
Numerical Studies of the Impact of Electromagnetic Field of Radiation on Valine

Abstract
:1. Introduction
2. Materials and Methods
2.1. Atoms/Molecules in Electromagnetic Fields
2.2. Anisotropic Gaussian-Type Orbital Basis Sets for Atoms in Intermediate Magnetic Fields
2.3. AGTO Construction Scheme
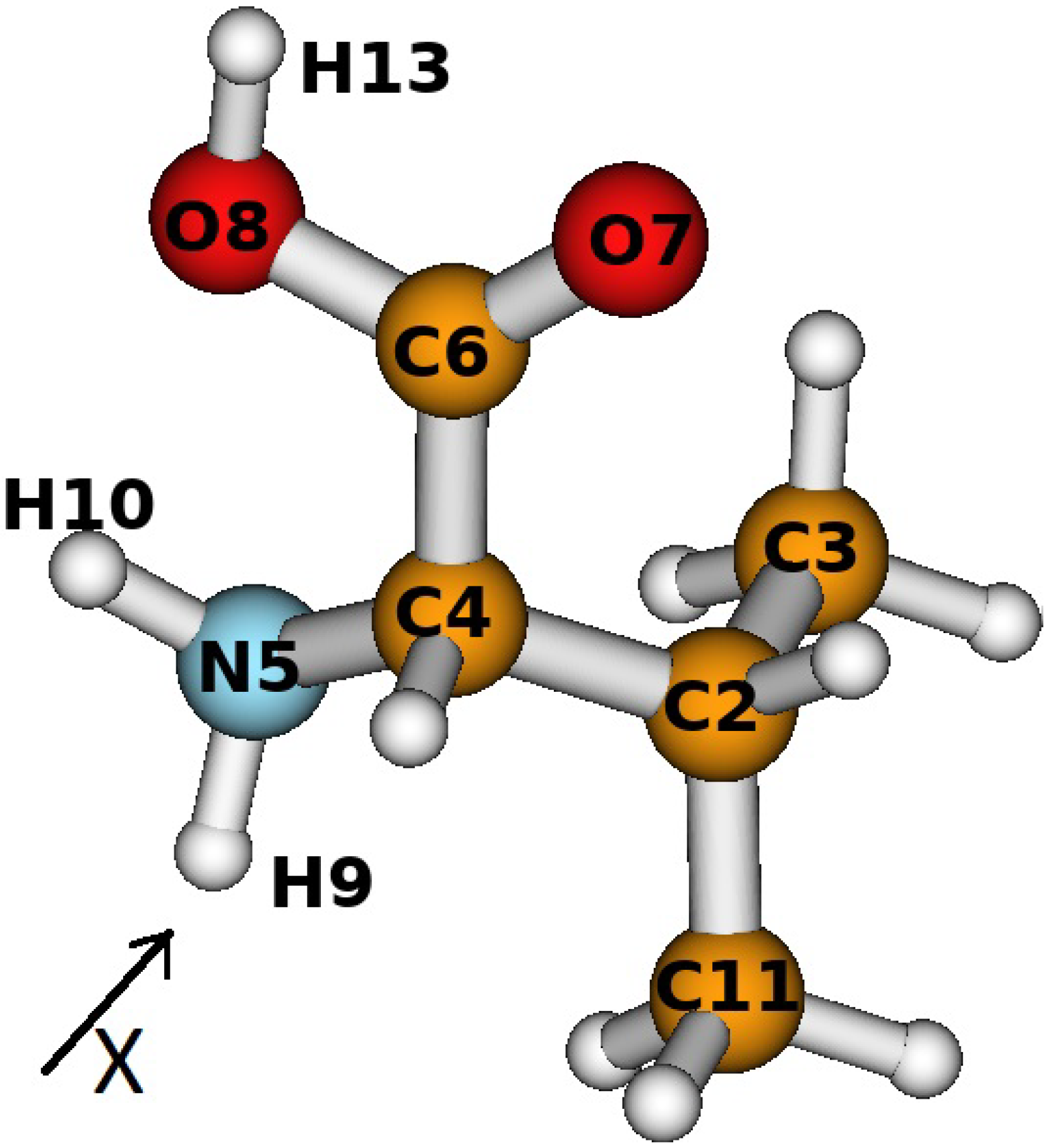
3. Results
- no new peaks were observed in the 5 and 20 kGy spectra, in comparison to the one measured without interaction with a high-energy electron;
- the intensities of the peaks, with respect to that of the m/z = 72 fragment (corresponding to ), significantly changed;
- the peak intensity of the m/z = 27–29 fragments decreased differently with increase of the irradiation dose;
- the intensity of the m/z = 45 fragment increased, because the molecule joined the H atoms formed due to irradiation, whereas the m/z = 55 and 56 fragments resulted from the decomposition of not only the parent valine molecule, but also the or fragments formed under irradiation.
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| EM | electromagnetic |
| AGTO | anisotropic Gaussian-type orbital |
| KL | Kravchenko and Liberman |
References
- Farajmand, B.; Bahrami, H. Electron ionization of serine and threonine: A discussion about peak intensities. Phys. Chem. Res. 2016, 4, 539–551. [Google Scholar]
- Ros, R.; Muñoz-Bertomeu, J.; Krueger, S. Serine in plants: Biosynthesis, metabolism, and functions. Trends Plant Sci. 2014, 19, 564–569. [Google Scholar] [CrossRef]
- Tabatabaie, L.; Klomp, L.W.; Berger, R.; de Koning, T.J. L-Serine synthesis in the central nervous system: A review on serine deficiency disorders. Mol. Genet. Metab. 2010, 99, 256–262. [Google Scholar] [CrossRef]
- Savelieva, K.V.; Zhao, S.; Pogorelov, V.M.; Rajan, I.; Yang, Q.; Cullinan, E.; Lanthorn, T.H. Genetic disruption of both tryptophan hydroxylase genes dramatically reduces serotonin and affects behavior in models sensitive to antidepressants. PLoS ONE 2008, 10, e3301. [Google Scholar] [CrossRef] [Green Version]
- Hankes, L.V.; Brown, R.R.; Leklem, J.; Schmaeler, M.; Jesseph, J. Metabolism of C14 Labeled Enantiomers of Tryptophan, Kynurenine and Hydroxykynurenine in Humans with Scleroderma. J. Investig. Dermatol. 1972, 58, 85–95. [Google Scholar] [CrossRef] [Green Version]
- Abdoul-Carime, H.; Gohlke, S.; Illenberger, E. Fragmentation of tryptophan by low-energy electrons. Chem. Phys. Lett. 2005, 402, 497–502. [Google Scholar] [CrossRef]
- Fuciarelli, A.F.; Zimbrick, J.D. Radiation Damage in DNA: Structure/Function Relationship at Early Times; Battelle Press: Columbus, OH, USA, 1995. [Google Scholar]
- Papp, P.; Shchukin, P.; Kočíšek, J.; Matejčík, Š. Electron ionization and dissociation of aliphatic amino acids. J. Chem. Phys. 2012, 137, 105101. [Google Scholar] [CrossRef]
- Tamuliene, J.; Romanova, L.G.; Vukstich, V.S.; Snegursky, A.V. Electron-impact and thermal fragmentation of amino acid molecules: Mechanisms and structure of the molecules. Nucl. Instrum. Methods Phys. Pes. Sect. B 2012, 279, 128–134. [Google Scholar] [CrossRef]
- Borkowski, E.J.; Cecati, F.M.; Suvire, F.D.; Ruiz, D.M.; Ardanaz, C.E.; Romanelli, G.P.; Enriz, R.D. Mass spectrometry and theoretical calculations about the loss of methyl radical from methoxilated coumarins. J. Mol. Struc. 2015, 1093, 49–58. [Google Scholar] [CrossRef]
- Grimme, S. Towards first principles calculation of electron impact mass spectra of molecules. Angew. Chem. Int. Ed. 2013, 52, 6306–6312. [Google Scholar] [CrossRef]
- Tamulienė, J.; Romanova, L.; Vukstich, V.; Snegursky, A. High-energy electron impact influence on the amino acid fragmentation. In Horizons in World Physics; Reimer, A., Ed.; Nova Science Publishers: New York, NY, USA, 2021; Volume 305, pp. 1–92. [Google Scholar]
- Schmelcher, P.; Cederbaum, L.S. Molecules in strong magnetic fields: Properties of atomic orbitals. Phys. Rev. A 1988, 37, 672–681. [Google Scholar] [CrossRef]
- Kravchenko, Y.P.; Liberman, M.A. Application of Gaussian-type basis sets to ab initio calculations in strong magnetic fields. Int. J. Quantum Chem. 1988, 64, 513–522. [Google Scholar] [CrossRef]
- Zhu, W.; Zhang, L.; Trickey, S.B. Comparative studies of density-functional approximations for light atoms in strong magnetic field. Phys. Rev. A 2014, 90, 022504. [Google Scholar] [CrossRef] [Green Version]
- Zhu, W.; Trickey, S.B. Accurate and balanced anisotropic Gaussian type orbital basis sets for atoms in strong magnetic fields. J. Chem. Phys. 2017, 147, 244108. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, M.W.; Ruedenberg, K. Effective convergence to complete orbital bases and to the atomic Hartree–Fock limit through systematic sequences of Gaussian primitives. J. Chem. Phys. 1979, 71, 3951. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. I. The effect of the exchange-only gradient correction. J. Chem. Phys. 1992, 96, 2155–2160. [Google Scholar] [CrossRef] [Green Version]
- Becke, A.D. Density-functional thermochemistry. II. The effect of the Perdew–Wang generalized-gradient correlation correction. J. Chem. Phys. 1992, 97, 9173–9177. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. II. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revis. A.02; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]


{kind=link}
| Bond Length, A | ||||
|---|---|---|---|---|
| Bond | Valine | Valine in E Field | Valine in EM Field (s, p Orbitals) | Valine in EM Field (p Orbitals) |
| C2-C11 | 1.53 | 1.54 | 1.53 | 1.53 |
| C2-C3 | 1.53 | 1.53 | 1.53 | 1.53 |
| C2-C4 | 1.55 | 1.55 | 1.55 | 1.54 |
| C4-C6 | 1.52 | 1.56 | 1.55 | 1.55 |
| C4-N5 | 1.46 | 1.47 | 1.47 | 1.47 |
| C6-O7 | 1.20 | 1.21 | 1.20 | 1.20 |
| C6-O8 | 1.36 | 1.31 | 1.31 | 1.31 |
| N5-H9 | 1.01 | 1.02 | 1.02 | 1.02 |
| N5-H10 | 1.01 | 1.02 | 1.02 | 1.02 |
| O8-H13 | 0.97 | 0.98 | 0.98 | 0.98 |
| Condense-to-Atom-All Electrons (Bond Order) | ||||
| Bond | Valine | Valine in E Field | Valine in EM Field (s, p Orbitals) | Valine in EM Field (p Orbitals) |
| C2-C11 | 0.680 | 0.416 | 0.418 | 0.418 |
| C2-C3 | 0.638 | 0.642 | 0.642 | 0.642 |
| C2-C4 | 0.554 | 0.300 | 0.296 | 0.296 |
| C4-C6 | 0.442 | 0.586 | 0.576 | 0.576 |
| C4-N5 | 0.530 | 0.502 | 0.492 | 0.492 |
| C6-O7 | 1.566 | 1.458 | 1.468 | 1.468 |
| C6-O8 | 0.898 | 1.008 | 1.008 | 1.008 |
| N5-H9 | 0.718 | 0.744 | 0.742 | 0.742 |
| N5-H10 | 0.716 | 0.772 | 0.778 | 0.778 |
| O8-H13 | 0.630 | 0.668 | 0.672 | 0.672 |
| Bond Angle Degree, Degrees | ||||
| Angle | Valine | Valine in E Field | Valine in EM Field (s, p Orbitals) | Valine in EM Field (p Orbitals) |
| C3-C2-C11 | 111.333 | 110.914 | 111.032 | 111.383 |
| C11-C2-C4 | 110.463 | 110.737 | 110.068 | 110.489 |
| C3-C2-C4 | 112.481 | 112.614 | 112.325 | 112.183 |
| C2-C4-C6 | 110.862 | 109.778 | 110.093 | 109.046 |
| C2-C4-N5 | 111.925 | 111.760 | 111.162 | 111.967 |
| C4-C6-O7 | 154.415 | 121.109 | 121.195 | 120.897 |
| C4-C6-O8 | 113.037 | 110.454 | 110.108 | 109.871 |
| O7-C6-O8 | 121.984 | 128.428 | 128.694 | 129.232 |
| C6-O8-H13 | 106.546 | 114.158 | 113.803 | 112.937 |
| C4-N5-H10 | 111.020 | 108.986 | 108.335 | 107.031 |
| C4-N5-H9 | 110.244 | 107.918 | 107.447 | 112.937 |
| Dihedral Angle Degree, Degrees | ||||
| Angle | Valine | Valine in E field | Valine in EM Field (s, p Orbitals) | Valine in EM Field (p Orbitals) |
| C3-C2-C11-H17 | 60.321 | 51.561 | 50.052 | 49.582 |
| C11-C2-C4-N5 | −64.581 | −65.939 | −64.983 | −64.276 |
| C3-C2-C4-N5 | 60.493 | 58.895 | 59.284 | 60.662 |
| C2-C4-N5-H9 | 73.831 | 158.145 | 158.979 | 160.783 |
| C2-C4-C6-O7 | −34.575 | −10.922 | −6.738 | −15.142 |
| C2-C4-C6-O8 | 148.704 | 170.052 | 173.869 | 164.766 |
| C4-C6-O8-H13 | 177.167 | 176.189 | 177.042 | 176.120 |
| H9-N5-C4-C2 | 73.831 | 158.147 | 158.979 | 160.783 |
| H10-N5-C4-C2 | −166.996 | −91.199 | −91.500 | −93.784 |
| Dipole Moment, Debye | x | y | z | Total |
|---|---|---|---|---|
| Valine | −1.09 | 1.064 | −1.314 | 2.0097 |
| Valine in E field | −8.833 | 1.630 | 0.191 | 8.985 |
| Valine in EM field (s, p orbitals) | −8.872 | 1.629 | 0.146 | 9.022 |
| Valine in EM field (p orbitals) | −8.915 | 1.503 | 0.194 | 9.043 |
| E Field, a.u. | Valine in E Field, Energy, a.u. | Valine in EM Field (s, p Orbitals), Energy, a.u. | Valine in EM Field (p Orbitals), Energy, a.u. |
|---|---|---|---|
| 0.3 | stable | stable | stable |
| −402.586 | -402.562 | −402.546 | |
| 0.4 | unstable | unstable | unstable |
| −402.629 | −402.600 | −402.586 | |
| 0.5 | unstable | unstable | CO, , H |
| −402.662 | −402.650 | ||
| 0.6 | CO, H | CO, H | CHO, , H |
| CO, H | |||
| 0.7 | C, 2O, H | CO, , H |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kirova, T.; Tamuliene, J. Numerical Studies of the Impact of Electromagnetic Field of Radiation on Valine. Materials 2023, 16, 1814. https://doi.org/10.3390/ma16051814
Kirova T, Tamuliene J. Numerical Studies of the Impact of Electromagnetic Field of Radiation on Valine. Materials. 2023; 16(5):1814. https://doi.org/10.3390/ma16051814
Chicago/Turabian StyleKirova, Teodora, and Jelena Tamuliene. 2023. "Numerical Studies of the Impact of Electromagnetic Field of Radiation on Valine" Materials 16, no. 5: 1814. https://doi.org/10.3390/ma16051814