
Microwave Synthesis of Poly(Acrylic) Acid-Coated Magnetic Nanoparticles as Draw Solutes in Forward Osmosis
 , , ,
, , ,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
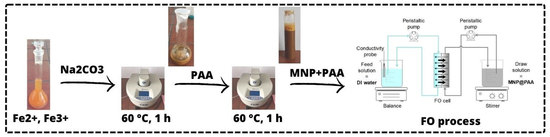
2.1. Particle Synthesis and Coating
2.2. X-ray Diffraction Analysis (XRD)
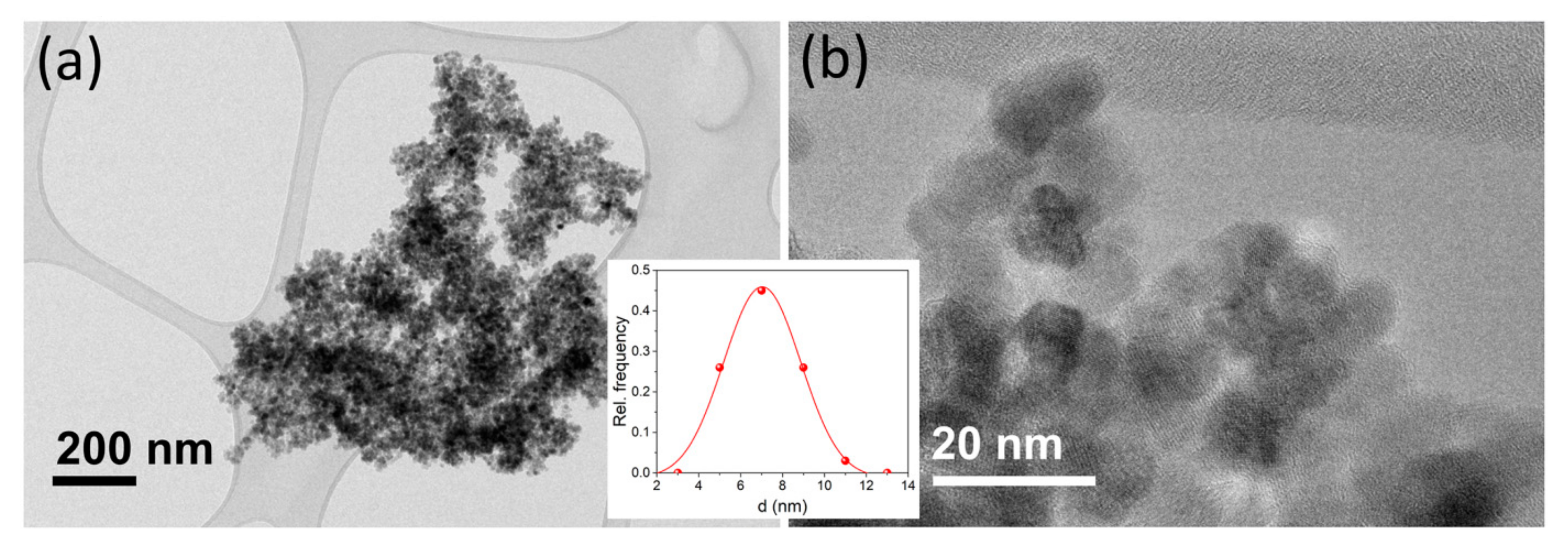
2.3. Transmission Electron Microscopy (TEM)
2.4. Fourier Transform Infrared Spectroscopy (FTIR)
2.5. Thermogravimetric Analysis (TGA)
2.6. Dynamic Light Scattering
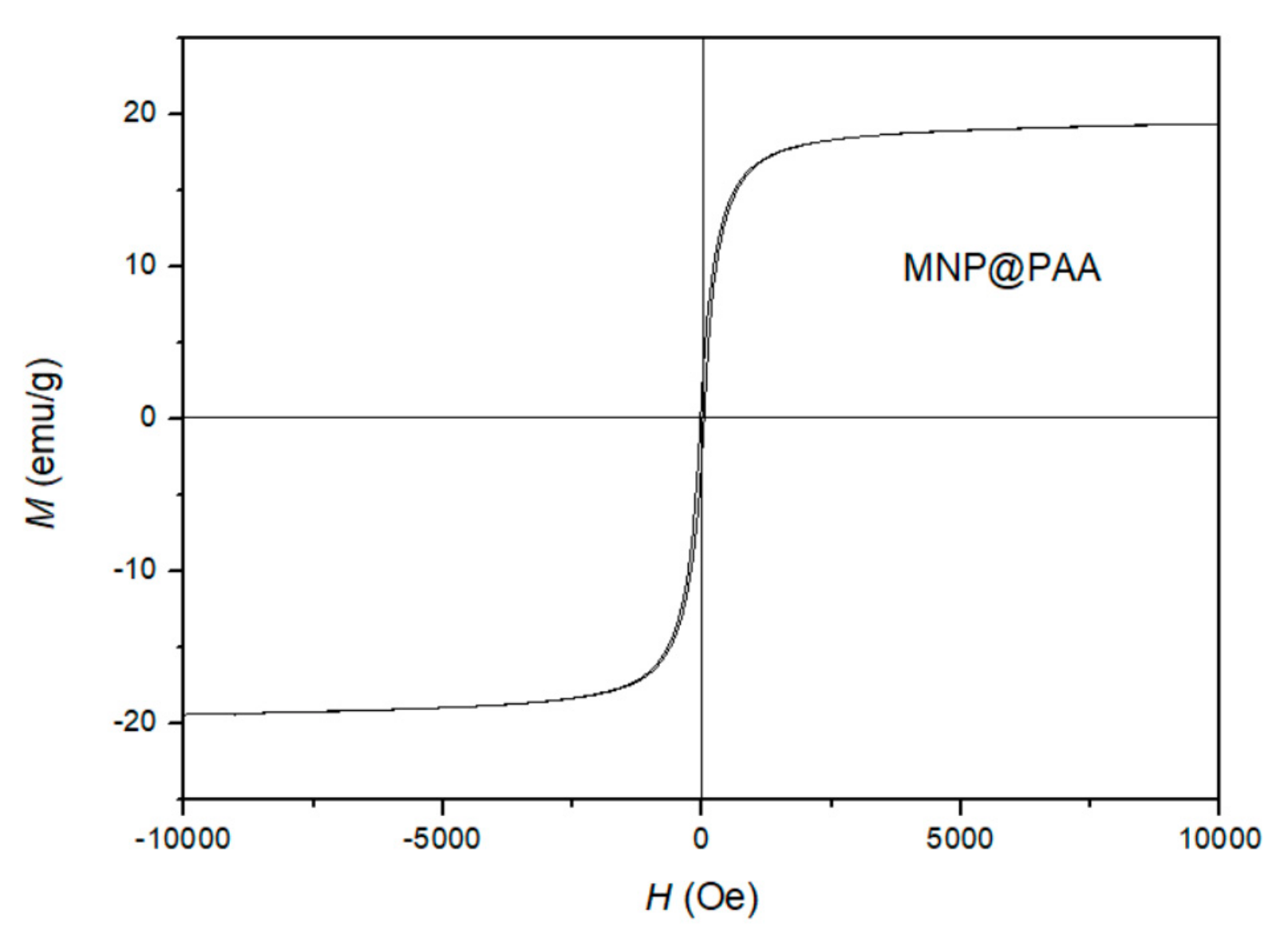
2.7. Magnetic Measurements
2.8. Osmotic Pressure
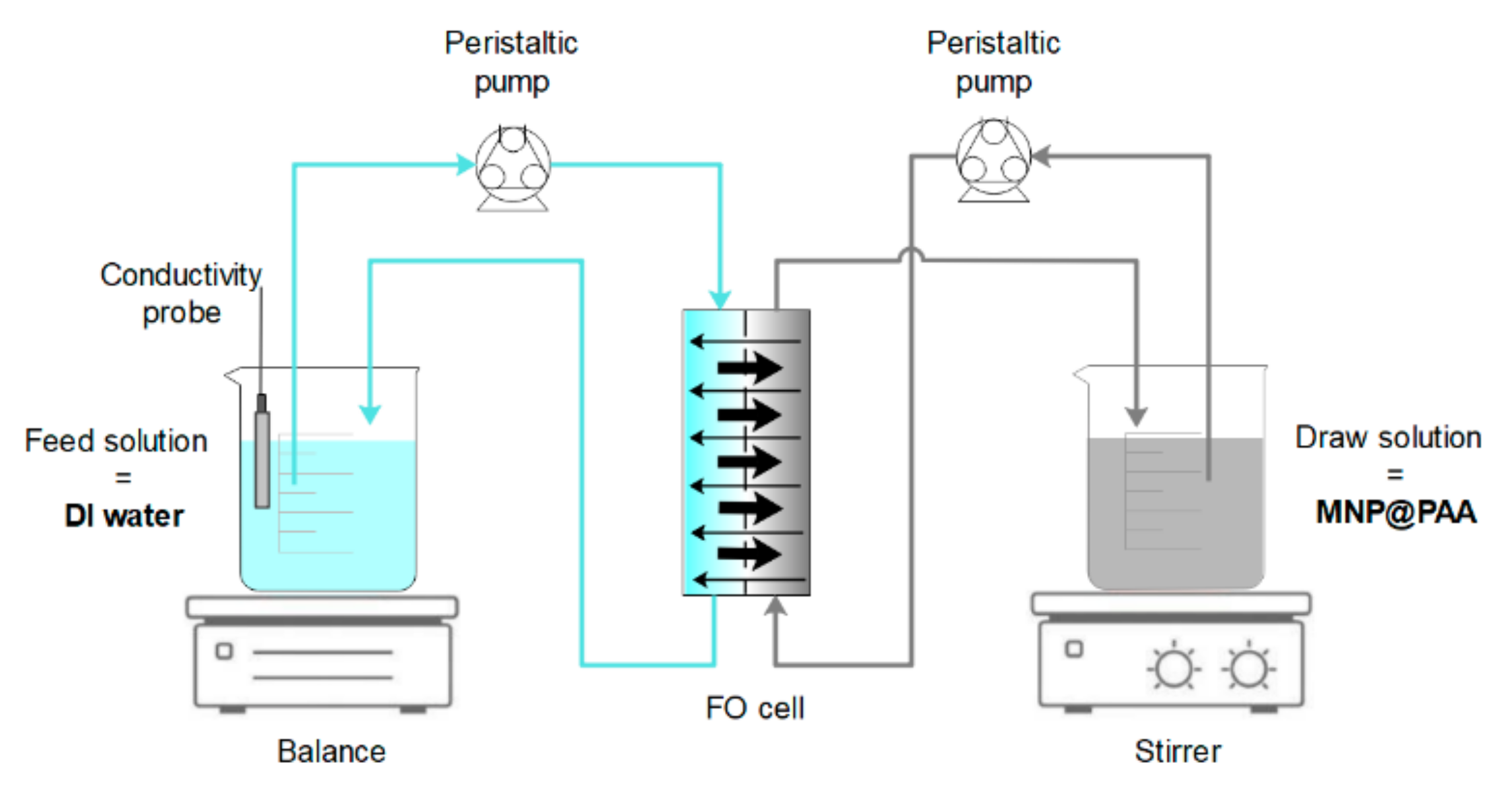
2.9. FO Filtration Experiments
3. Results and Discussion
3.1. Synthesis and Particle Size Distribution
3.2. Surface Characterization
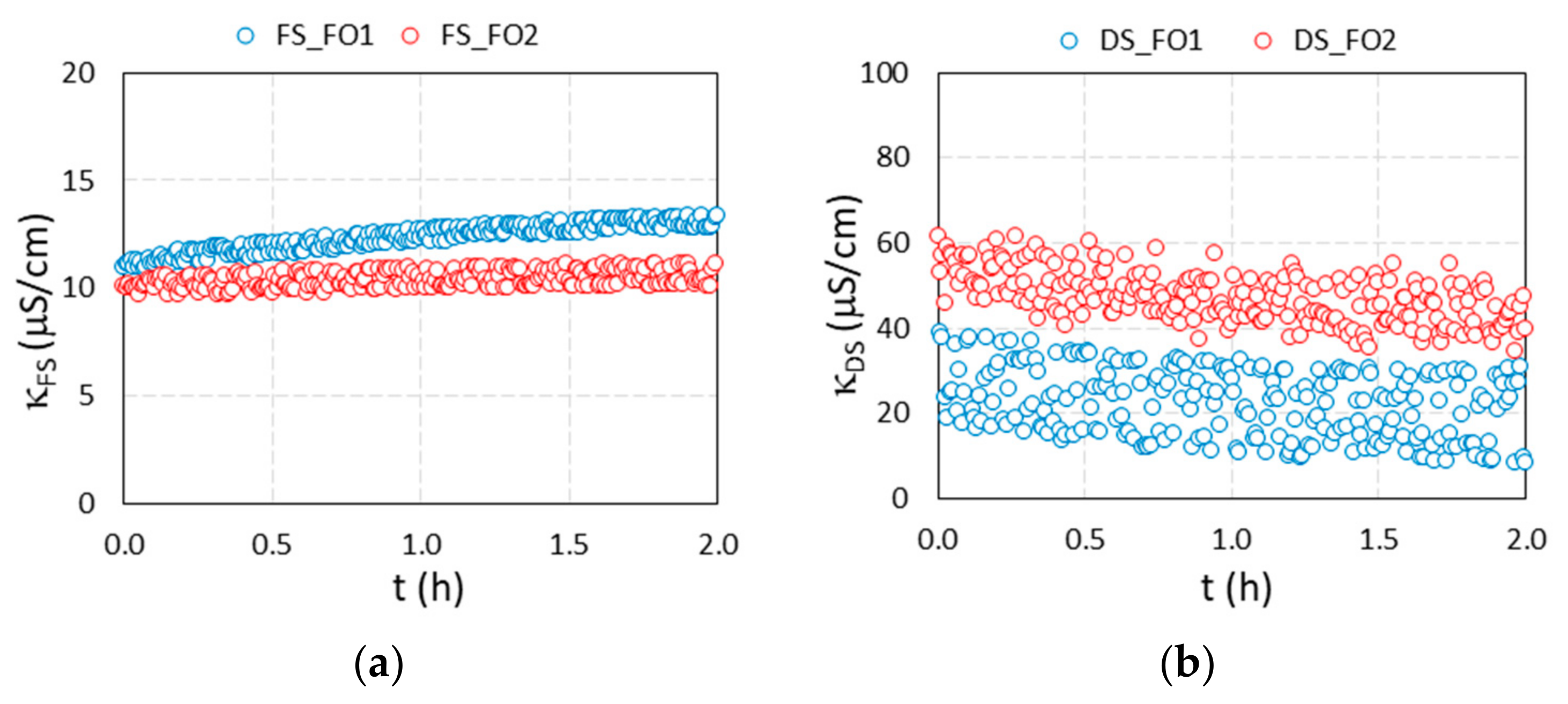
3.3. Results of FO Experiments
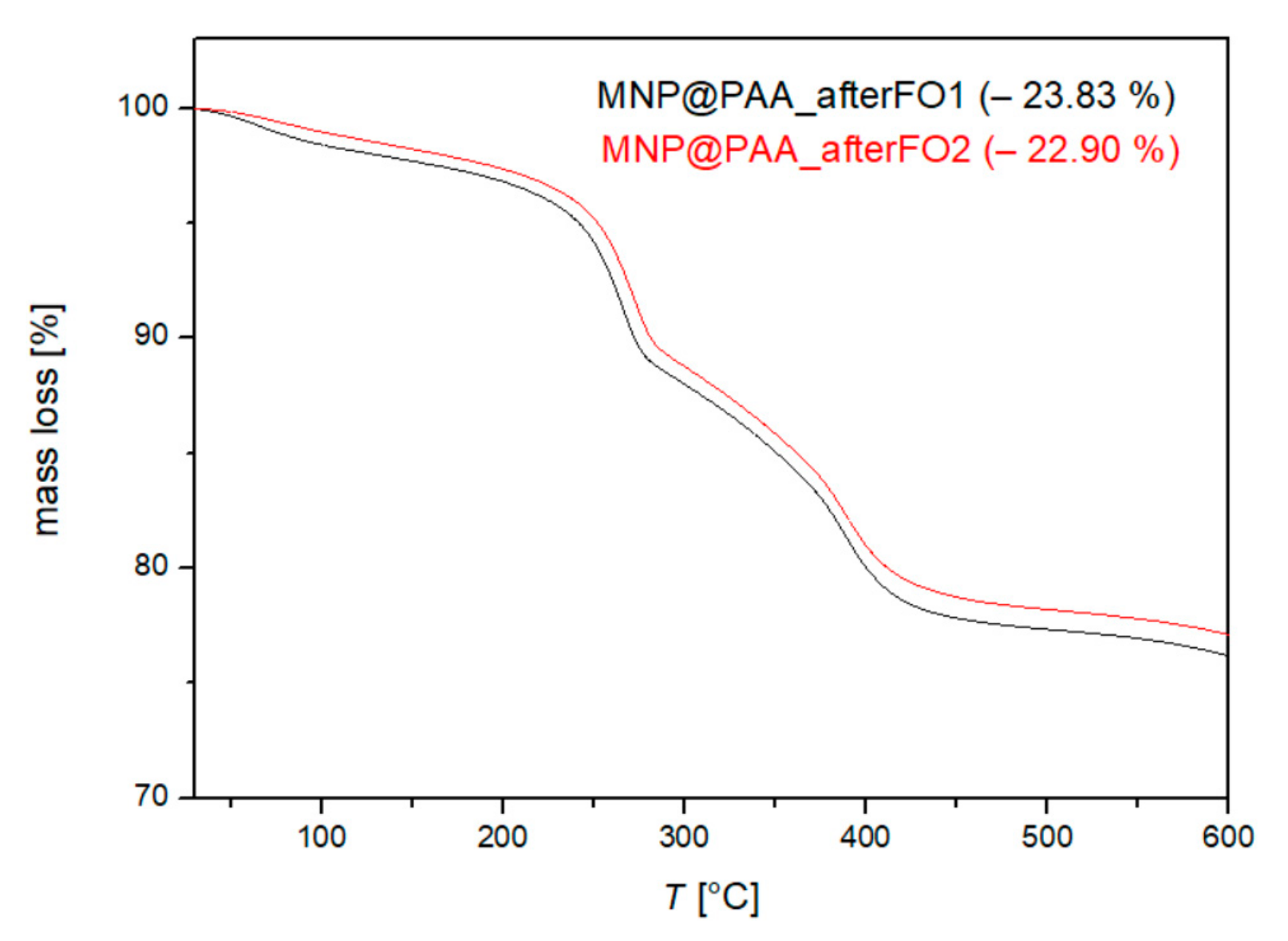
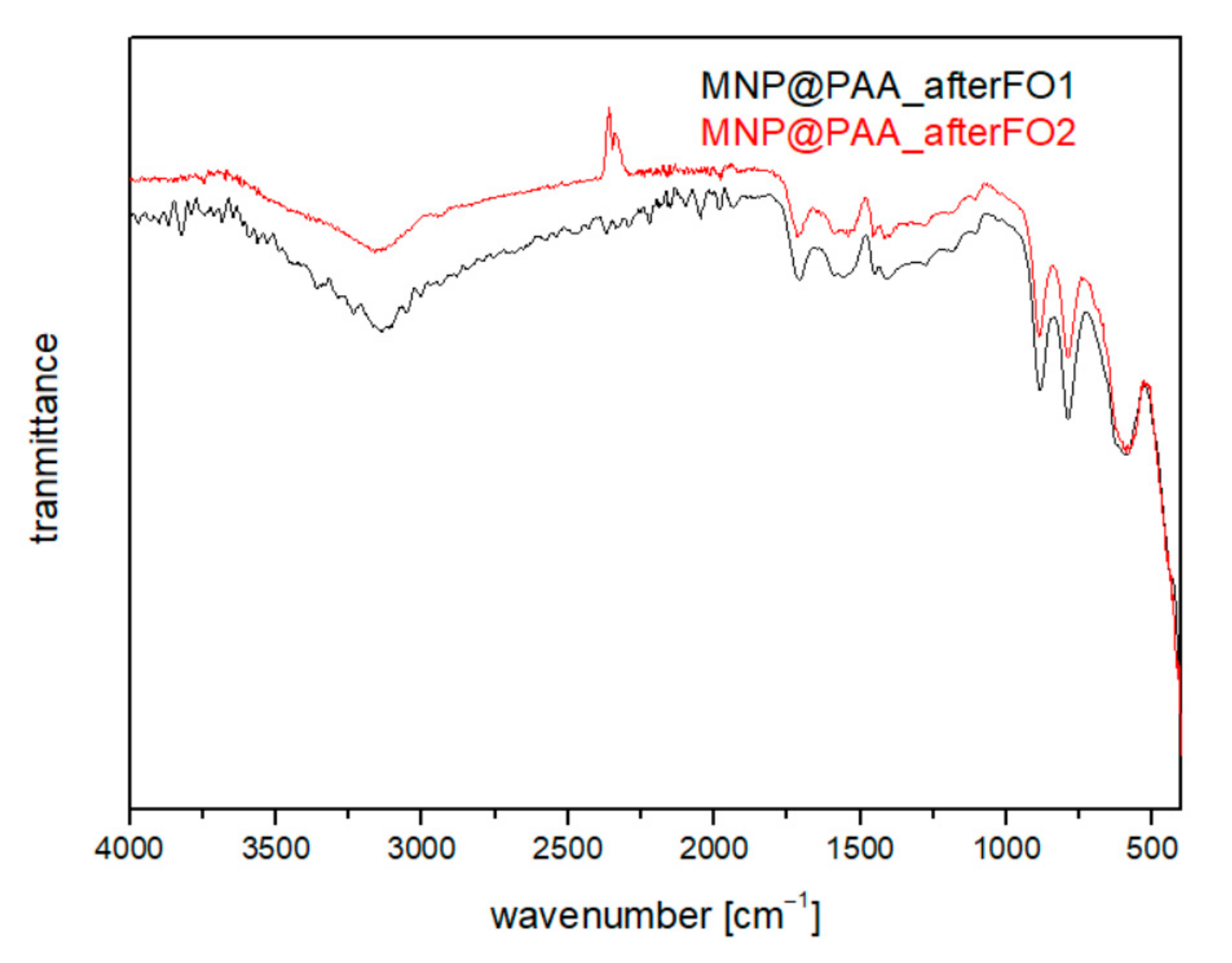
3.4. Characterization after the First and Second FO Processes
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wang, J.; Liu, X. Forward osmosis technology for water treatment: Recent advances and future perspectives. J. Clean. Prod. 2021, 280, 124354. [Google Scholar] [CrossRef]
- Salomon, E.; Tijani, J.; Badmus, K.; Pereao, O.; Onaneye-Babajide, O.; Zhang, C.; Shao, T.; Sosnin, E.; Tarasenko, V.; Fatoba, O.; et al. Removal of Pharmaceutical Residues from Water and Wastewater Using Dielectric Barrier Discharge Methods-A Review. Int. J. Environ. Res. Public Health 2021, 18, 1683. [Google Scholar]
- Semiat, R. Energy Issues in Desalination Processes. Environ. Sci. Technol. 2008, 42, 8193–8201. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Zhu, Y.; Chen, Z.; Zhu, J.; Chen, G. A Comprehensive Review on Forward Osmosis Water Treatment: Recent Advances and Prospects of Membranes and Draw Solutes. Int. J. Environ. Res. Public Health 2022, 19, 8215. [Google Scholar] [CrossRef] [PubMed]
- Cath, T.; Childress, A.; Elimelech, M. Forward osmosis: Principles, applications, and recent developments. J. Membr. Sci. 2006, 281, 70–87. [Google Scholar] [CrossRef]
- Johnson, D.; Suwaileh, W.; Mohammed, A.; Hilal, N. Osmotic’s potential: An overview of draw solutes for forward osmosis. Desalination 2018, 434, 100–120. [Google Scholar] [CrossRef]
- Al-Sakaji, B.; Al-Asheh, S.; Maraqa, M. Effects of Operating Conditions on the Performance of Forward Osmosis with Ultrasound for Seawater Desalination. Water 2022, 14, 2092. [Google Scholar] [CrossRef]
- Lugito, G.; Ariono, D.; Putra, M.; Zafra, Z. Progress, Challenges, and Prospects of Forward Osmosis (FO) and Pressure Retarded Osmosis (PRO) as an Alternative Solution for Water and Energy Crisis; IOP Conference Series: Materials Science and Engineering; IOP Publishing: Bristol, UK, 2021; p. 012060. [Google Scholar]
- Ezugbe, E.; Tetteh, E.K.; Rathilal, S.; Asante-Sackey, D.; Amo-Duodu, G. Desalination of municipal wastewater using forward osmosis. Membranes 2021, 11, 119. [Google Scholar] [CrossRef]
- Nijmeijer, K.; Oymaci, P.; Lubach, S.; Borneman, Z. Apple Juice, Manure and Whey Concentration with Forward Osmosis Using Electrospun Supported Thin-Film Composite Membranes. Membranes 2022, 12, 456. [Google Scholar] [CrossRef]
- Gosmann, L.; Geitner, C.; Wieler, N. Data-driven forward osmosis model development using multiple linear regression and artificial neural networks. Comput. Chem. Eng. 2022, 165, 107933. [Google Scholar] [CrossRef]
- Kim, D.; Gwak, G.; Zhan, M.; Hong, S. Sustainable dewatering of grapefruit juice through forward osmosis: Improving membrane performance, fouling control, and product quality. J. Membr. Sci. 2019, 578, 53–60. [Google Scholar] [CrossRef]
- Yazdanabad, S.; Samimi, A.; Shokrollahzadeh, S.; Kalhori, D.; Moazami, N.; González, M.; Sobczuk, T.; Grima, E. Microalgae biomass dewatering by forward osmosis: Review and critical challenges. Algal Res. 2021, 56, 102323. [Google Scholar] [CrossRef]
- Cao, D.-Q.; Liu, H.; Tian, F.; Zhang, W.-Y.; Hao, X.-D.; Iritani, E.; Katagiri, N. Dead-end forward osmosis as an alternative for deep sludge dewatering: Evaluation method and characteristics analysis. Chem. Eng. J. 2023, 143519. [Google Scholar] [CrossRef]
- Le, H.; Nguyen, T.; Chen, S.-S.; Duong, C.; Cao, T.-D.; Chang, H.-M.; Ray, S.; Nguyen, N. Application of progressive freezing on forward osmosis draw solute recovery. Environ. Sci. Pollut. Res. 2020, 27, 34664–34674. [Google Scholar] [CrossRef] [PubMed]
- Francis, L.; Ogunbiyi, O.; Saththasivam, J.; Lawler, J.; Liu, Z. A comprehensive review of forward osmosis and niche applications. Environ. Sci. Water Res. Technol. 2020, 6, 1986–2015. [Google Scholar] [CrossRef]
- Ibrar, I.; Altaee, A.; Zhou, J.; Naji, O.; Khanafer, D. Challenges and potentials of forward osmosis process in the treatment of wastewater. Crit. Rev. Environ. Sci. Technol. 2020, 50, 1339–1383. [Google Scholar] [CrossRef]
- Deniz, Ş. Forward Osmosis Membrane Technology in Wastewater Treatment. In Osmotically Driven Membrane Processes; Muharrem, I., Kaplan, I.O., Eds.; IntechOpen: Rijeka, Croatia, 2021; Chapter 3. [Google Scholar]
- Ibraheem, B.; Aani, S.; Alsarayreh, A.; Alsalhy, Q.; Salih, I. Forward Osmosis Membrane: Review of Fabrication, Modification. Challenges and Potential. Membranes 2023, 13, 374. [Google Scholar] [CrossRef]
- Mecha, A. Applications of Reverse and Forward Osmosis Processes in Wastewater Treatment: Evaluation of Membrane Fouling; IntechOpen: Rijeka, Croatia, 2017. [Google Scholar]
- Chekli, L.; Phuntsho, S.; Shon, H.; Vigneswaran, S.; Kandasamy, J.; Chanan, A. A review of draw solutes in forward osmosis process and their use in modern applications. Desalination Water Treat. 2012, 43, 167–184. [Google Scholar] [CrossRef]
- Chaoui, I.; Abderafi, S.; Vaudreuil, S.; Bounahmidi, T. Water desalination by forward osmosis: Draw solutes and recovery methods–review. Environ. Technol. Rev. 2019, 8, 25–46. [Google Scholar] [CrossRef]
- McCutcheon, J.; McGinnis, R.; Elimelech, M. Desalination by ammonia–carbon dioxide forward osmosis: Influence of draw and feed solution concentrations on process performance. J. Membr. Sci. 2006, 278, 114–123. [Google Scholar] [CrossRef]
- Long, Q.; Shen, L.; Chen, R.-Y.; Huang, J.; Xiong, S.; Wang, Y. Synthesis and Application of Organic Phosphonate Salts as Draw Solutes in Forward Osmosis for Oil-Water Separation. Environ. Sci. Technol. 2016, 50, 12022–12029. [Google Scholar] [CrossRef]
- Tharayil, J.; Manaf, A. Effect of different inorganic draw solutes on SiNPs-TFN membrane for forward osmosis desalination. Int. J. Environ. Sci. Technol. 2021, 19, 289–298. [Google Scholar] [CrossRef]
- Alejo, T.; Arruebo, M.; Carcelen, V.; Monsalvo, V.; Sebastian, V. Advances in draw solutes for forward osmosis: Hybrid organic-inorganic nanoparticles and conventional solutes. Chem. Eng. J. 2017, 309, 738–752. [Google Scholar] [CrossRef]
- Nizam, A.; Yeo, W. Effect of Draw Solution on the Treatment of Humic Acid in Forward Osmosis Process. In Proceedings of the 8th Malaysian Technical Universities Conference on Engineering & Technology (MUCET 2014), Malacca, Malaysia, 10–11 November 2014. [Google Scholar]
- Alaswad, S.; Al-aibi, S.; Alpay, E.; Sharif, A. Efficiency of Organic Draw Solutions in a Forward Osmosis Process Using Nano-filtration Flat Sheet Membrane. J. Chem. Eng. Process Technol. 2018, 9, 1–10. [Google Scholar] [CrossRef]
- Maouia, D.; Boubakri, A.; Hafiane, A.; Bouguecha, S.-T. Aluminum Sulfate as an Innovative Draw Solute for Forward Osmosis Desalination. Chem. Afr. 2020, 3, 141–152. [Google Scholar] [CrossRef]
- Xu, P.; Zeng, G.; Huang, D.; Feng, C.; Hu, S.; Zhao, M.; Lai, C.; Wei, Z.; Huang, C.; Xie, G.; et al. Use of iron oxide nanomaterials in wastewater treatment: A review. Sci. Total Environ. 2012, 424, 1–10. [Google Scholar] [CrossRef]
- Lin, C.; Lee, C.; Chiu, W. Preparation and properties of poly(acrylic acid) oligomer stabilized superparamagnetic ferrofluid. J. Colloid Interface Sci. 2005, 291, 411–420. [Google Scholar] [CrossRef]
- Hafiz, M.; Hassanein, A.; Talhami, M.; Al-Ejji, M.; Hassan, M.; Hawari, A. Magnetic nanoparticles draw solution for forward osmosis: Current status and future challenges in wastewater treatment. J. Environ. Chem. Eng. 2022, 10, 108955. [Google Scholar] [CrossRef]
- Hafiz, M.; Talhami, M.; Ba-Abbad, M.; Hawari, A. Optimization of Magnetic Nanoparticles Draw Solution for High Water Flux in Forward Osmosis. Water 2021, 13, 3653. [Google Scholar] [CrossRef]
- Dey, P.; Izake, E. Magnetic nanoparticles boosting the osmotic efficiency of a polymeric FO draw agent: Effect of polymer conformation. Desalination 2015, 373, 79–85. [Google Scholar] [CrossRef]
- Ban, I.; Drofenik, M.; Bukšek, H.; Petrinic, I.; Helix-Nielsen, C.; Vohl, S.; Gyergyek, S.; Stergar, J. Synthesis of magnetic nanoparticles with covalently bonded polyacrylic acid for use as forward osmosis draw agents. Environ. Sci. Water Res. Technol. 2023, 9, 442–453. [Google Scholar] [CrossRef]
- Shin, J.; An, G.; Choi, S.-C. Influence of Carboxylic Modification Using Polyacrylic Acid on Characteristics of Fe3O4 Nanoparticles with Cluster Structure. Processes 2021, 9, 1795. [Google Scholar] [CrossRef]
- Zhao, P.; Liu, R.; Liu, H.; Peng, J.; Qu, J. Introduction of poly(acrylic acid) sodium into traditional draw solution to enhance its driving capacity in forward osmosis process. Environ. Sci. Pollut. Res. 2022, 30, 19224–19233. [Google Scholar] [CrossRef] [PubMed]
- Schneider, T.; Löwa, A.; Karagiozov, S.; Sprenger, L.; Gutiérrez, L.; Esposito, T.; Marten, G.; Saatchi, K.; Häfeli, U. Facile microwave synthesis of uniform magnetic nanoparticles with minimal sample processing. J. Magn. Magn. Mater. 2017, 421, 283–291. [Google Scholar] [CrossRef]
- Pineda, R.; Tomás, M.; Moya, M.; Foix, F.; Moreno, R. Comparison in mechanical properties of zirconium titanate (ZrTiO4) synthetized by alternative routes and sintered by microwave (MW). In Proceedings of the 17th International Conference on Microwave and High Frequency Heating, Valencia, Spain, 9–12 September 2019. [Google Scholar]
- Liu, M.; Sun, M.; Wang, Z. Synthesis, Optical Properties and Applications of Ternary Oxide Nanoparticles by a Microwave Technique. J. Nanosci. Nanotechnol. 2021, 21, 5307–5311. [Google Scholar] [CrossRef]
- Li, C.; Wei, Y.; Liivat, A.; Zhu, Y.; Zhu, J. Microwave-solvothermal synthesis of Fe3O4 magnetic nanoparticles. Mater. Lett. 2013, 107, 23–26. [Google Scholar] [CrossRef]
- Petrinic, I.; Stergar, J.; Bukšek, H.; Drofenik, M.; Gyergyek, S.; Hélix-Nielsen, C.; Ban, I. Superparamagnetic Fe3O4@CA Nanoparticles and Their Potential as Draw Solution Agents in Forward Osmosis. Nanomaterials 2021, 11, 2965. [Google Scholar] [CrossRef]
- Ferk, G.; Ban, I.; Stergar, J.; Makovec, D.; Hamler, A.; Jagličić, Z.; Drofenik, M. A Facile Route to the Synthesis of Coated Maghemite Nanocomposites for Hyperthermia Applications. Acta Chim. Slov. 2012, 59, 366–374. [Google Scholar] [PubMed]
- Blanco-Andujar, C.; Ortega, D.; Pankhurst, Q.; Thanh, N. Elucidating the morphological and structural evolution of iron oxide nanoparticles formed by sodium carbonate in aqueous medium. J. Mater. Chem. 2012, 22, 12498–12506. [Google Scholar] [CrossRef]
- Ban, I.; Markuš, S.; Gyergyek, S.; Drofenik, M.; Korenak, J.; Helix-Nielsen, C.; Petrinic, I. Synthesis of Poly-Sodium-Acrylate (PSA) Coated Magnetic Nanoparticles for Use in Forward Osmosis Draw Solutions. Nanomaterials 2019, 9, 1238. [Google Scholar] [CrossRef] [PubMed]
- Piñeiro-Redondo, Y.; Bañobre-López, M.; Pardiñas-Blanco, I.; Goya, G.; López-Quintela, M.; Rivas, J. The influence of colloidal parameters on the specific power absorption of PAA-coated magnetite nanoparticles. Nanoscale Res. Lett. 2011, 6, 383. [Google Scholar] [CrossRef] [PubMed]
- Datsyuk, V.; Billon, L.; Guerret-Piecourt, C.; Dagréou, S.; Passade-Boupatt, N.; Bourrigaud, S.; Guerret, O.; Couvreur, L. In situ nitroxide-mediated polymerized poly(acrylic acid) as a stabilizer/compatibilizer carbon nanotube/polymer composite. J. Nanomater 2007, 2007, 5. [Google Scholar] [CrossRef]













Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vohl, S.; Ban, I.; Drofenik, M.; Buksek, H.; Gyergyek, S.; Petrinic, I.; Hélix-Nielsen, C.; Stergar, J. Microwave Synthesis of Poly(Acrylic) Acid-Coated Magnetic Nanoparticles as Draw Solutes in Forward Osmosis. Materials 2023, 16, 4138. https://doi.org/10.3390/ma16114138
Vohl S, Ban I, Drofenik M, Buksek H, Gyergyek S, Petrinic I, Hélix-Nielsen C, Stergar J. Microwave Synthesis of Poly(Acrylic) Acid-Coated Magnetic Nanoparticles as Draw Solutes in Forward Osmosis. Materials. 2023; 16(11):4138. https://doi.org/10.3390/ma16114138
Chicago/Turabian StyleVohl, Sabina, Irena Ban, Miha Drofenik, Hermina Buksek, Sašo Gyergyek, Irena Petrinic, Claus Hélix-Nielsen, and Janja Stergar. 2023. "Microwave Synthesis of Poly(Acrylic) Acid-Coated Magnetic Nanoparticles as Draw Solutes in Forward Osmosis" Materials 16, no. 11: 4138. https://doi.org/10.3390/ma16114138