
Interesterification of Glyceryl Trioctanoate Catalyzed by Sulfonic Silica-Based Materials: Insight into the Role of Catalysts on the Reaction Mechanism



Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Catalysts Synthesis
2.2.1. Mesoporous Silica Supports
2.2.2. Propyl-Sulfonic (Pr-SO3H) Catalysts
2.3. Catalyst Characterization
2.4. Interesterification Reaction and Products Analysis
3. Results and Discussion
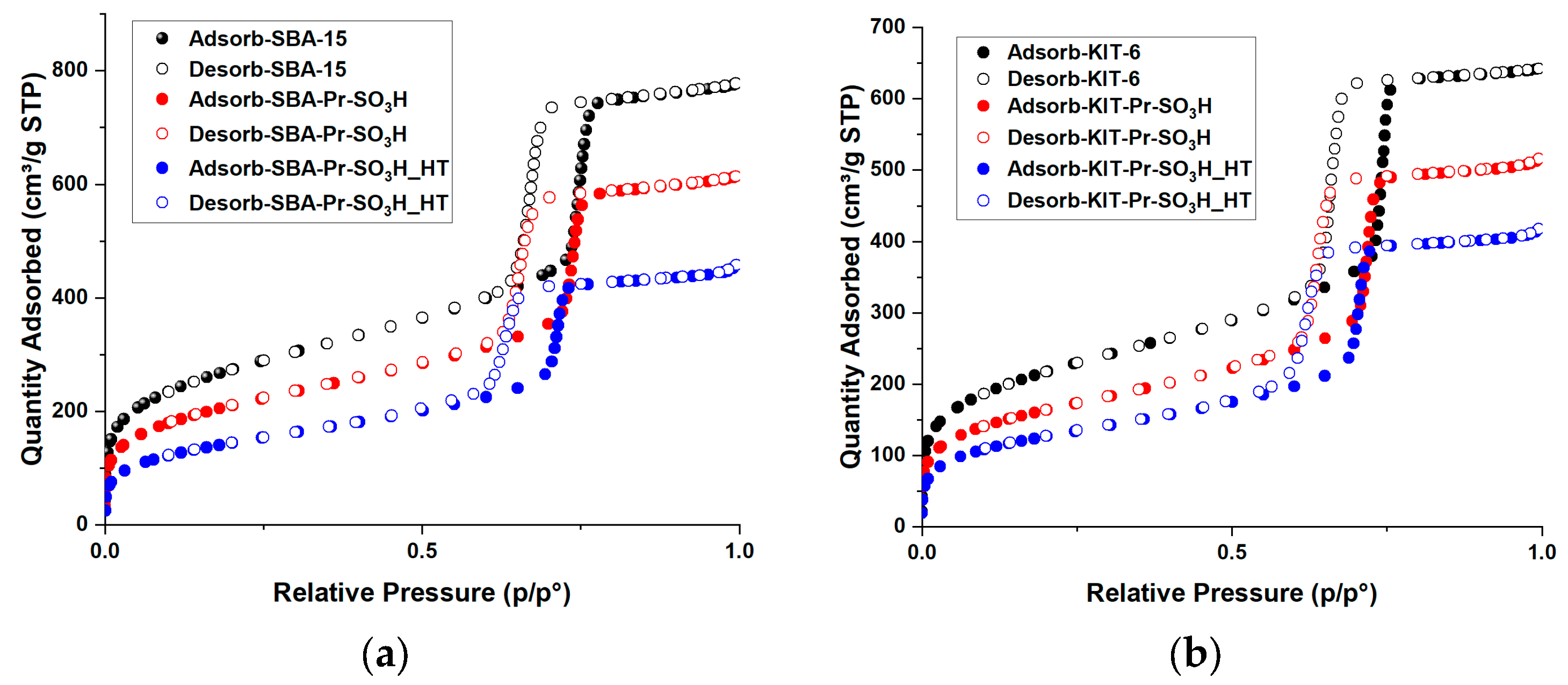
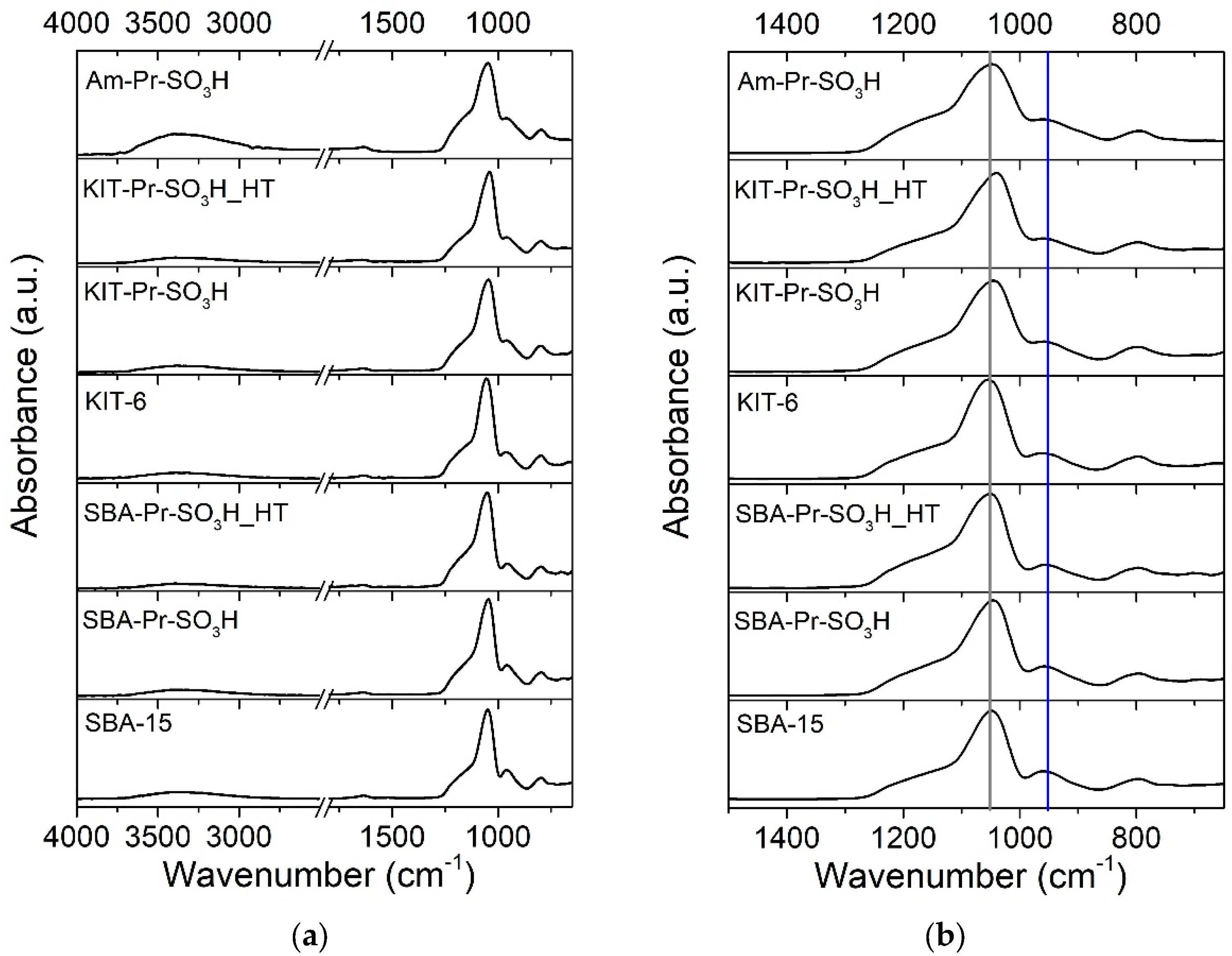
3.1. Catalyst Characterization
3.2. Preliminary Catalytic Tests
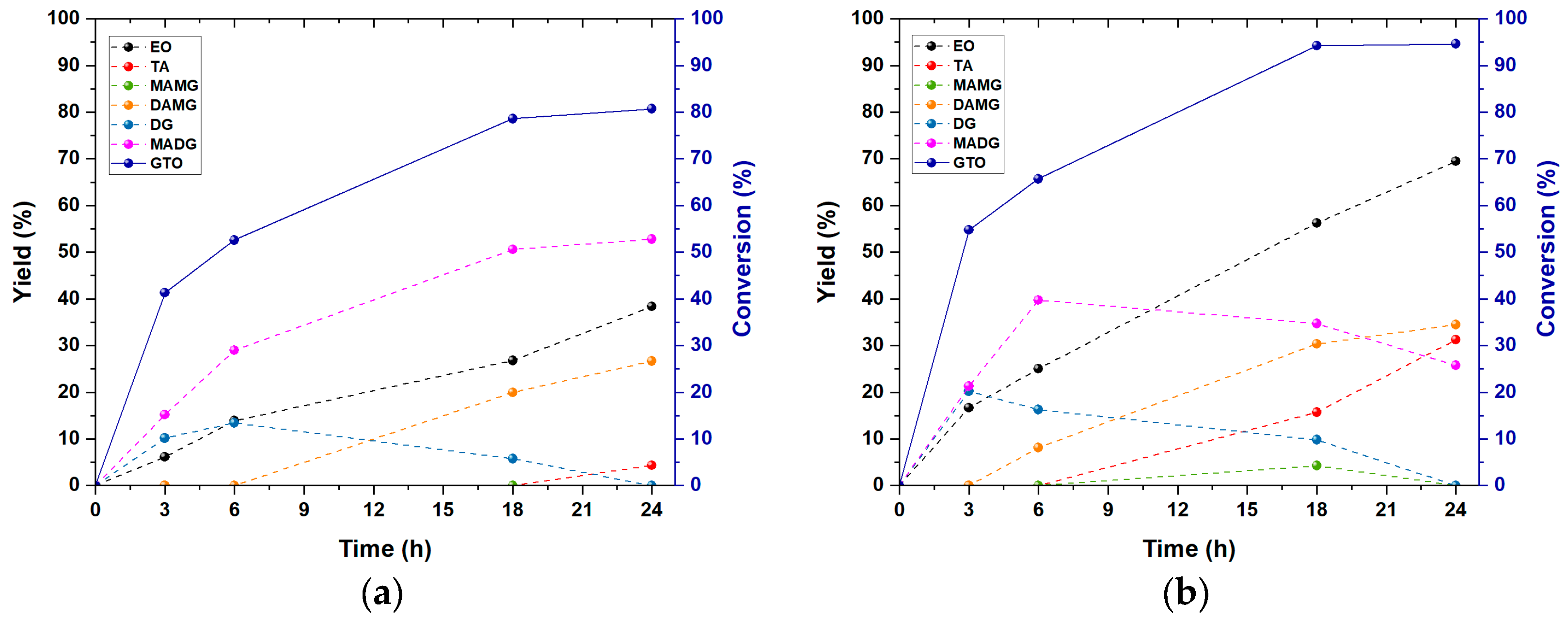
3.3. Effect of Time and Ethanol on the Interesterification Process
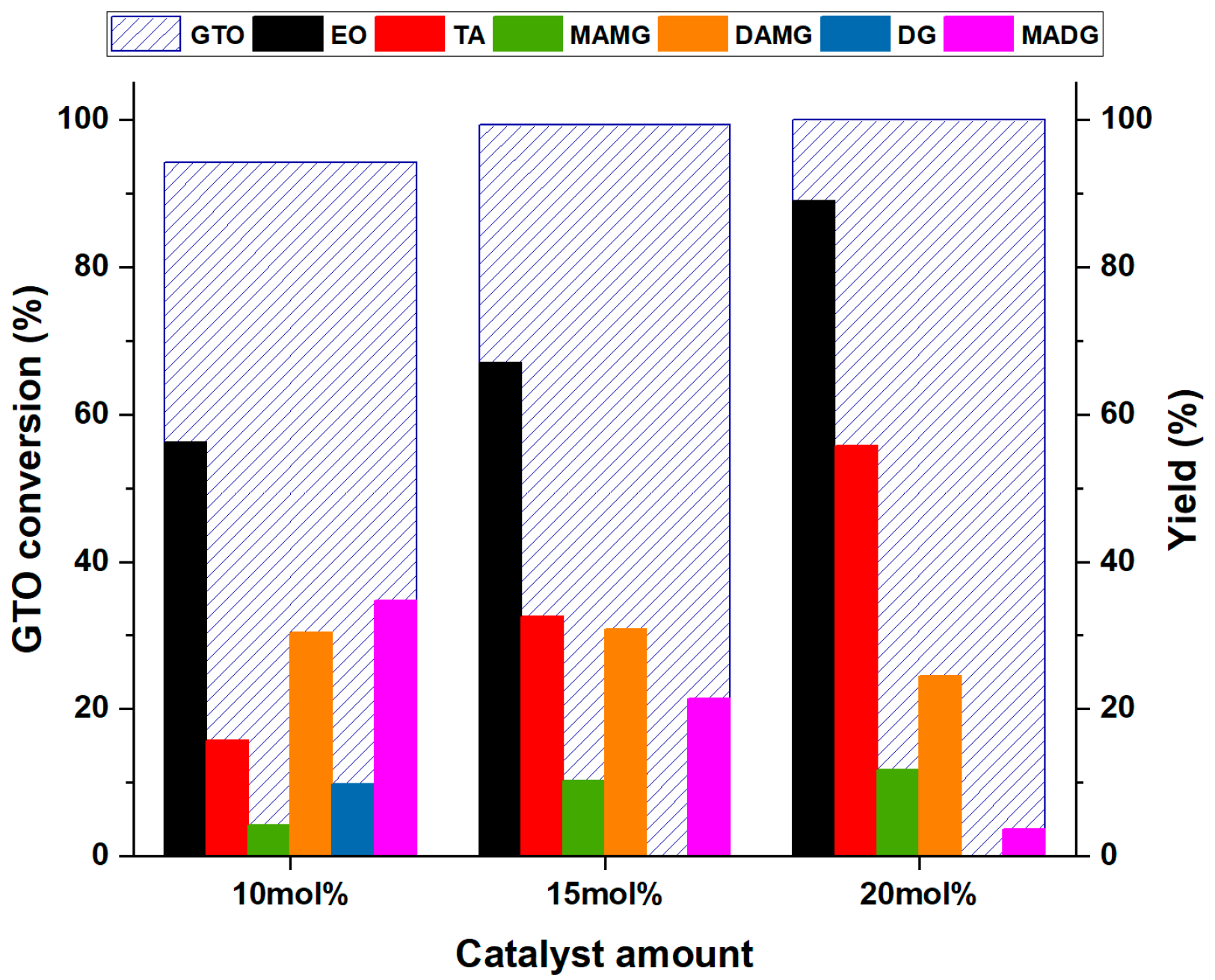
3.4. Effect of Catalyst Loading on Conversion and Yields
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- DIRECTIVE (EU) 2015/1513 of the European Parliament and of the Council-of 9 September 2015-Amending Directive 98/70/EC Relating to the Quality of Petrol and Diesel Fuels and Amending Directive 2009/28/EC on the Promotion of the Use of Energy from Renewable Sources. Available online: https://european-union.europa.eu/index_en (accessed on 1 July 2021).
- Vicente, G.; Martínez, M.; Aracil, J. Integrated biodiesel production: A comparison of different homogeneous catalysts systems. Bioresour. Technol. 2004, 92, 297–305. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.F.; Bennett, J.A.; Manayil, J.C.; Wilson, K. Heterogeneous catalysis for sustainable biodiesel production via esterification and transesterification. Chem. Soc. Rev. 2014, 43, 7887–7916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Norjannah, B.; Ong, H.C.; Masjuki, H.H.; Juan, J.C.; Chong, W.T. Enzymatic transesterification for biodiesel production: A comprehensive review. RSC Adv. 2016, 6, 60034–60055. [Google Scholar] [CrossRef]
- Leoneti, A.B.; Aragão-Leoneti, V.; de Oliveira, S.V.W.B. Glycerol as a by-product of biodiesel production in Brazil: Alternatives for the use of unrefined glycerol. Renew. Energy 2012, 45, 138–145. [Google Scholar] [CrossRef]
- Haas, M.J.; McAloon, A.J.; Yee, W.C.; Foglia, T.A. A process model to estimate biodiesel production costs. Bioresour. Technol. 2006, 97, 671–678. [Google Scholar] [CrossRef] [PubMed]
- Abdullah, A.; Abdullah, A.Z.; Ahmed, M.; Khan, J.; Shahadat, M.; Umar, K.; Alim, A. A review on recent developments and progress in sustainable acrolein production through catalytic dehydration of bio-renewable glycerol. J. Clean. Prod. 2022, 341, 130876. [Google Scholar] [CrossRef]
- Enweremadu, C.C.; Mbarawa, M.M. Technical aspects of production and analysis of biodiesel from used cooking oil—A review. Renew. Sustain. Energy Rev. 2009, 13, 2205–2224. [Google Scholar] [CrossRef]
- Borges, M.; Díaz, L. Recent developments on heterogeneous catalysts for biodiesel production by oil esterification and transesterification reactions: A review. Renew. Sustain. Energy Rev. 2012, 16, 2839–2849. [Google Scholar] [CrossRef]
- Ruhul, A.M.; Kalam, M.A.; Masjuki, H.H.; Fattah, I.M.R.; Reham, S.S.; Rashed, M.M. State of the art of biodiesel production processes: A review of the heterogeneous catalyst. RSC Adv. 2015, 5, 101023–101044. [Google Scholar] [CrossRef]
- Coman, S.M.; Parvulescu, V.I. Heterogeneous Catalysis for Biodiesel Production. In The Role of Catalysis for the Sustainable Production of Bio-Fuels and Bio-Chemicals; Elsevier Inc.: Amsterdam, The Netherlands, 2013; pp. 93–136. ISBN 9780444563309. [Google Scholar]
- Tan, X.; Sudarsanam, P.; Tan, J.; Wang, A.; Zhang, H.; Li, H.; Yang, S. Sulfonic acid-functionalized heterogeneous catalytic materials for efficient biodiesel production: A review. J. Environ. Chem. Eng. 2020, 9, 104719. [Google Scholar] [CrossRef]
- Shagufta; Ahmad, I.; Dhar, R. Sulfonic Acid-Functionalized Solid Acid Catalyst in Esterification and Transesterification Reactions. Catal. Surv. Asia 2017, 21, 53–69. [Google Scholar] [CrossRef]
- Kijenski, J.A.; Lipkowski, A.W.; Walisiewicz-Niedbalska, W.; Gwardiak, H.; Rozycki, K.; Pawlak, I. A Biofuel for Compression-Ignition Engines and a Method for Preparing the Biofuel. European Patent EP 1 580 255 A1, 28 September 2005. [Google Scholar]
- Casas, A.; Ramos, M.J.; Pérez, Á. New trends in biodiesel production: Chemical interesterification of sunflower oil with methyl acetate. Biomass Bioenergy 2011, 35, 1702–1709. [Google Scholar] [CrossRef]
- Casas, A.; Ruiz, J.R.; Ramos, M.J.; Pérez, Á. Effects of Triacetin on Biodiesel Quality. Energy Fuels 2010, 24, 4481–4489. [Google Scholar] [CrossRef]
- Leggieri, P.A.; Senra, M.; Soh, L. Cloud point and crystallization in fatty acid ethyl ester biodiesel mixtures with and without additives. Fuel 2018, 222, 243–249. [Google Scholar] [CrossRef]
- Esan, A.O.; Olabemiwo, O.M.; Smith, S.M.; Ganesan, S. A concise review on alternative route of biodiesel production via interesterification of different feedstocks. Int. J. Energy Res. 2021, 45, 12614–12637. [Google Scholar] [CrossRef]
- Trentini, C.P.; de Mello, B.T.F.; Postaue, N.; Stevanato, N.; Cardozo-Filho, L.; da Silva, C. Interesterification of grease trap waste lipids using methyl acetate under supercritical conditions. J. Supercrit. Fluids 2020, 164, 104896. [Google Scholar] [CrossRef]
- de Mello, B.T.F.; Trentini, C.P.; Postaue, N.; da Silva, C. Sequential process for obtaining methyl esters and triacetin from crambe oil using pressurized methyl acetate. Ind. Crop. Prod. 2020, 147, 112233. [Google Scholar] [CrossRef]
- Postaue, N.; Trentini, C.P.; de Mello, B.T.F.; Cardozo-Filho, L.; da Silva, C. Continuous catalyst-free interesterification of crambe oil using methyl acetate under pressurized conditions. Energy Convers. Manag. 2019, 187, 398–406. [Google Scholar] [CrossRef]
- Goembira, F.; Saka, S. Advanced supercritical Methyl acetate method for biodiesel production from Pongamia pinnata oil. Renew. Energy 2015, 83, 1245–1249. [Google Scholar] [CrossRef]
- Goembira, F.; Saka, S. Factors Affecting Biodiesel Yield in Interesterification of Rapeseed Oil by Supercritical Methyl Acetate. In Zero-Carbon Energy Kyoto 2011, Green Energy and Technology; Takeshi, Y., Ed.; Springer: Tokyo, Japan, 2012; pp. 147–152. [Google Scholar] [CrossRef]
- Komintarachat, C.; Sawangkeaw, R.; Ngamprasertsith, S. Continuous production of palm biofuel under supercritical ethyl acetate. Energy Convers. Manag. 2015, 93, 332–338. [Google Scholar] [CrossRef]
- Goembira, F.; Matsuura, K.; Saka, S. Biodiesel production from rapeseed oil by various supercritical carboxylate esters. Fuel 2012, 97, 373–378. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Du, W.; Liu, D. Study on the kinetics of enzymatic interesterification of triglycerides for biodiesel production with methyl acetate as the acyl acceptor. J. Mol. Catal. B Enzym. 2005, 32, 241–245. [Google Scholar] [CrossRef]
- Duraiarasan, S.; Razack, S.A.; Manickam, A.; Munusamy, A.; Syed, M.B.; Ali, M.Y.; Ahmed, G.M.; Mohiuddin, S. Direct conversion of lipids from marine microalga C. salina to biodiesel with immobilised enzymes using magnetic nanoparticle. J. Environ. Chem. Eng. 2016, 4, 1393–1398. [Google Scholar] [CrossRef]
- Surendhiran, D.; Sirajunnisa, A.R.; Vijay, M. An alternative method for production of microalgal biodiesel using novel Bacillus lipase. 3 Biotech 2015, 5, 715–725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kashyap, S.S.; Gogate, P.R.; Joshi, S.M. Ultrasound assisted synthesis of biodiesel from karanja oil by interesterification: Intensification studies and optimization using RSM. Ultrason. Sonochem. 2019, 50, 36–45. [Google Scholar] [CrossRef]
- Maddikeri, G.L.; Pandit, A.B.; Gogate, P.R. Ultrasound assisted interesterification of waste cooking oil and methyl acetate for biodiesel and triacetin production. Fuel Process. Technol. 2013, 116, 241–249. [Google Scholar] [CrossRef]
- Kashyap, S.S.; Gogate, P.R.; Joshi, S.M. Ultrasound assisted intensified production of biodiesel from sustainable source as karanja oil using interesterification based on heterogeneous catalyst (γ-alumina). Chem. Eng. Process. Process Intensif. 2019, 136, 11–16. [Google Scholar] [CrossRef]
- Ansori, A.; Mahfud, M. Ultrasound assisted interesterification for biodiesel production from palm oil and methyl acetate: Optimization using RSM. J. Phys. Conf. Ser. 2021, 1747, 012044. [Google Scholar] [CrossRef]
- Casas, A.; Ramos, M.J.; Pérez, Á. Kinetics of chemical interesterification of sunflower oil with methyl acetate for biodiesel and triacetin production. Chem. Eng. J. 2011, 171, 1324–1332. [Google Scholar] [CrossRef]
- Sustere, Z.; Murnieks, R.; Kampars, V. Chemical interesterification of rapeseed oil with methyl, ethyl, propyl and isopropyl acetates and fuel properties of obtained mixtures. Fuel Process. Technol. 2016, 149, 320–325. [Google Scholar] [CrossRef]
- Battistel, E.; Calaprice, C.; Gualdi, E.; Rebesco, E.; Usai, E.M. Co-production of butyrate methyl ester and triacetylglycerol from tributyrin and methyl acetate. Appl. Catal. A Gen. 2011, 394, 149–157. [Google Scholar] [CrossRef]
- Ribeiro, J.D.S.; Celante, D.; Simões, S.S.; Bassaco, M.M.; da Silva, C.; de Castilhos, F. Efficiency of heterogeneous catalysts in interesterification reaction from macaw oil (Acrocomia aculeata) and methyl acetate. Fuel 2017, 200, 499–505. [Google Scholar] [CrossRef]
- Interrante, L.; Bensaid, S.; Galletti, C.; Pirone, R.; Schiavo, B.; Scialdone, O.; Galia, A. Interesterification of rapeseed oil catalysed by a low surface area tin (II) oxide heterogeneous catalyst. Fuel Process. Technol. 2018, 177, 336–344. [Google Scholar] [CrossRef]
- Prestigiacomo, C.; Biondo, M.; Galia, A.; Monflier, E.; Ponchel, A.; Prevost, D.; Scialdone, O.; Tilloy, S.; Bleta, R. Interesterification of triglycerides with methyl acetate for biodiesel production using a cyclodextrin-derived SnO@γ-Al2O3 composite as heterogeneous catalyst. Fuel 2022, 321, 124026. [Google Scholar] [CrossRef]
- Tian, Y.; Xiang, J.; Verni, C.C.; Soh, L. Fatty acid methyl ester production via ferric sulfate catalyzed interesterification. Biomass Bioenergy 2018, 115, 82–87. [Google Scholar] [CrossRef]
- Wong, W.; Lim, S.; Pang, Y.; Chen, W.; Lam, M.; Tan, I. Synthesis of glycerol-free fatty acid methyl ester using interesterification reaction based on solid acid carbon catalyst derived from low-cost biomass wastes. Int. J. Energy Res. 2022, 46, 147–162. [Google Scholar] [CrossRef]
- Wong, W.-Y.; Lim, S.; Pang, Y.-L.; Shuit, S.-H.; Chen, W.-H.; Lee, K.-T. Synthesis of renewable heterogeneous acid catalyst from oil palm empty fruit bunch for glycerol-free biodiesel production. Sci. Total Environ. 2020, 727, 138534. [Google Scholar] [CrossRef]
- Usai, E.M.; Sini, M.F.; Meloni, D.; Solinas, V.; Salis, A. Sulfonic acid-functionalized mesoporous silicas: Microcalorimetric characterization and catalytic performance toward biodiesel synthesis. Microporous Mesoporous Mater. 2013, 179, 54–62. [Google Scholar] [CrossRef]
- Peixoto, A.F.; Soliman, M.M.; Pinto, T.V.; Silva, S.M.; Costa, P.; Alegria, E.C.; Freire, C. Highly active organosulfonic aryl-silica nanoparticles as efficient catalysts for biomass derived biodiesel and fuel additives. Biomass Bioenergy 2021, 145, 105936. [Google Scholar] [CrossRef]
- Testa, M.L.; La Parola, V. Sulfonic Acid-Functionalized Inorganic Materials as Efficient Catalysts in Various Applications: A Minireview. Catalysts 2021, 11, 1143. [Google Scholar] [CrossRef]
- Drago, C.; Liotta, L.F.; La Parola, V.; Testa, M.L.; Nicolosi, G. One-pot microwave assisted catalytic transformation of vegetable oil into glycerol-free biodiesel. Fuel 2013, 113, 707–711. [Google Scholar] [CrossRef]
- Testa, M.L.; La Parola, V.; Liotta, L.F.; Venezia, A.M. Screening of different solid acid catalysts for glycerol acetylation. J. Mol. Catal. A Chem. 2013, 367, 69–76. [Google Scholar] [CrossRef]
- Aguado-Deblas, L.; Estevez, R.; Russo, M.; La Parola, V.; Bautista, F.M.; Testa, M.L. Sustainable microwave-assisted solketal synthesis over sulfonic silica-based catalysts. J. Environ. Chem. Eng. 2022, 10, 108628. [Google Scholar] [CrossRef]
- Casas, A.; Ramos, M.J.; Pérez, A. Methanol-enhanced chemical interesterification of sunflower oil with methyl acetate. Fuel 2013, 106, 869–872. [Google Scholar] [CrossRef]
- Cattaneo, A.S.; Ferrara, C.; Villa, D.C.; Angioni, S.; Milanese, C.; Capsoni, D.; Grandi, S.; Mustarelli, P.; Allodi, V.; Mariotto, G.; et al. SBA-15 mesoporous silica highly functionalized with propylsulfonic pendants: A thorough physico-chemical characterization. Microporous Mesoporous Mater. 2016, 219, 219–229. [Google Scholar] [CrossRef]
- Wawrzyńczak, A.; Jarmolińska, S.; Nowak, I. Nanostructured KIT-6 materials functionalized with sulfonic groups for catalytic purposes. Catal. Today 2022, 397–399, 526–539. [Google Scholar] [CrossRef]
- Testa, M.L.; La Parola, V.; Venezia, A.M. Transesterification of short chain esters using sulfonic acid-functionalized hybrid silicas: Effect of silica morphology. Catal. Today 2014, 223, 115–121. [Google Scholar] [CrossRef]
- Díaz, I.; Mohino, F.; Blasco, T.; Sastre, E.; Pérez-Pariente, J. Influence of the alkyl chain length of HSO3-R-MCM-41 on the esterification of glycerol with fatty acids. Microporous Mesoporous Mater. 2005, 80, 33–42. [Google Scholar] [CrossRef]
- Brunauer, S.; Emmett, P.H.; Teller, E. Adsorption of Gases in Multimolecular Layers. J. Am. Chem. Soc. 1938, 60, 309–319. [Google Scholar] [CrossRef]
- Testa, M.L.; Miroddi, G.; Russo, M.; La Parola, V.; Marcì, G. Dehydration of Fructose to 5-HMF over Acidic TiO2 Catalysts. Materials 2020, 13, 1178. [Google Scholar] [CrossRef] [Green Version]
- Sing, K.S.W.; Everett, D.H.; Haul, R.A.W.; Moscou, L.; Pierotti, R.A.; Rouquerol, J.; Siemieniewska, T. Reporting physisorption data for gas/solid systems with special reference to the determination of surface area and porosity (Recommendations 1984). Pure Appl. Chem. 1985, 57, 603–619. [Google Scholar] [CrossRef]
- Kruk, M.; Jaroniec, M.; Ko, C.H.; Ryoo, R. Characterization of the Porous Structure of SBA-15. Chem. Mater. 2000, 12, 1961–1968. [Google Scholar] [CrossRef]
- Dutt, M.; Suhasini, K.; Ratan, A.; Shah, J.; Kotnala, R.K.; Singh, V. Mesoporous silica mediated synthesis of α-Fe2O3 porous structures and their application as humidity sensors. J. Mater. Sci. Mater. Electron. 2018, 29, 20506–20516. [Google Scholar] [CrossRef]
- Sun, X.; Yu, W.; Yan, J.; Li, J.; Jin, G.; Feng, J.; Guo, Z.; Liang, X. Mesoporous silica–carbon composites fabricated by a universal strategy of hydrothermal carbonization: Controllable synthesis and applications. RSC Adv. 2018, 8, 27207–27215. [Google Scholar] [CrossRef]
- Kaya, H.; Ngo, D.; Gin, S.; Kim, S.H. Spectral changes in Si–O–Si stretching band of porous glass network upon ingress of water. J. Non Cryst. Solids 2020, 527, 119722. [Google Scholar] [CrossRef]
- Loganathan, S.; Tikmani, M.; Ghoshal, A.K. Novel Pore-Expanded MCM-41 for CO2 Capture: Synthesis and Characterization. Langmuir 2013, 29, 3491–3499. [Google Scholar] [CrossRef]
- Testa, M.L.; Tummino, M.L.; Agostini, S.; Avetta, P.; Deganello, F.; Montoneri, E.; Magnacca, G.; Prevot, A.B. Synthesis, characterization and environmental application of silica grafted photoactive substances isolated from urban biowaste. RSC Adv. 2015, 5, 47920–47927. [Google Scholar] [CrossRef]
- Dubey, R.; Rajesh, Y.; More, M. Synthesis and Characterization of SiO2 Nanoparticles via Sol-gel Method for Industrial Applications. Mater. Today Proc. 2015, 2, 3575–3579. [Google Scholar] [CrossRef]
- Morrow, B.A.; McFarlan, A.J. Surface vibrational modes of silanol groups on silica. J. Phys. Chem. 1992, 96, 1395–1400. [Google Scholar] [CrossRef]
- Erdem, B.; Erdem, S.; Öksüzoğlu, R.M. Catalytic Applications of Large Pore Sulfonic Acid-Functionalized SBA-15 Mesoporous Silica for Esterification. Open Chem. 2018, 16, 1233–1241. [Google Scholar] [CrossRef]
- Barbosa, S.L.; Ottone, M.; De Almeida, M.T.; Lage, G.L.C.; Almeida, M.A.R.; Nelson, D.L.; Dos Santos, W.T.P.; Clososki, G.C.; Lopes, N.P.; Klein, S.I.; et al. Ketalization of Ketones to 1,3-Dioxolanes and Concurring Self-Aldolization Catalyzed by an Amorphous, Hydrophilic SiO2-SO3H Catalyst under Microwave Irradiation. J. Braz. Chem. Soc. 2018, 29, 1663–1671. [Google Scholar] [CrossRef]
- Wang, X.; Lin, K.S.K.; Chan, J.C.C.; Cheng, S. Direct Synthesis and Catalytic Applications of Ordered Large Pore Aminopropyl-Functionalized SBA-15 Mesoporous Materials. J. Phys. Chem. B 2005, 109, 1763–1769. [Google Scholar] [CrossRef] [PubMed]
- Martina, P.; Gayathri, R.; Raja Pugalenthi, M.; Cao, G.; Liu, C.; Ramesh Prabhu, M. Nanosulfonated Silica In-corporated SPEEK/SPVdF-HFP Polymer Blend Membrane for PEM Fuel Cell Application. Ionics 2020, 26, 3447–3458. [Google Scholar] [CrossRef]
- Dyker, G. Silica-Mediated Monohydrolysis of Dicarboxylic Esters. Eur. J. Org. Chem. 2021, 2021, 6773–6776. [Google Scholar] [CrossRef]
- Liu, L. How is chemical interesterification initiated: Nucleophilic substitution or α-proton abstraction? J. Am. Oil Chem. Soc. 2004, 81, 331–337. [Google Scholar] [CrossRef]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Catalyst | Acid Capacities (mmol H+ g−1) a | TGA (Loading %) | BET | |
|---|---|---|---|---|---|
| SSA (m2 g−1) | Vp (cm3 g−1) | ||||
| 1 | SBA-15 | - | - | 988 | 1.09 |
| 2 | KIT-6 | - | - | 785 | 0.92 |
| 3 | Am-Pr-SO3H | 2.0 | 15.5 | 306 | 0.34 |
| 4 | SBA-Pr-SO3H | 0.28 | 9.3 | 761 | 0.88 |
| 5 | SBA-Pr-SO3H_HT | 0.60 | 13.7 | 526 | 0.67 |
| 6 | KIT-Pr-SO3H | 0.43 | 10.2 | 589 | 0.76 |
| 7 | KIT-Pr-SO3H_HT | 0.72 | 14.8 | 458 | 0.63 |
| Catalyst | T (°C) | t (h) | Catalyst Loading (wt%) | Acetyl Donor:TG d | XTG (mol%) | YTA (mol%) | YFAEE/FAME (mol%) | Ref. |
|---|---|---|---|---|---|---|---|---|
| Am-Pr-SO3H | 120 | 6 | 11 | 30 | 52 | 0 | 14 | This work |
| Am-Pr-SO3H a | 120 | 6 | 11 | 30 | 66 | 0 | 25 | This work |
| Am-Pr-SO3H a | 120 | 18 | 22 b | 30 | 100 | 56 | 89 | This work |
| SBA-15-Propyl-SO3H | 130 | 6 | 13 | 20 e | 6 | - | 0 g | [42] |
| SBA-15-Phenyl-SO3H | 130 | 6 | 13 | 20 e | 20 | - | 19 g | [42] |
| Amberlyst-15® | 120 | 20 | 5–15 c | 20 f | 9 | 0 | 4 h | [35] |
| Nafion SAC-13® | 130 | 20 | 5–15 c | 20 f | 98 | 60 | 83 h | [35] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Testa, M.L.; Tummino, M.L.; Venezia, A.M.; Russo, M. Interesterification of Glyceryl Trioctanoate Catalyzed by Sulfonic Silica-Based Materials: Insight into the Role of Catalysts on the Reaction Mechanism. Materials 2023, 16, 5121. https://doi.org/10.3390/ma16145121
Testa ML, Tummino ML, Venezia AM, Russo M. Interesterification of Glyceryl Trioctanoate Catalyzed by Sulfonic Silica-Based Materials: Insight into the Role of Catalysts on the Reaction Mechanism. Materials. 2023; 16(14):5121. https://doi.org/10.3390/ma16145121
Chicago/Turabian StyleTesta, Maria Luisa, Maria Laura Tummino, Anna Maria Venezia, and Marco Russo. 2023. "Interesterification of Glyceryl Trioctanoate Catalyzed by Sulfonic Silica-Based Materials: Insight into the Role of Catalysts on the Reaction Mechanism" Materials 16, no. 14: 5121. https://doi.org/10.3390/ma16145121