

Resistance of Pastes from Carbonated, Low-Lime Calcium Silica Cements to External Sulfate Attack †

Abstract
:1. Introduction
2. Description of the Analytical Methods
2.1. Thermogravimetric Analysis (TGA)
2.2. X-ray Diffraction (XRD) Analysis
2.3. Fourier Transform Infrared (FTIR) Analysis
2.4. Soak Solution Analysis
3. Results of Characterization of Cements and Reference Pastes
3.1. Low-Lime Calcium Silicate Cements
3.2. Carbonated Reference Pastes
3.2.1. Preparation of the Pastes
3.2.2. Results of the Thermogravimetric Analysis (TGA) of the Pastes
3.2.3. Results of the X-ray Diffraction (XRF) Analysis of the Pastes
3.2.4. Results of the FTIR Analysis of the Pastes
4. Sulfate Exposure Experiment
4.1. Experimental Setup
4.2. Post-Exposure Analysis
5. Results of Sulfate Exposure Experiments
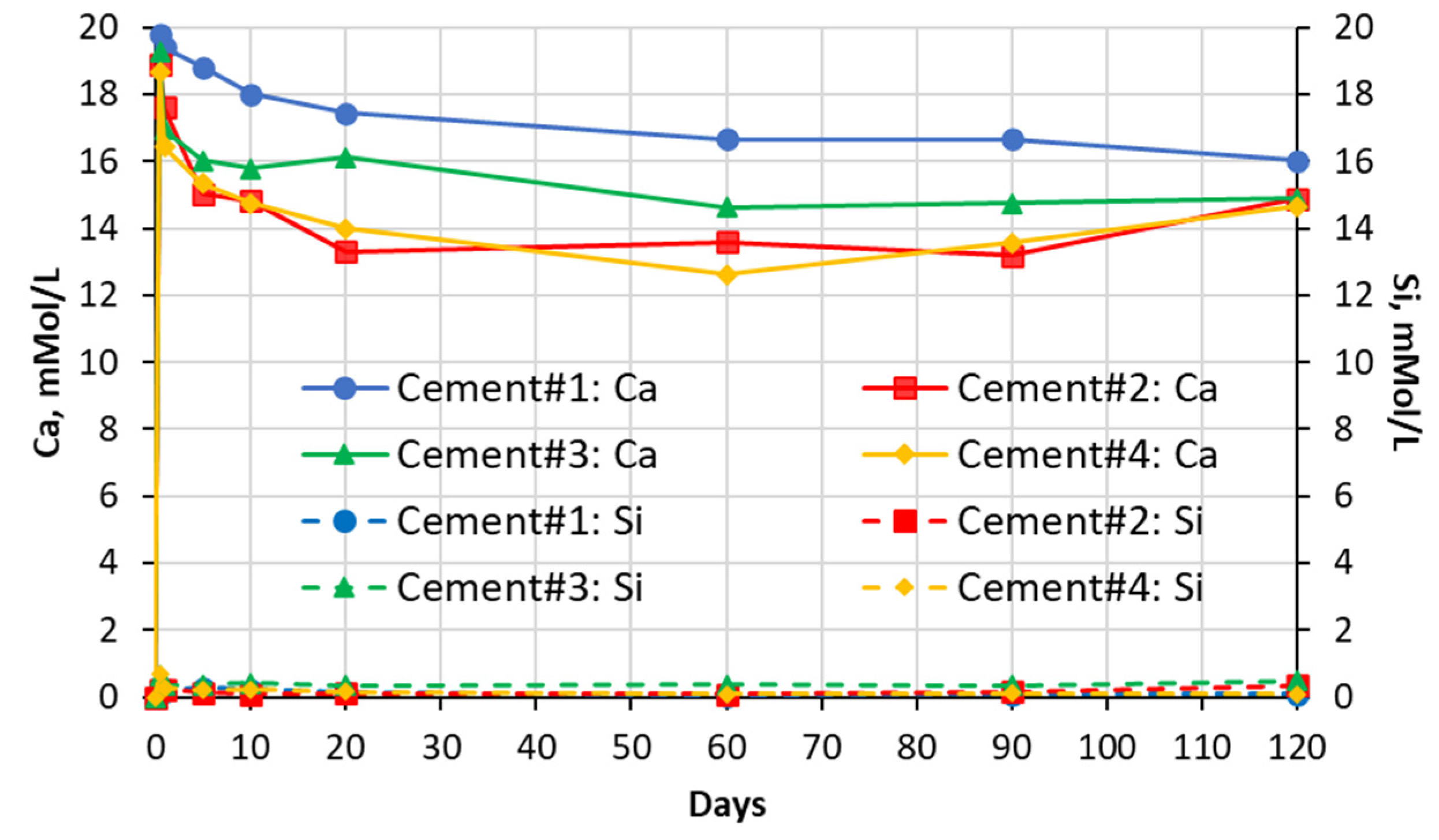
5.1. Soak Solution Chemistry Results
- -
- Sodium sulfate: 8–9 for cements #1, #2, and #4 paste samples and 10 for cement #3 paste sample.
- -
- Magnesium sulfate: ~8 for all cement paste samples.
- -
- Aluminum sulfate: ~7.5–7.8 for all cement paste samples.
- -
- Deionized water: ~9–10 for all cement pastes.
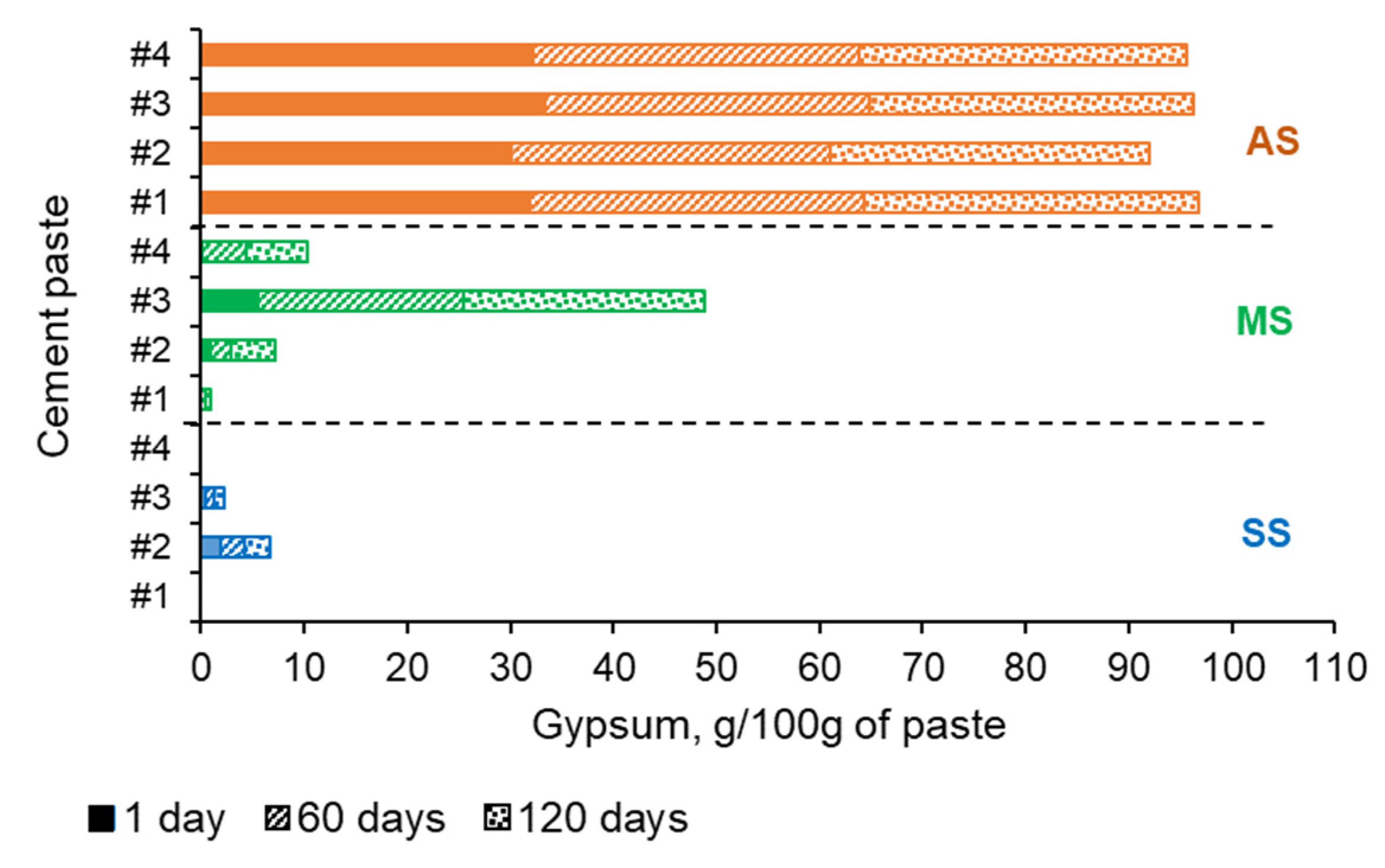
5.2. XRD Analysis Results
5.3. Thermal Analysis Results
5.4. FTIR Analysis Results
6. Discussion
6.1. General Features of the Low-Lime CCS Systems Exposed to Sulfate Solutions
6.2. Effect of the Type of Low-Lime Calcium Silicate on the Sulfate Resistance of the CCS System
6.3. Effect of the Type of the Sulfate Solution on the Sulfate Resistance of the CCS Systems
7. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

CaCO3 (aq.) + SO42− + 2H2O ↔ CaSO4∙2H2O + CO32−
100 g/mol 96 g/mol

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cement Paste # | Consumed CaCO3 | |
|---|---|---|
| Calculated | Measured | |
| 1 | 59.4 | 61.5 |
| 2 | 61 | 53.4 |
| 3 | 90.1 | 44.6 |
| 4 | 75.8 | 60.7 |
References
- Tokpatayeva, R.; Olek, J.; Jain, J.; Seth, A.; DeCristofaro, N. Sulfate Resistance Study of Carbonated Low-Calcium Silicate Systems. In Proceedings of the 6th International Conference on Durability of Concrete Structures, Leeds, UK, 18–20 July 2018; pp. 408–420. [Google Scholar]
- Jain, J.A.; Seth, A.; DeCristofaro, N. Environmental impact and durability of carbonated calcium silicate concrete. Proc. Inst. Civ. Eng. Constr. Mater. 2019, 172, 179–191. [Google Scholar] [CrossRef] [Green Version]
- Tokpatayeva, R.; Olek, J.; Sahu, S.; Jain, J. Study of Exposure of Carbonated Low-Lime Calcium Silicate Cements (CCSCs) Pastes to Sulfate Solutions at Low Temperatures. In Proceedings of the 12th International Symposium of Brittle Matrix Composites, Warsaw, Poland, 23–24 September 2019. [Google Scholar]
- Qin, L.; Gao, X.; Su, A.; Li, Q. Effect of Carbonation Curing on Sulfate Resistance of Cement-Coal Gangue Paste. J. Clean. Prod. 2021, 278, 123897. [Google Scholar] [CrossRef]
- Zhang, S.; Ghouleh, Z.; Azar, A.; Shao, Y. Improving concrete resistance to low temperature sulfate attack through carbonation curing. Mater. Struct. 2021, 54, 37. [Google Scholar] [CrossRef]
- Milkey, R. IR Spectra of Some Tectosilicates. Am. Mineral. 1960, 45, 990–1007. [Google Scholar]
- Huang, C.K.; Kerr, P.F. IR Study of Carbonate Minerals. Am. Mineral. 1960, 45, 311–324. [Google Scholar]
- Hunt, J.M.; Turner, D.S. Determination of Mineral Constituents of Rocks by IR. Anal. Chem. 1953, 25, 1169–1174. [Google Scholar] [CrossRef]
- Ihli, J.; Wong, W.; Noel, E.; Kim, Y.; Christenson, H.K.; Duer, M.; Meldrum, F. Dehydration and Crystallization of Amorphous Calcium Carbonate in Solution and Air. Nat. Commun. 2014, 5, 2–10. [Google Scholar] [CrossRef] [Green Version]
- Kirboga, S.; Oner, M. Investigation of Calcium Carbonate Precipitation in the Presence of Carboxymethyl Inulin. CrystEngComm 2013, 15, 3678–3686. [Google Scholar] [CrossRef]
- Ashraf, W.; Olek, J. Carbonation Behavior of Hydraulic and Non-hydraulic Calcium Silicates: Potential of Utilizing Low-Lime Calcium Silicates in Cement-based Materials. J. Mater. Sci. 2016, 51, 6173–6191. [Google Scholar] [CrossRef]
- Kalousek, G.L.; Roy, R. Crystal chemistry of Hydrous Calcium Silicates: II. Characterization of Interlayer Water. J. Am. Cer. Soc. 1957, 40, 236–239. [Google Scholar] [CrossRef]
- Hunt, J.M.; Wisherd, M.P.; Bonham, L.C. IR Absorption Spectra of Minerals and Other Inorganic Compounds. Anal. Chem. 1950, 22, 1478–1497. [Google Scholar] [CrossRef]
- Tettenhorst, R. Cation Migration in montmorillonite. Am. Mineral. 1962, 47, 769–773. [Google Scholar]
- Stubican, V.; Roy, R. IR Spectra of Layer-Structure Silicates. J. Am. Cer. Soc. 1961, 44, 625–627. [Google Scholar] [CrossRef]
- Launer, P.J. Regularities in the IR Absorption Spectra of Silicate Minerals. Am. Mineral. 1957, 42, 764–784. [Google Scholar]
- Yu, P.; Kirkpatrick, R.J.; Poe, B.; McMillan, P.F.; Cong, X. Structure of calcium silicate hydrate (C-S-H): Near-, Mid-, and Far-infrared spectroscopy. J. Am. Cer. Soc. 1999, 82, 742–748. [Google Scholar] [CrossRef]
- Hart, P.W.; Colson, G.W.; Burris, J. Application of Carbon Dioxide to Reduce Water-Side Lime Scale in Heat Exchangers. J. Sci. Technol. For. Prod. Process. 2011, 1, 67–70. [Google Scholar]
- Kim, T.; Olek, J. Effects of Sample Preparation and Interpretation of Thermogravimetric Curves on Calcium Hydroxide in Hydrated Pastes and Mortars. Transp. Res. Rec. 2012, 2290, 10–18. [Google Scholar] [CrossRef]
- Evangelou, V.P. Pyrite Oxidation and Its Control; CRC Press: Boca Raton, FL, USA, 1995. [Google Scholar]
- Harris, D.C. Quantitative Chemical Analysis, 6th ed.; W.H. Freeman and Company: New York, NY, USA, 2003. [Google Scholar]
- Adams, F. Ionic Concentration and Activities in Soil Solutions. Soil Soc. Am. Proc. 1971, 35, 420–426. [Google Scholar] [CrossRef]
- Hasset, J.J.; Jurinak, J.J. Effect of Mg2+ ion on the Solubility of Solid Carbonates. Soil Soc. Am. Proc. 1971, 35, 403–406. [Google Scholar] [CrossRef]
- Bonen, D.; Cohen, M.D. Magnesium Sulfate Attack on Portland Cement Paste—I. Microstructural Analysis. Cem. Concr. Res. 1992, 22, 169–180. [Google Scholar] [CrossRef]
- Bonen, D.; Cohen, M.D. Magnesium Sulfate Attack on Portland Cement Paste—II. Chemical and Mineralogical Analysis. Cem. Concr. Res. 1992, 22, 707–718. [Google Scholar] [CrossRef]
- Santhanam, M.; Cohen, M.D.; Olek, J. Mechanism of Sulfate Attack: A Fresh Look Part 1: Summary of Experimental Results. Cem. Concr. Res. 2002, 32, 915–921. [Google Scholar] [CrossRef]












| Cement Paste | Calcite/Total Amount of Carbonates, g/100 g of the Paste Sample | Presence of the Hump | Hump Temp. Range, °C |
|---|---|---|---|
| #1 | 41.5 | No | - |
| #2 | 40.5/42 | Yes | 567–619 |
| #3 | 27.2/34 | Yes | 450–633 |
| #4 | 32.5/36 | Yes | 500–632 |
| Ion | C*i, mol/L | Zi | Ci·Zi2 | I, mol/L |
|---|---|---|---|---|
| Sodium sulfate solution | ||||
| Na+ | 0.679 | 1 | 0.679 | 1.055 |
| SO42− | 0.358 | 2 | 1.430 | |
| Magnesium sulfate solution | ||||
| Mg2+ | 0.370 | 2 | 1.480 | 1.444 |
| SO42− | 0.352 | 2 | 1.408 | |
| Aluminum sulfate solution | ||||
| Al3+ | 0.240 | 3 | 2.158 | 1.829 |
| SO42− | 0.375 | 2 | 1.5 | |
| Reaction Equation | Ion-Pairing Constant (K) [22] | Comments |
| Ions pairing with sulfate ion | ||
| NaSO4− (aq.) ↔ Na+ + SO42− | 2.4 × 10−1 | Increased likelihood of magnesium and aluminum producing pairs with sulfates. |
| MgSO40 (aq.) ↔ Mg2+ + SO42− | 5.88 × 10−3 | |
| AlSO4+ (aq.) ↔ Al3+ + SO42− | 6.3 × 10−4 | |
| Ions pairing with carbonate ion | ||
| NaCO3− (aq.) ↔ Na+ + CO32− | 5.35 × 10−2 | It is more likely that MgCO3 (aq.) will be present. |
| MgCO30 (aq.) ↔ Mg2+ + CO32− | 4 × 10−4 | |
| For Al3+: N/A | N/A | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tokpatayeva, R.; Olek, J.; Sahu, S. Resistance of Pastes from Carbonated, Low-Lime Calcium Silica Cements to External Sulfate Attack. Materials 2023, 16, 4276. https://doi.org/10.3390/ma16124276
Tokpatayeva R, Olek J, Sahu S. Resistance of Pastes from Carbonated, Low-Lime Calcium Silica Cements to External Sulfate Attack. Materials. 2023; 16(12):4276. https://doi.org/10.3390/ma16124276
Chicago/Turabian StyleTokpatayeva, Raikhan, Jan Olek, and Sadananda Sahu. 2023. "Resistance of Pastes from Carbonated, Low-Lime Calcium Silica Cements to External Sulfate Attack" Materials 16, no. 12: 4276. https://doi.org/10.3390/ma16124276