Morsy, M.A.; Gupta, S.; Attimarad, M.; Sreeharsha, N.; et al. Clarithromycin solid lipid nanoparticles for topical ocular therapy: Optimization, evaluation and in vivo studies. Pharmaceutics 2021, 13, 523. [Google Scholar] [CrossRef]
- Khalil, R.M.; El-Bary, A.A.; Kassem, M.A.; Ghorab, M.M.; Basha, M. Influence of formulation parameters on the physicochemical properties of meloxicam-loaded solid lipid nanoparticles. Egypt. Pharm. J. 2013, 12, 63–72. [Google Scholar]
- Chokshi, N.V.; Khatri, H.N.; Patel, M.M. Formulation, optimization, and characterization of rifampicin-loaded solid lipid nanoparticles for the treatment of tuberculosis. Drug Dev. Ind. Pharm. 2018, 44, 1975–1989. [Google Scholar] [CrossRef]
- Ojha, S.; Kumar, B. Preparation and statistical modeling of solid lipid nanoparticles of dimethyl fumarate for better management of multiple sclerosis. Adv. Pharm. Bull. 2018, 8, 225–233. [Google Scholar] [CrossRef]
- Sharma, M.; Sharma, V.; Panda, A.K.; Majumdar, D.K. Development of enteric submicron particles formulation of α-amylase for oral delivery. Pharm. Dev. Technol. 2013, 18, 560–569. [Google Scholar] [CrossRef] [PubMed]
- Rao, N.N.M.; Sharma, S.; Kaduba, P.; Sadhu, V.; Sharma, M.; Sainath, A.V.S. Rationally designed curcumin laden glycopolymeric nanoparticles: Implications on cellular uptake and anticancer efficacy. J. Appl. Polym. Sci. 2020, 137, 48954. [Google Scholar] [CrossRef]
- Sharaf, M.; Hamouda, H.I.; Shabana, S.; Khan, S.; Arif, M.; Rozan, H.E.; Abdalla, M.; Chi, Z.; Liu, C. Design of lipid-based nanocarrier for drug delivery has a double therapy for six common pathogens eradication. Colloids Surf. A Physicochem. Eng. Asp. 2021, 625, 126662. [Google Scholar] [CrossRef]
- Solleti, V.S.; Alhariri, M.; Halwani, M.; Omri, A. Antimicrobial properties of liposomal azithromycin for Pseudomonas infections in cystic fibrosis patients. J. Antimicrob. Chemother. 2015, 70, 784–796. [Google Scholar] [CrossRef]
- Moreno-Sastre, M.; Pastor, M.; Esquisabel, A.; Sans, E.; Viñas, M.; Fleischer, A.; Palomino, E.; Bachiller, D.; Pedraz, J.L. Pulmonary delivery of tobramycin-loaded nanostructured lipid carriers for Pseudomonas aeruginosa infections associated with cystic fibrosis. Int. J. Pharm. 2016, 498, 263–273. [Google Scholar] [CrossRef]
- Xie, S.; Tao, Y.; Pan, Y.; Qu, W.; Cheng, G.; Huang, L.; Chen, D.; Wang, X.; Liu, Z.; Yuan, Z. Biodegradable nanoparticles for intracellular delivery of antimicrobial agents. J. Control. Release. 2014, 187, 101–117. [Google Scholar] [CrossRef]
- Shukla, P.; Dwivedi, P.; Gupta, P.K.; Mishra, P.R. Optimization of novel tocopheryl acetate nanoemulsions for parenteral delivery of curcumin for therapeutic intervention of sepsis. Expert Opin. Drug Deliv. 2014, 11, 1697–1712. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, S.; Jiang, H.; Sun, A.; Wang, Y.; Zou, Y.; Ge, J.; Chen, H. Effects of statin therapy on inflammatory markers in chronic heart failure: A meta-analysis of randomized controlled trials. Arch. Med. Res. 2010, 41, 464–471. [Google Scholar] [CrossRef]
- Singhal, A.; Aliouat, E.M.; Hervé, M.; Mathys, V.; Kiass, M.; Creusy, C.; Delaire, B.; Tsenova, L.; Fleurisse, L.; Bertout, J.; et al. Experimental tuberculosis in the wistar rat: A model for protective immunity and control of infection. PLoS ONE 2011, 6, e18632. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| F. Code | Compritol (mg) | Stearic Acid (mg) | PF-68 (% w/v) | Drug (mg) | Sonication Time (min) | External Phase Volume (mL) |
|---|---|---|---|---|---|---|
| F1 | 100 | 200 | 0.5 | 20 | 10 | 25 |
| F2 | 100 | 100 | 0.5 | 20 | 10 | 25 |
| F3 | 200 | 100 | 0.5 | 20 | 10 | 25 |
| F4 | 100 | 100 | 0.5 | 20 | 10 | 50 |
| F5 | 100 | 100 | 0.5 | 10 | 10 | 25 |
| F6 | 100 | 100 | 0.5 | 30 | 10 | 25 |
| F7 | 100 | 100 | 0.25 | 20 | 10 | 25 |
| F8 | 100 | 100 | 1.0 | 20 | 10 | 25 |
| F9 | 100 | 100 | 0.5 | 20 | 7 | 25 |
| F10 | 100 | 100 | 0.5 | 20 | 15 | 25 |
| F. Code | Particle Size (nm ± SD) | PDI (PDI ± SD) | Zeta Potential (mV ± SD) | Entrapment Efficiency (% ± SD) |
|---|---|---|---|---|
| F1 | 316.21 ± 36.97 | 0.24 ± 0.02 | −26.17 ± 2.14 | 74.87 ± 3.27 |
| F2 | 326.19 ± 24.14 | 0.22 ± 0.04 | −31.34 ± 2.87 | 85.78 ± 4.01 |
| F3 | 424.54 ± 22.74 | 0.38 ± 0.03 | −25.62 ± 2.25 | 81.03 ± 3.38 |
| F4 | 506.17 ± 27.34 | 0.43 ± 0.02 | −17.85 ± 2.14 | 65.35 ± 6.45 |
| F5 | 315.34 ± 24.15 | 0.31 ± 0.03 | −23.28 ± 2.94 | 75.27 ± 7.14 |
| F6 | 392.43 ± 27.27 | 0.46 ± 0.04 | −22.14 ± 2.15 | 77.31 ± 6.25 |
| F7 | 501.14 ± 29.64 | 0.52 ± 0.03 | −20.32 ± 1.96 | 75.78 ± 5.56 |
| F8 | 329.29 ± 25.41 | 0.36± 0.04 | −20.87 ± 2.83 | 71.94 ± 4.87 |
| F9 | 579.24 ± 28.87 | 0.68 ± 0.04 | −17.81 ± 2.19 | 61.64 ± 6.47 |
| F10 | 464.42 ± 25.54 | 0.47 ± 0.03 | −20.37 ± 3.15 | 71.01 ± 5.46 |
| Parameter | Clarithromycin | Formulation |
|---|---|---|
| Cmax (ng mL−1) | 405.59 ± 64.89 | 942.36 ± 77.71 |
| Tmax (h) | 1.00 ± 0.11 | 2.00 ± 0.13 |
| Ke (h−1) | 0.38 ± 0.07 | 0.12 ± 0.04 |
| t1/2 (h) | 1.38 ± 0.42 | 3.96 ± 0.67 |
| MRT (h) | 2.01 ± 0.53 | 3.69 ± 0.69 |
| AUC (ngh2mL−1) | 1058.67 ± 124.57 | 6278.47 ± 283.65 |
| Treatment | CFU Count (Mean ± SD) | |||
|---|---|---|---|---|
| Blood | Lung | Liver | Spleen | |
| E. coli (1 × 1010) (+ve control) | 5.16 ± 0.84 × 1010 | 6.89 ± 0.56 × 108 | 5.45 ± 0.25 × 109 | 3.04 ± 0.75 × 108 |
| Pure drug (5 mg/kg) | 4.17 ± 0.94 × 107 | 5.65 ± 0.47 × 107 | 5.27 ± 0.53 × 107 | 1.69 ± 0.64 × 106 |
| CLA-DLCs (equivalent to 5 mg/kg) | 3.28 ± 0.36 × 103 | 4.45 ± 0.18 × 103 | 5.62 ± 0.24 × 103 | 2.14 ± 0.79 × 103 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rawat, R.; Chouhan, R.S.; Sadhu, V.; Sharma, M. Clarithromycin-Loaded Submicron-Sized Carriers: Pharmacokinetics and Pharmacodynamic Evaluation. Materials 2023, 16, 3593. https://doi.org/10.3390/ma16093593
Rawat R, Chouhan RS, Sadhu V, Sharma M. Clarithromycin-Loaded Submicron-Sized Carriers: Pharmacokinetics and Pharmacodynamic Evaluation. Materials. 2023; 16(9):3593. https://doi.org/10.3390/ma16093593
Chicago/Turabian StyleRawat, Reetika, Raghuraj Singh Chouhan, Veera Sadhu, and Manu Sharma. 2023. "Clarithromycin-Loaded Submicron-Sized Carriers: Pharmacokinetics and Pharmacodynamic Evaluation" Materials 16, no. 9: 3593. https://doi.org/10.3390/ma16093593