
Viscosity Measurement of CO2–Solvent Mixtures for the Study of the Morphology and Size of Crystalline Particles Obtained Using Supercritical Antisolvent Precipitation

Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
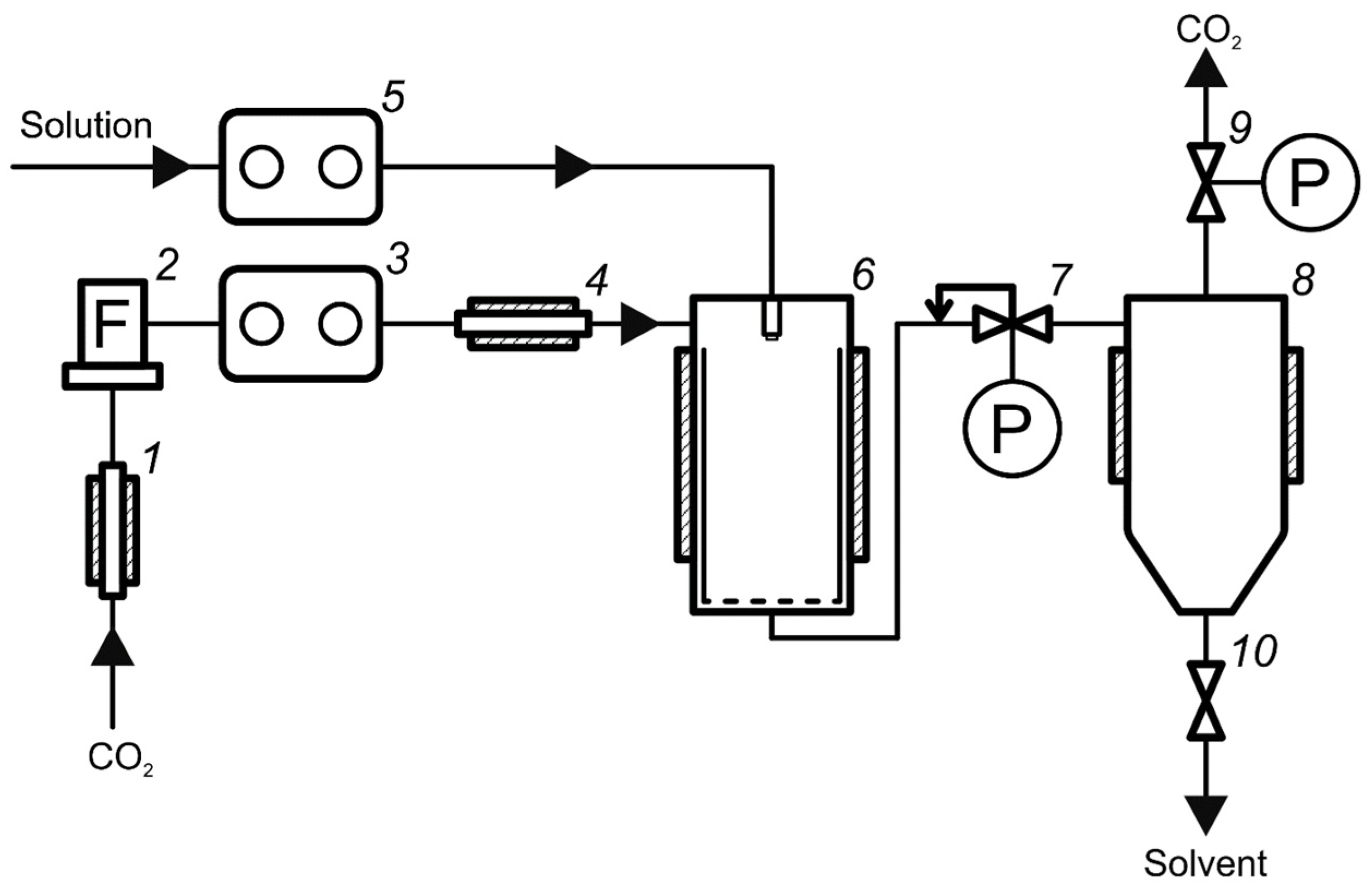
2.2.1. Installations for Viscosity and Density Measurements
- CO2 and co-solvent pump Acquity ccBSM;
- Back pressure regulator Acquity Convergence Manager.

2.2.2. Measurement of the Mixture Density
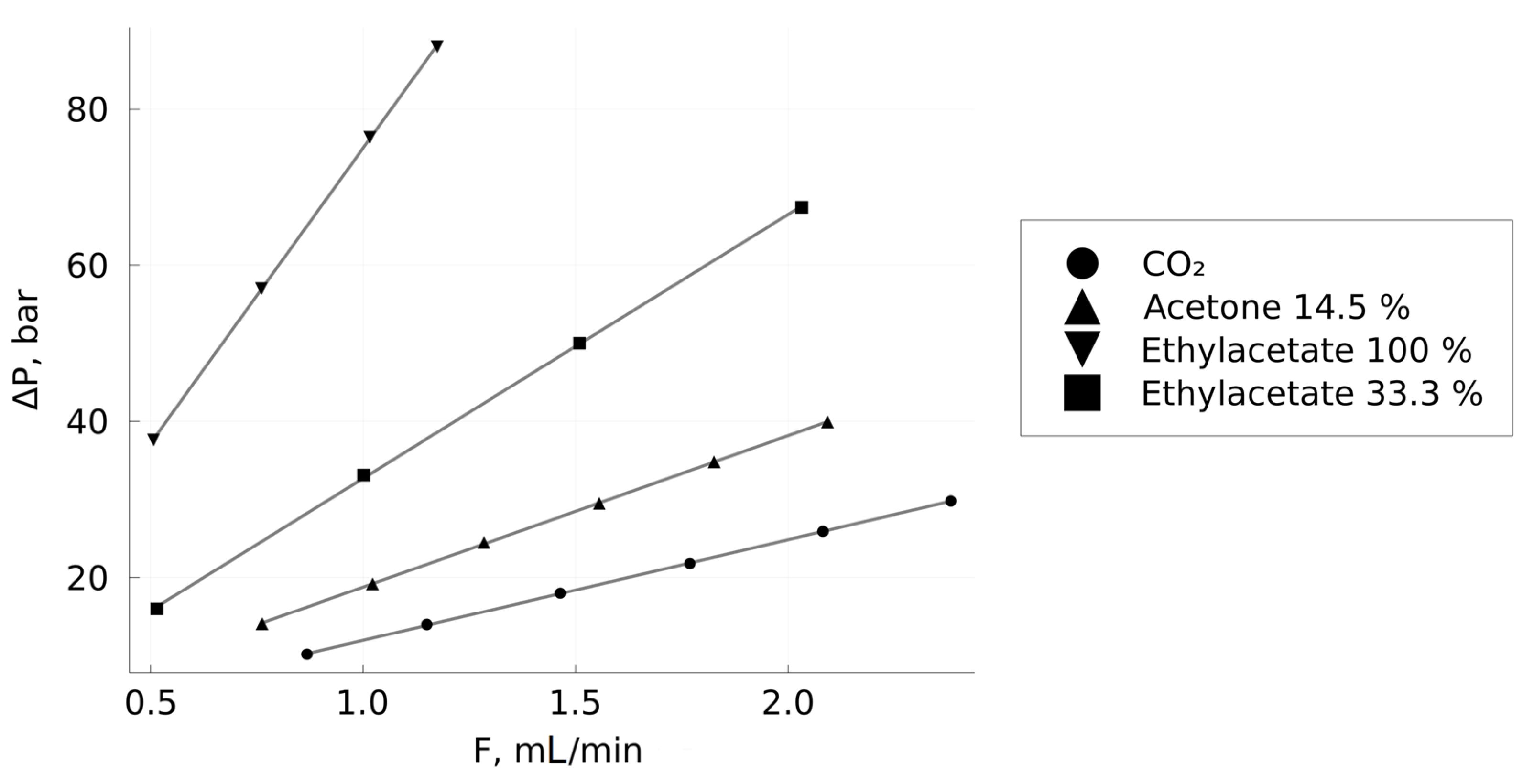
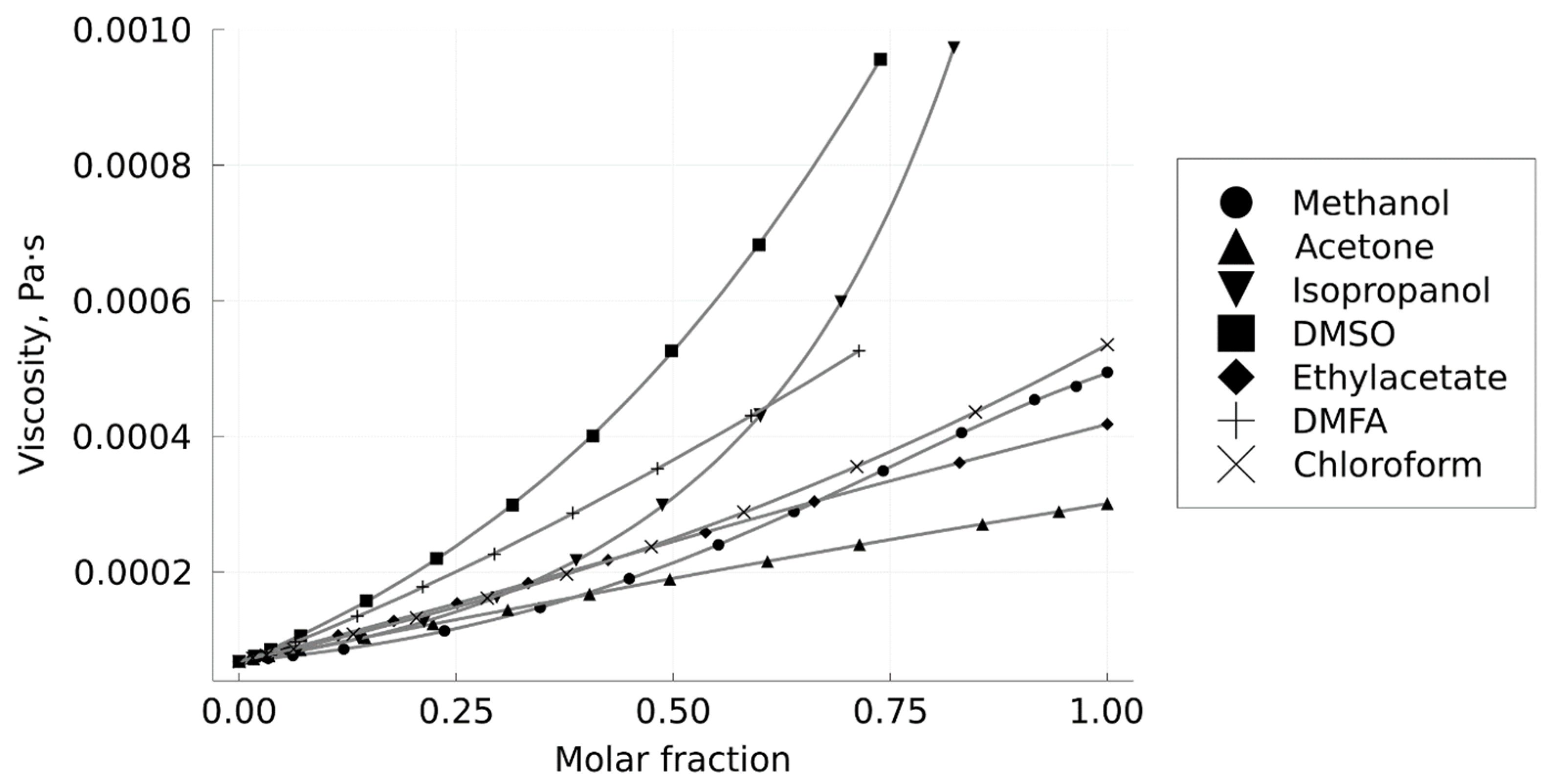
2.2.3. Measurement of the Mixtures Viscosity
- The liquid thermostat was turned on and the temperature value was set to 313 K. The chromatograph was prepared: the pump block and the back pressure regulator were turned on and the liquid pump was washed with the selected solvent.
- The flow meter was connected to position 3a (Figure 1) and the system was washed with a stream of pure CO2.
- Using the control program, the volume content of the solvent in the mixture was varied in the range of 0–100% with a step of 2.5–10%. At each step, the system was maintained for about 15 min to enter the stationary mode (constant readings of pressure sensors at the ends of the column). Then, the back pressure at the outlet of the system was manually adjusted to obtain an average pressure in the column of 150 bar; the values of liquid solvent mass flow and column pressure drop were recorded.
- The system was stopped, the flow meter was switched to position 3b (Figure 1), and the measurements were repeated similarly to point 3, but the values of the mixture total flow in the system were recorded.
2.2.4. Supercritical Antisolvent Precipitation
2.2.5. Scanning Electron Microscopy
3. Results and Discussion
3.1. Density and Viscosity of CO2–Solvent Mixtures
3.2. Size and Morphology of Micronized Particles
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Reverchon, E. Supercritical antisolvent precipitation of micro-and nano-particles. J. Supercrit. Fluids 1999, 15, 1–21. [Google Scholar] [CrossRef]
- Clercq, S.; Mouahid, A.; Gérard, P.; Badens, E. Investigation of crystallization mechanisms for polymorphic and habit control from the Supercritical AntiSolvent process. J. Supercrit. Fluids 2018, 141, 29–38. [Google Scholar] [CrossRef]
- Franco, P.; De Marco, I. Supercritical antisolvent process for pharmaceutical applications: A review. Processes 2020, 8, 938. [Google Scholar] [CrossRef]
- Liu, G.; Li, J.; Deng, S. Applications of Supercritical Anti-Solvent Process in Preparation of Solid Multicomponent Systems. Pharmaceutics 2021, 13, 475. [Google Scholar] [CrossRef]
- Vorobei, A.M.; Fedorovskiy, A.G.; Kostenko, M.O.; Zuev, Y.I.; Parenago, O.O. Micronization of Levofloxacin Hydrochloride by the Supercritical Antisolvent Precipitation Method from Single-Phase and Two-Phase CO2–Acetone Mixtures. Russ. J. Phys. Chem. B 2022, 16, 1416–1421. [Google Scholar] [CrossRef]
- Nesterov, N.S.; Paharukova, V.P.; Yakovlev, V.A.; Martyanov, O.N. The facile synthesis of Ni–Cu catalysts stabilized in SiO2 framework via a supercritical antisolvent approach. J. Supercrit. Fluids 2016, 112, 119–127. [Google Scholar] [CrossRef]
- Nesterov, N.S.; Shalygin, A.S.; Pakharukova, V.P.; Glazneva, T.S.; Martyanov, O.N. Mesoporous aerogel-like Al-Si oxides obtained via supercritical antisolvent precipitation of alumina and silica sols. J. Supercrit. Fluids 2019, 149, 110–119. [Google Scholar] [CrossRef]
- Gavrikov, A.V.; Loktev, A.S.; Ilyukhin, A.B.; Mukhin, I.E.; Bykov, M.A.; Vorobei, A.M.; Parenago, O.O.; Cherednichenko, K.A.; Sadovnikov, A.A.; Dedov, A.G. Partial oxidation of methane to syngas over SmCoO3-derived catalysts: The effect of the supercritical fluid assisted modification of the perovskite precursor. Int. J. Hydrogen Energy 2022, 48, 2998–3012. [Google Scholar] [CrossRef]
- Gavrikov, A.V.; Loktev, A.S.; Ilyukhin, A.; Mukhin, I.E.; Bykov, M.A.; Maslakov, K.I.; Vorobei, A.M.; Parenago, O.O.; Sadovnikov, A.; Dedov, A.G. Supercritical fluid-assisted modification combined with the resynthesis of SmCoO3 as an effective tool to enhance the long-term performance of SmCoO3-derived catalysts for the dry reforming of methane to syngas. Dalton Trans. 2022, 51, 18446–18461. [Google Scholar] [CrossRef]
- Quintana, S.E.; Villanueva-Bermejo, D.; Reglero, G.; García-Risco, M.R.; Fornari, T. Supercritical antisolvent particle precipitation and fractionation of rosemary (Rosmarinus officinalis L.) extracts. J. CO2 Util. 2019, 34, 479–489. [Google Scholar] [CrossRef]
- Villalva, M.; Jaime, L.; Villanueva-Bermejo, D.; Lara, B.; Fornari, T.; Reglero, G.; Santoyo, S. Supercritical anti-solvent fractionation for improving antioxidant and anti-inflammatory activities of an Achillea millefolium L. extract. Food Res. Int. 2019, 115, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Pokrovskiy, O.; Vorobei, A.; Zuev, Y.; Kostenko, M.; Lunin, V. Investigation of precipitation selectivity and particle size concentration dependences in supercritical antisolvent method via online supercritical fluid chromatography. Adv. Powder Technol. 2020, 31, 2257–2266. [Google Scholar] [CrossRef]
- Vorobei, A.M.; Parenago, O.O. Using supercritical fluid technologies to prepare micro-and nanoparticles. Russ. J. Phys. Chem. 2021, 95, 407–417. [Google Scholar] [CrossRef]
- Reverchon, E.; De Marco, I. Mechanisms controlling supercritical antisolvent precipitate morphology. Chem. Eng. J. 2011, 169, 358–370. [Google Scholar] [CrossRef]
- Reverchon, E.; Torino, E.; Dowy, S.; Braeuer, A.; Leipertz, A. Interactions of phase equilibria, jet fluid dynamics and mass transfer during supercritical antisolvent micronization. Chem. Eng. J. 2010, 156, 446–458. [Google Scholar] [CrossRef]
- De Marco, I.; Knauer, O.; Cice, F.; Braeuer, A.; Reverchon, E. Interactions of phase equilibria, jet fluid dynamics and mass transfer during supercritical antisolvent micronization: The influence of solvents. Chem. Eng. J. 2012, 203, 71–80. [Google Scholar] [CrossRef]
- Shekunov, B.Y.; Baldyga, J.; York, P. Particle formation by mixing with supercritical antisolvent at high Reynolds numbers. Chem. Eng. Sci. 2001, 56, 2421–2433. [Google Scholar] [CrossRef]
- Tenorio, A.; Jaeger, P.; Gordillo, M.D.; Pereyra, C.M.; de la Ossa, E.J.M. On the selection of limiting hydrodynamic conditions for the supercritical antisolvent (SAS) process. Ind. Eng. Chem. Res. 2009, 48, 9224–9232. [Google Scholar] [CrossRef]
- Martın, A.; Cocero, M.J. Numerical modeling of jet hydrodynamics, mass transfer, and crystallization kinetics in the supercritical antisolvent (SAS) process. J. Supercrit. Fluids 2004, 32, 203–219. [Google Scholar] [CrossRef]
- Clercq, S.; Mouahid, A.; Pèpe, G.; Badens, E. Prediction of crystal–solvent interactions in a supercritical medium: A possible way to control crystal habit at high supersaturations with molecular modeling. Cryst. Growth Des. 2020, 20, 6863–6876. [Google Scholar] [CrossRef]
- Reverchon, E.; Adami, R.; Caputo, G.; De Marco, I. Spherical microparticles production by supercritical antisolvent precipitation: Interpretation of results. J. Supercrit. Fluids 2008, 47, 70–84. [Google Scholar] [CrossRef]
- Rossmann, M.; Braeuer, A.; Leipertz, A.; Schluecker, E. Manipulating the size, the morphology and the polymorphism of acetaminophen using supercritical antisolvent (SAS) precipitation. J. Supercrit. Fluids 2013, 82, 230–237. [Google Scholar] [CrossRef]
- Rossmann, M.; Braeuer, A.; Dowy, S.; Gallinger, T.G.; Leipertz, A.; Schluecker, E. Solute solubility as criterion for the appearance of amorphous particle precipitation or crystallization in the supercritical antisolvent (SAS) process. J. Supercrit. Fluids 2012, 66, 350–358. [Google Scholar] [CrossRef]
- Rodrigues, M.A.; Tiago, J.M.; Duarte, A.; Geraldes, V.; Matos, H.A.; Gomes Azevedo, E. Polymorphism in pharmaceutical drugs by supercritical CO2 processing: Clarifying the role of the antisolvent effect and atomization enhancement. Cryst. Growth Des. 2016, 16, 6222–6229. [Google Scholar] [CrossRef]
- Careno, S.; Boutin, O.; Badens, E. Drug recrystallization using supercritical anti-solvent (SAS) process with impinging jets: Effect of process parameters. J. Cryst. Growth 2012, 342, 34–41. [Google Scholar] [CrossRef]
- Weber Brun, G.; Martín, Á.; Cassel, E.; Vargas, R.M.F.; Cocero, M.J. Crystallization of caffeine by supercritical antisolvent (SAS) process: Analysis of process parameters and control of polymorphism. Cryst. Growth Des. 2012, 12, 1943–1951. [Google Scholar] [CrossRef]
- Brittain, H.G. Polymorphism in Pharmaceutical Solids; CRC Press: Boca Raton, FL, USA, 2016; 654p. [Google Scholar]
- Perlovich, G.L. Thermodynamic characteristics of cocrystal formation and melting points for rational design of pharmaceutical two-component systems. CrystEngComm 2015, 17, 7019–7028. [Google Scholar] [CrossRef]
- Zuev, Y.I.; Vorobei, A.M.; Gavrikov, A.V.; Parenago, O.O. Micronization of Adipic Acid via Supercritical Antisolvent Precipitation. Russ. J. Phys. Chem. B 2022, 16, 1242–1252. [Google Scholar] [CrossRef]
- Fields, P.R.; Chester, T.L.; Stalcup, A.M. Viscosity estimation in binary and ternary supercritical fluid mixtures containing carbon dioxide using a supercritical fluid chromatograph. J. Liq. Chromatogr. Relat. Technol. 2011, 34, 995–1003. [Google Scholar] [CrossRef]
- Farkas, T.; Zhong, G.; Guiochon, G. Validity of Darcy’s law at low flow-rates in liquid chromatography. J. Chromatogr. A 1999, 849, 35. [Google Scholar] [CrossRef]
- De Pauw, R.; Choikhet, K.; Desmet, G.; Broeckhoven, K. Occurrence of turbulent flow conditions in supercritical fluid chromatography. J. Chromatogr. A 2014, 1361, 277–285. [Google Scholar] [CrossRef] [PubMed]
- Peper, S.; Fonseca, J.M.S.; Dohrn, R. High-pressure fluid-phase equilibria: Trends, recent developments, and systems investigated (2009–2012). Fluid Phase Equilibria 2019, 484, 126–224. [Google Scholar] [CrossRef]
- Fonseca, J.M.S.; Dohrn, R.; Peper, S. High-pressure fluid-phase equilibria: Experimental methods and systems investigated (2005–2008). Fluid Phase Equilibria 2011, 300, 1–69. [Google Scholar] [CrossRef]
- Dohrn, R.; Peper, S.; Fonseca, J.M.S. High-pressure fluid-phase equilibria: Experimental methods and systems investigated (2000–2004). Fluid Phase Equilibria 2010, 288, 1–54. [Google Scholar] [CrossRef]
- Lemon, E.W.; McLinden, M.O.; Friend, D.G.; Linstrom, P.J.; Mallard, W.G. Thermophysical properties of fluid systems. In NIST Chemistry Webbook; NIST Standart Reference Database No. 69; National Institute of Standarts and Technology: Gaithersburg, MD, USA, 2017. [Google Scholar]
- Zuev, Y.I.; Vorobei, A.M.; Parenago, O.O. Russ. J. Preparation of Polyurethane–Carbon Nanotube Composites via Suspension Supercritical Antisolvent Precipitation. Phys. Chem. B 2021, 15, 1107–1112. [Google Scholar]
- Vorobei, A.M.; Pokrovskiy, O.I.; Ustinovich, K.B.; Parenago, O.O.; Lunin, V.V.; Miroshnichenko, A.G. Micronization of salbutamol sulfate by supercritical antisolvent precipitation: The effect of process parameters on the size and morphology of particles. Russ. J. Phys. Chem. B 2018, 12, 1240–1248. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Solvent | Mean Particle Size, µm | Aspect Ratio |
|---|---|---|
| DMF | 31 ± 2 | 3.1 |
| chloroform | 2.8 ± 0.2 | 1.6 |
| methanol | 18 ± 1 | 6.1 |
| ethyl acetate | 1.6 ± 0.1 | 1.2 |
| acetone | 3.3 ± 0.2 | 1.5 |
| Solvent | Mean Particle Size, µm | Aspect Ratio |
|---|---|---|
| DMSO | 6.7 ± 0.5 | 2.1 |
| DMF | 7 ± 1 | 2.8 |
| chloroform | 1.4 ± 0.4 | 1 |
| methanol | 3.7 | 4.1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vorobei, A.M.; Kostenko, M.O.; Parenago, O.O. Viscosity Measurement of CO2–Solvent Mixtures for the Study of the Morphology and Size of Crystalline Particles Obtained Using Supercritical Antisolvent Precipitation. Materials 2023, 16, 6151. https://doi.org/10.3390/ma16186151
Vorobei AM, Kostenko MO, Parenago OO. Viscosity Measurement of CO2–Solvent Mixtures for the Study of the Morphology and Size of Crystalline Particles Obtained Using Supercritical Antisolvent Precipitation. Materials. 2023; 16(18):6151. https://doi.org/10.3390/ma16186151
Chicago/Turabian StyleVorobei, Anton M., Mikhail O. Kostenko, and Olga O. Parenago. 2023. "Viscosity Measurement of CO2–Solvent Mixtures for the Study of the Morphology and Size of Crystalline Particles Obtained Using Supercritical Antisolvent Precipitation" Materials 16, no. 18: 6151. https://doi.org/10.3390/ma16186151