
Structural, Electronic, and Mechanical Properties of Zr2SeB and Zr2SeN from First-Principle Investigations
 ,
,
Abstract
:1. Introduction
2. Computational Details
3. Results and Discussions
3.1. Analysis of Crystal Structure of Zr2SeX (X = B, N)
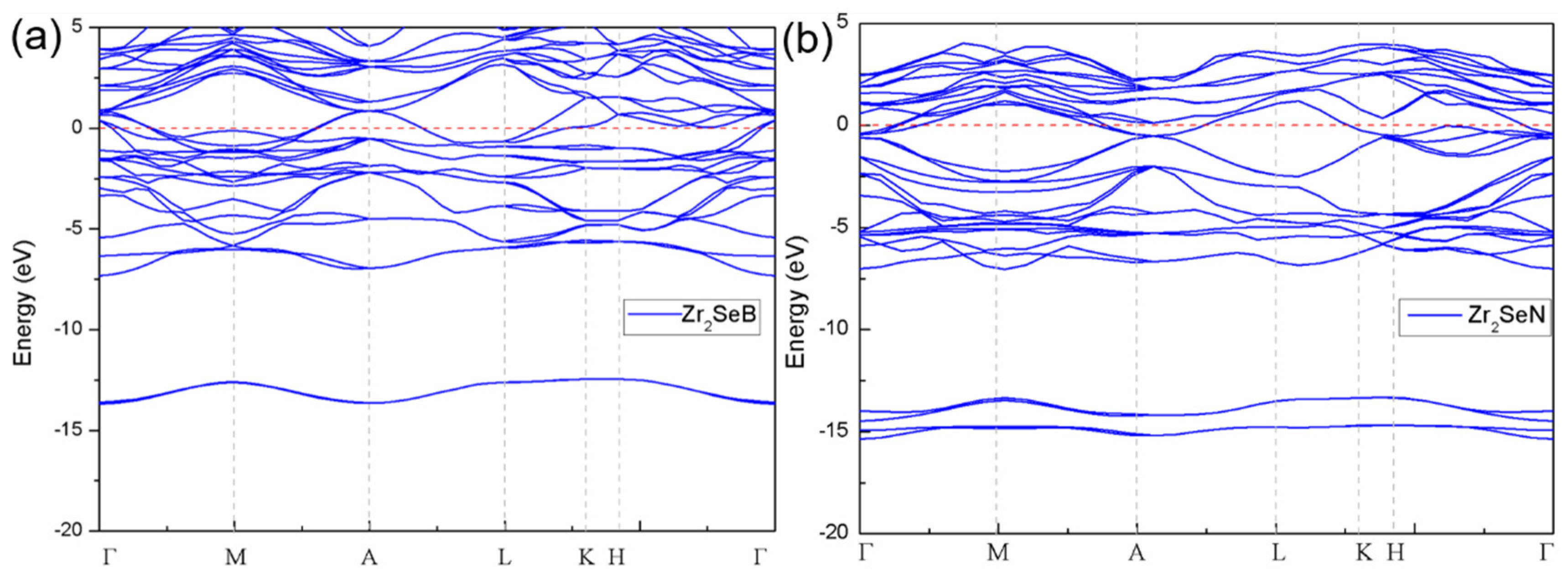
3.2. Electronic Properties
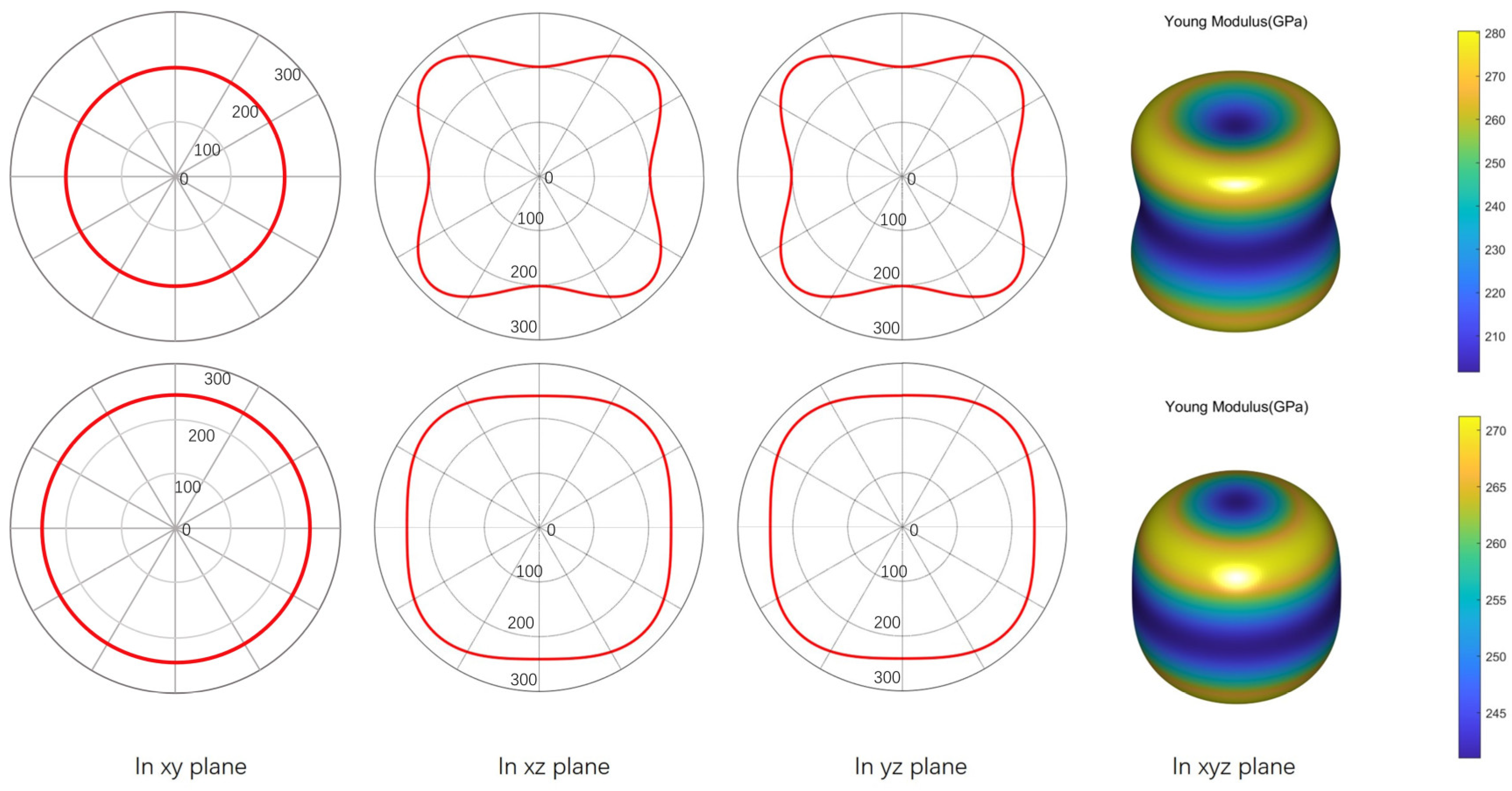
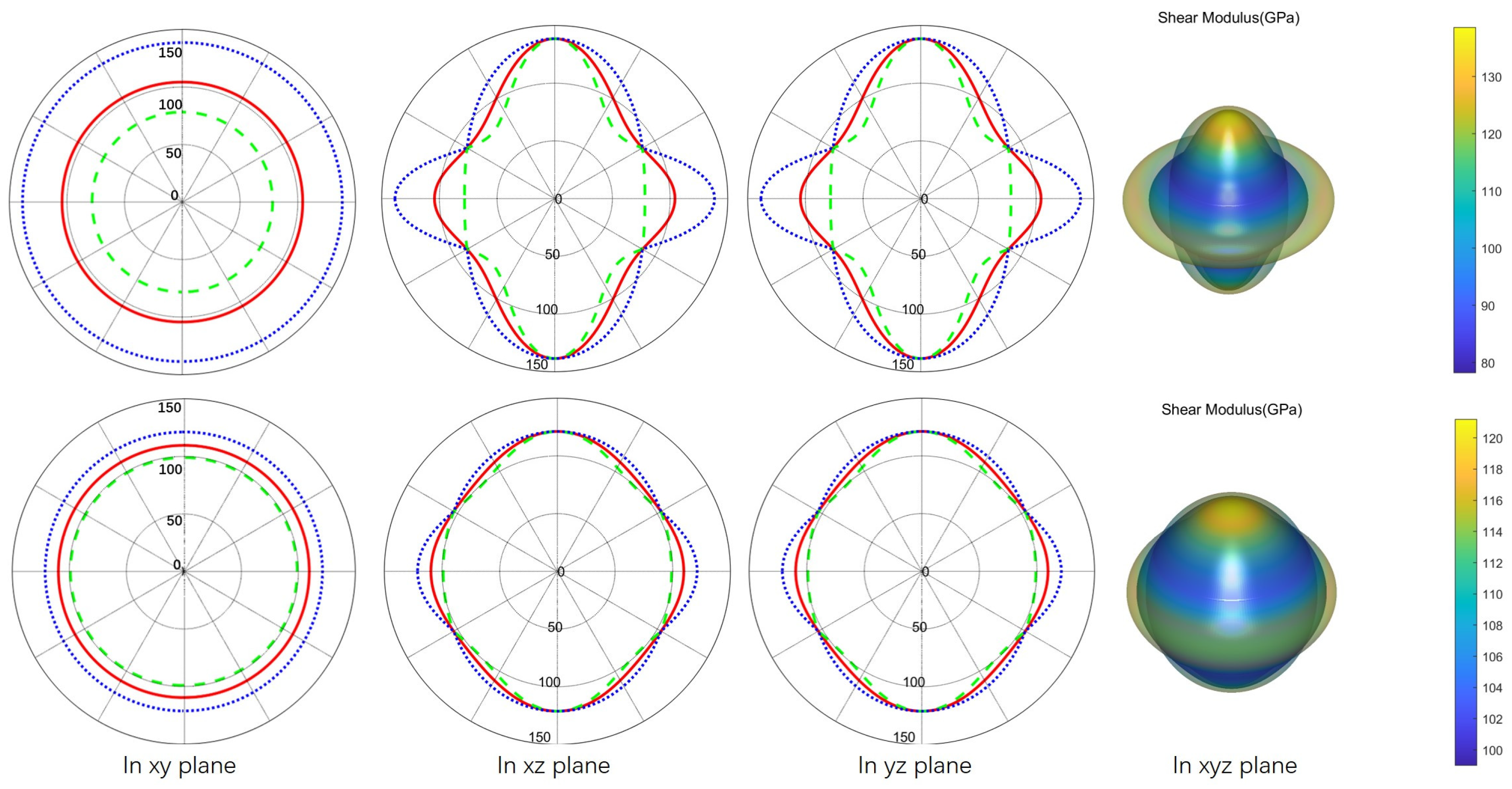
3.3. Mechanical Properties
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Jeitschko, W.; Nowotny, H.; Benesovsky, F. Carbides of formula T2MC. J. Less Common Met. 1964, 7, 133–138. [Google Scholar] [CrossRef]
- Wolfsgruber, H.; Nowotny, H.; Benesovsky, F. Die Kristallstruktur von Ti3GeC2. Monatshefte Chem. Verwandte Teile Anderer Wiss. 1967, 98, 2403–2405. [Google Scholar] [CrossRef]
- Nowotny, V.H. Strukturchemie einiger Verbindungen der Übergangsmetalle mit den elementen C, Si, Ge, Sn. Prog. Solid State Chem. 1971, 5, 27–70. [Google Scholar] [CrossRef]
- Nowotny, H.; Rogl, P.; Schuster, J.C. Structural chemistry of complex carbides and related compounds. J. Solid State Chem. 1982, 44, 126–133. [Google Scholar] [CrossRef]
- Barsoum, M.W.; El-Raghy, T. Synthesis and Characterization of a Remarkable Ceramic: Ti3SiC2. J. Am. Ceram. Soc. 1996, 79, 1953–1956. [Google Scholar] [CrossRef]
- Barsoum, M.W. The MN+1AXN phases: A new class of solids: Thermodynamically stable nanolaminates. Prog. Solid State Chem. 2000, 28, 201–281. [Google Scholar] [CrossRef]
- Barsoum, M.W. MAX Phases: Properties of Machinable Ternary Carbides and Nitrides, 2nd ed.; John Wiley & Sons, Ltd.: Weinheim, Germany, 2013; pp. 1–421. [Google Scholar] [CrossRef]
- Sokol, M.; Natu, V.; Kota, S.; Barsoum, M.W. On the Chemical Diversity of the MAX Phases. Trends Chem. 2019, 1, 210–223. [Google Scholar] [CrossRef]
- Gonzalez-Julian, J. Processing of MAX phases: From synthesis to applications. J. Am. Ceram. Soc. 2021, 104, 659–690. [Google Scholar] [CrossRef]
- Kurakevych, O. Superhard Phases of Simple Substances and Binary Compounds of the B-C-N-O System: From Diamond to the Latest Results (a Review). J. Superhard Mater. 2011, 31, 139–157. [Google Scholar] [CrossRef] [Green Version]
- Khazaei, M.; Arai, M.; Sasaki, T.; Estili, M.; Sakka, Y. Trends in electronic structures and structural properties of MAX phases: A first-principles study on M2AlC (M = Sc, Ti, Cr, Zr, Nb, Mo, Hf, or Ta), M2AlN, and hypothetical M2AlB phases. J. Phys. Condens. Matter 2014, 26, 505503. [Google Scholar] [CrossRef]
- Gencer, A.; Surucu, G. Electronic and Lattice Dynamical Properties of Ti2SiB MAX Phase. Mater. Res. Express 2018, 5, 76303. [Google Scholar] [CrossRef] [Green Version]
- Surucu, G. Investigation of structural, electronic, anisotropic elastic, and lattice dynamical properties of MAX phases borides: An Ab-initio study on hypothetical M2AB (M = Ti, Zr, Hf; A = Al, Ga, In) compounds. Mater. Chem. Phys. 2018, 203, 106–117. [Google Scholar] [CrossRef]
- Miao, N.; Wang, J.; Gong, Y.; Wu, J.; Niu, H.; Wang, S.; Li, K.; Oganov, A.R.; Tada, T.; Hosono, H. Computational Prediction of Boron-Based MAX Phases and MXene Derivatives. Chem. Mater. 2020, 32, 6947–6957. [Google Scholar] [CrossRef]
- Rackl, T.; Eisenburger, L.; Niklaus, R.; Johrendt, D. Syntheses and physical properties of the MAX phase boride Nb2SB and the solid solutions Nb2SBxC1−x (x= 0–1). Phys. Rev. Mater. 2019, 3, 54001. [Google Scholar] [CrossRef]
- Surucu, G.; Gencer, A.; Wang, X.; Surucu, O. Lattice dynamical and thermo-elastic properties of M2AlB (M = V, Nb, Ta) MAX phase borides. J. Alloys Compd. 2020, 819, 153256. [Google Scholar] [CrossRef]
- Rackl, T.; Johrendt, D. The MAX phase borides Zr2SB and Hf2SB. Solid State Sci. 2020, 106, 106316. [Google Scholar] [CrossRef]
- Ali, M.A.; Hossain, M.M.; Uddin, M.M.; Hossain, M.A.; Islam, A.K.M.A.; Naqib, S.H. Physical properties of new MAX phase borides M2SB (M = Zr, Hf and Nb) in comparison with conventional MAX phase carbides M2SC (M = Zr, Hf and Nb): Comprehensive insights. J. Mater. Res. Technol. 2020, 11, 1000–1018. [Google Scholar] [CrossRef]
- Liu, Y.Y.; Wang, Y.X.; Yan, Z.X.; Liu, W.; Zhou, G.L.; Xiong, K.Z.; Gu, J.B. Pressure effects on structure, mechanical properties and thermal conductivity of V2SnC: A first-principles study. Philos. Mag. 2022, 102, 228–243. [Google Scholar] [CrossRef]
- Chen, K.; Bai, X.; Mu, X.; Yan, P.; Qiu, N.; Li, Y.; Zhou, J.; Song, Y.; Zhang, Y.; Du, S.; et al. MAX phase Zr2SeC and its thermal conduction behavior. J. Eur. Ceram. Soc. 2021, 41, 4447–4451. [Google Scholar] [CrossRef]
- Ali, M.A.; Qureshi, M.W. Newly synthesized MAX phase Zr2SeC: DFT insights into physical properties towards possible applications. RSC Adv. 2021, 11, 16892–16905. [Google Scholar] [CrossRef]
- Ali, M.A.; Qureshi, M.W. DFT insights into the new Hf-based chalcogenide MAX phase Hf2SeC. Vacuum 2022, 201, 111072. [Google Scholar] [CrossRef]
- Hohenberg, P.; Kohn, W. Inhomogeneous Electron Gas. Phys. Rev. 1964, 136, B864. [Google Scholar] [CrossRef] [Green Version]
- Clark, S.J.; Segall, M.D.; Pickard, C.J.; Hasnip, P.J.; Probert, M.I.J.; Refson, K.; Payne, M.C. First principles methods using CASTEP. Z. Krist.-Cryst. Mater. 2005, 220, 567–570. [Google Scholar] [CrossRef] [Green Version]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Kohn, W.; Sham, L.J. Self-ConsistentEquations Including Exchange and Correlation Effects. Phys. Rev. 1965, 140, A1133. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Zhou, Y.; San, X.; Li, W.; Bao, Y.; Feng, Q.; Grasso, S.; Hu, C. Zr2SeB and Hf2SeB: Two new MAB phase compounds with the Cr2AlC-type MAX phase (211 phase) crystal structures. J. Adv. Ceram. 2022, 11, 1764–1776. [Google Scholar] [CrossRef]
- Baroni, S.; Giannozzi, P.; Isaev, E. Thermal properties of materials from ab-initio quasi-harmonic phonons. Rev. Miner. Geochem. 2011, 71, 39–57. [Google Scholar] [CrossRef] [Green Version]
- Misra, R.D. On the stability of crystal lattices. II. Math. Proc. Camb. Philos. Soc. 1940, 36, 173–182. [Google Scholar] [CrossRef]
- Watt, J.P.; Peselnick, L. Clarification of the Hashin-Shtrikman bounds on the effective elastic moduli of polycrystals with hexagonal, trigonal, and tetragonal symmetries. J. Appl. Phys. 1980, 51, 1525–1531. [Google Scholar] [CrossRef]
- Schreiber, E.; Anderson, O.; Soga, N.; Bell, J. Elastic Constants and Their Measurement. J. Appl. Mech. 1975, 42, 747–748. [Google Scholar] [CrossRef] [Green Version]
- Anderson, O.L. A simplified method for calculating the debye temperature from elastic constants. J. Phys. Chem. Solids 1963, 24, 909–917. [Google Scholar] [CrossRef]
- Bouhemadou, A.; Khenata, R. Structural, electronic and elastic properties of M2SC (M = Ti, Zr, Hf) compounds. Phys. Lett. A 2008, 372, 6448–6452. [Google Scholar] [CrossRef]
- Hadi, M.A.; Christopoulos, S.R.G.; Naqib, S.H.; Chroneos, A.; Fitzpatrick, M.E.; Islam, A.K.M.A. Physical properties and defect processes of M3SnC2 (M = Ti, Zr, Hf) MAX phases: Effect of M-elements. J. Alloys Compd. 2018, 748, 804–813. [Google Scholar] [CrossRef]
- Ledbetter, H. Elastic properties of zinc: A compilation and a review. J. Phys. Chem. Ref. Data 1977, 6, 1181–1203. [Google Scholar] [CrossRef] [Green Version]
- Islam, A.K.M.A.; Sikder, A.S.; Islam, F.N. NbB2: A density functional study. Phys. Lett. A 2006, 350, 288–292. [Google Scholar] [CrossRef]
- Wang, J.; Zhou, Y.; Liao, T.; Lin, Z. First-principles prediction of low shear-strain resistance of Al3BC3: A metal borocarbide containing short linear BC2 units. Appl. Phys. Lett. 2006, 89, 21917. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Methods | a (Å) | c (Å) | c/a | ||
|---|---|---|---|---|---|
| Zr2SeC | GGA-PBE | 3.487 | 12.631 | 3.622 | This work |
| Zr2SeB | GGA-PBE | 3.578 | 12.734 | 3.559 | This work |
| Zr2SeN | GGA-PBE | 3.478 | 12.371 | 3.557 | This work |
| Zr2SeC | Experiment | 3.491 | 12.556 | 3.615 | [20] |
| Zr2SeC | GGA-PBE | 3.465 | 12.540 | 3.618 | [21] |
| Zr2SB | GGA-PBE | 3.516 | 12.313 | 3.502 | [18] |
| Zr2SC | GGA-PBE | 3.420 | 12.205 | 3.568 | [18] |
| MAX | Ref. | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Zr2SeC | 276 | 75 | 93 | 289 | 126 | 100 | 151 | 109 | 264 | 0.20 | 512 | 1.38 | [20] |
| Zr2SeB | 264 | 107 | 105 | 303 | 138 | 78 | 162 | 106 | 260 | 0.23 | 498 | 1.54 | This work |
| Zr2SeN | 274 | 75 | 100 | 279 | 121 | 99 | 153 | 105 | 256 | 0.22 | 499 | 1.45 | This work |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bai, X.; Chen, K.; Luo, K.; Qiu, N.; Huang, Q.; Han, Q.; Liang, H.; Zhang, X.; Bai, C. Structural, Electronic, and Mechanical Properties of Zr2SeB and Zr2SeN from First-Principle Investigations. Materials 2023, 16, 5455. https://doi.org/10.3390/ma16155455
Bai X, Chen K, Luo K, Qiu N, Huang Q, Han Q, Liang H, Zhang X, Bai C. Structural, Electronic, and Mechanical Properties of Zr2SeB and Zr2SeN from First-Principle Investigations. Materials. 2023; 16(15):5455. https://doi.org/10.3390/ma16155455
Chicago/Turabian StyleBai, Xiaojing, Ke Chen, Kan Luo, Nianxiang Qiu, Qing Huang, Qi Han, Haijing Liang, Xiaohong Zhang, and Chengying Bai. 2023. "Structural, Electronic, and Mechanical Properties of Zr2SeB and Zr2SeN from First-Principle Investigations" Materials 16, no. 15: 5455. https://doi.org/10.3390/ma16155455