
Insight into the Exemplary Physical Properties of Zn-Based Fluoroperovskite Compounds XZnF3 (X = Al, Cs, Ga, In) Employing Accurate GGA Approach: A First-Principles Study


 , ,
, ,  ,
,
Abstract
:1. Introduction
2. Methodology
3. Results and Discussion
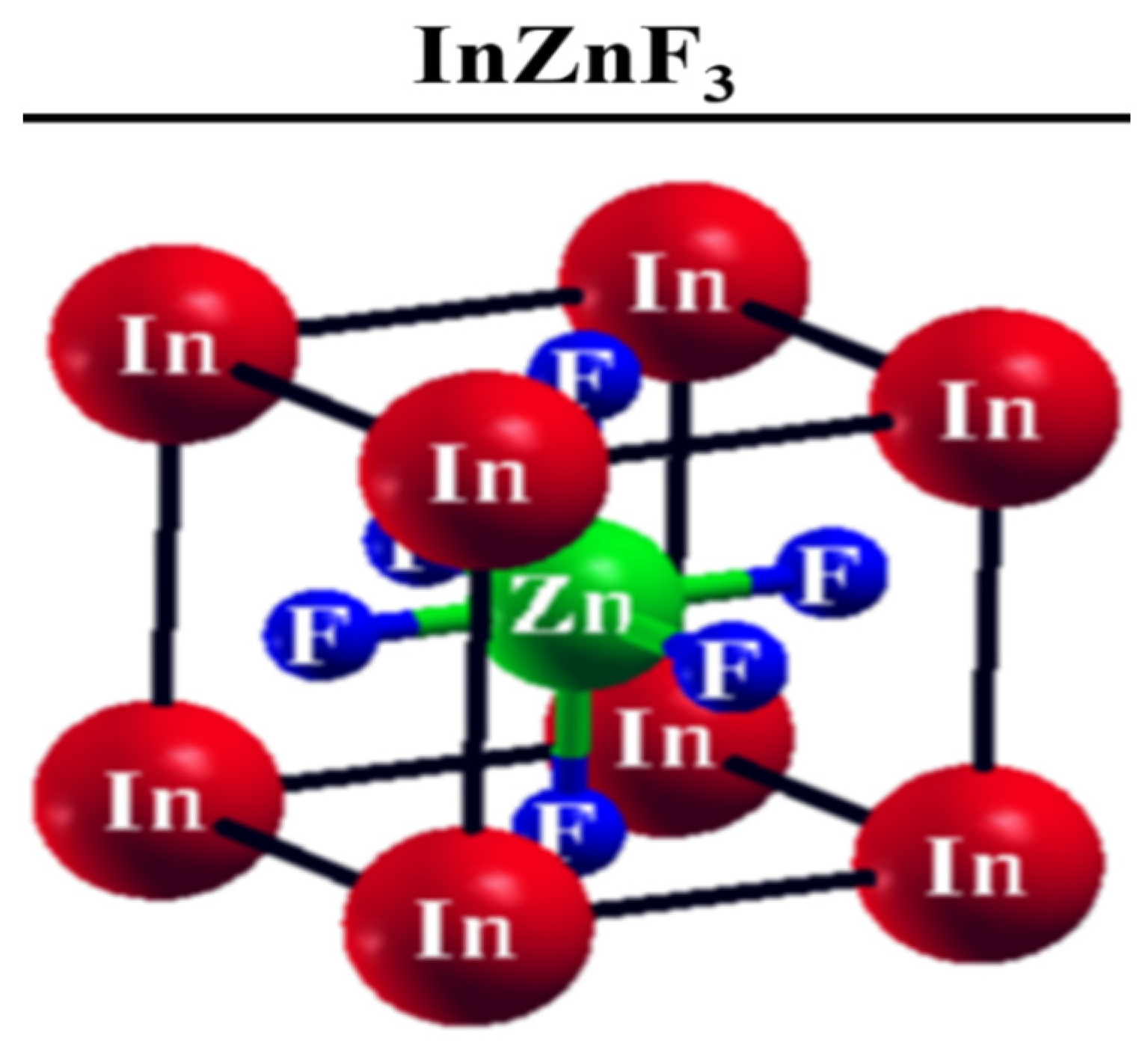
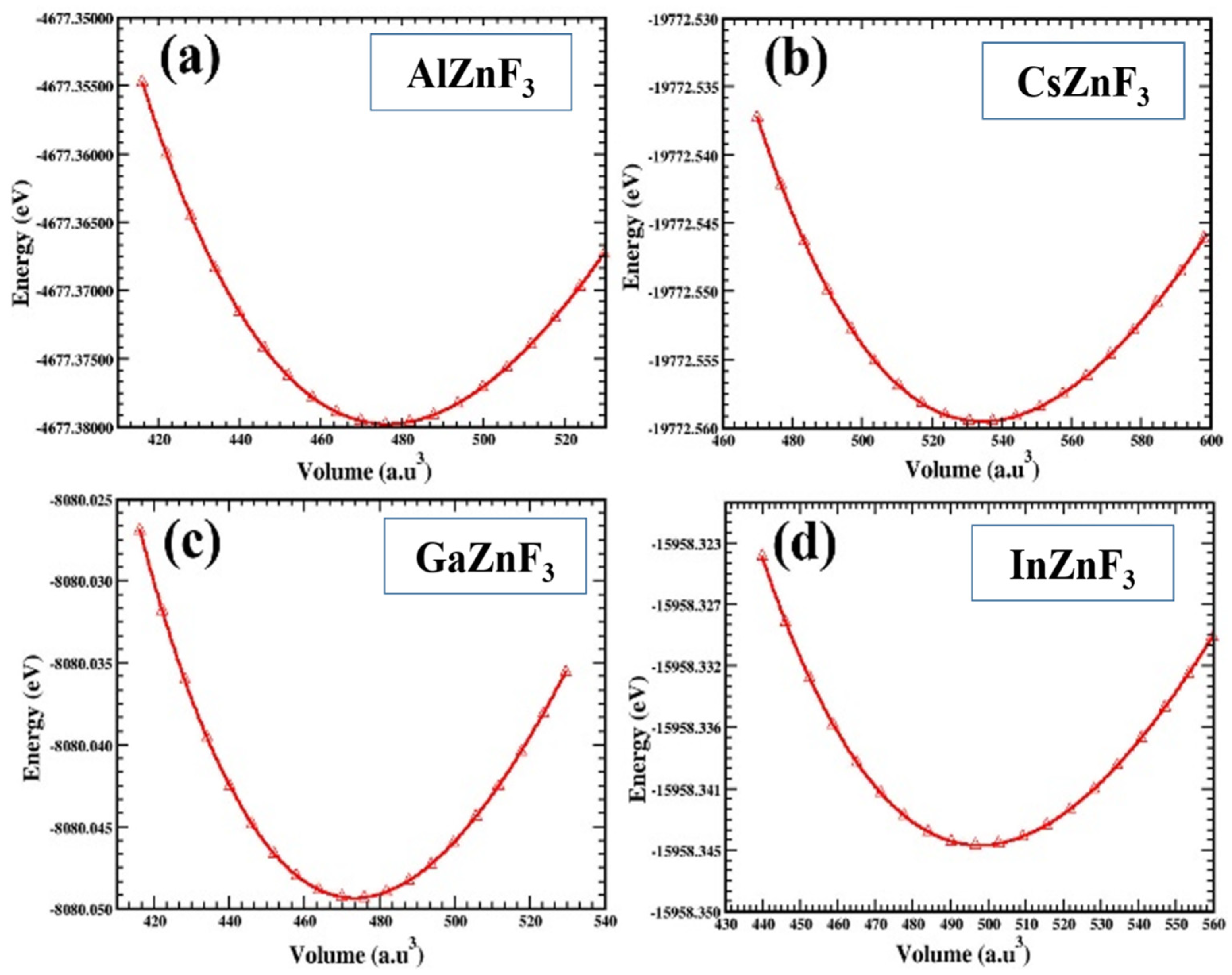
3.1. Structural Properties
3.2. Elastic Properties
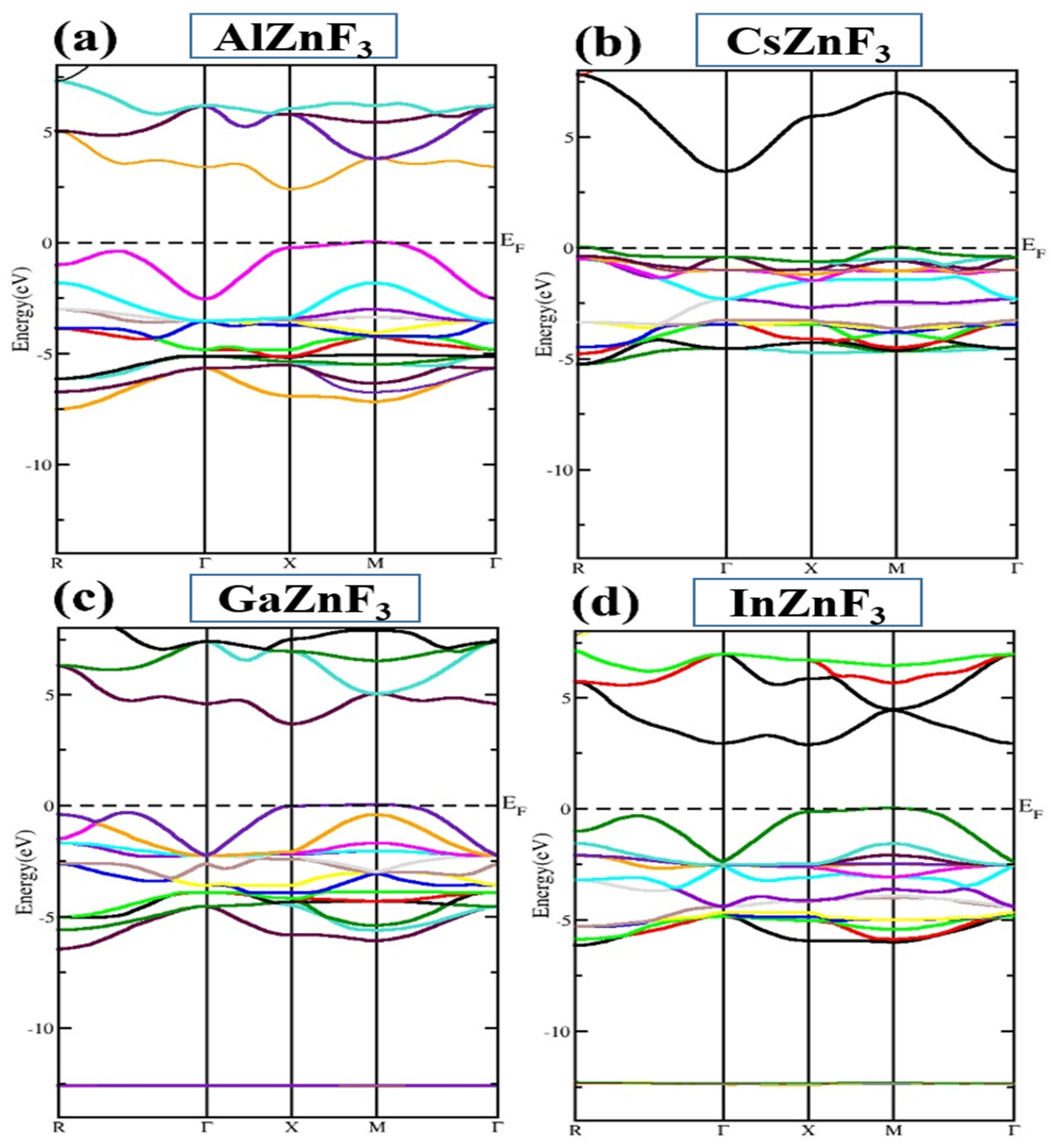
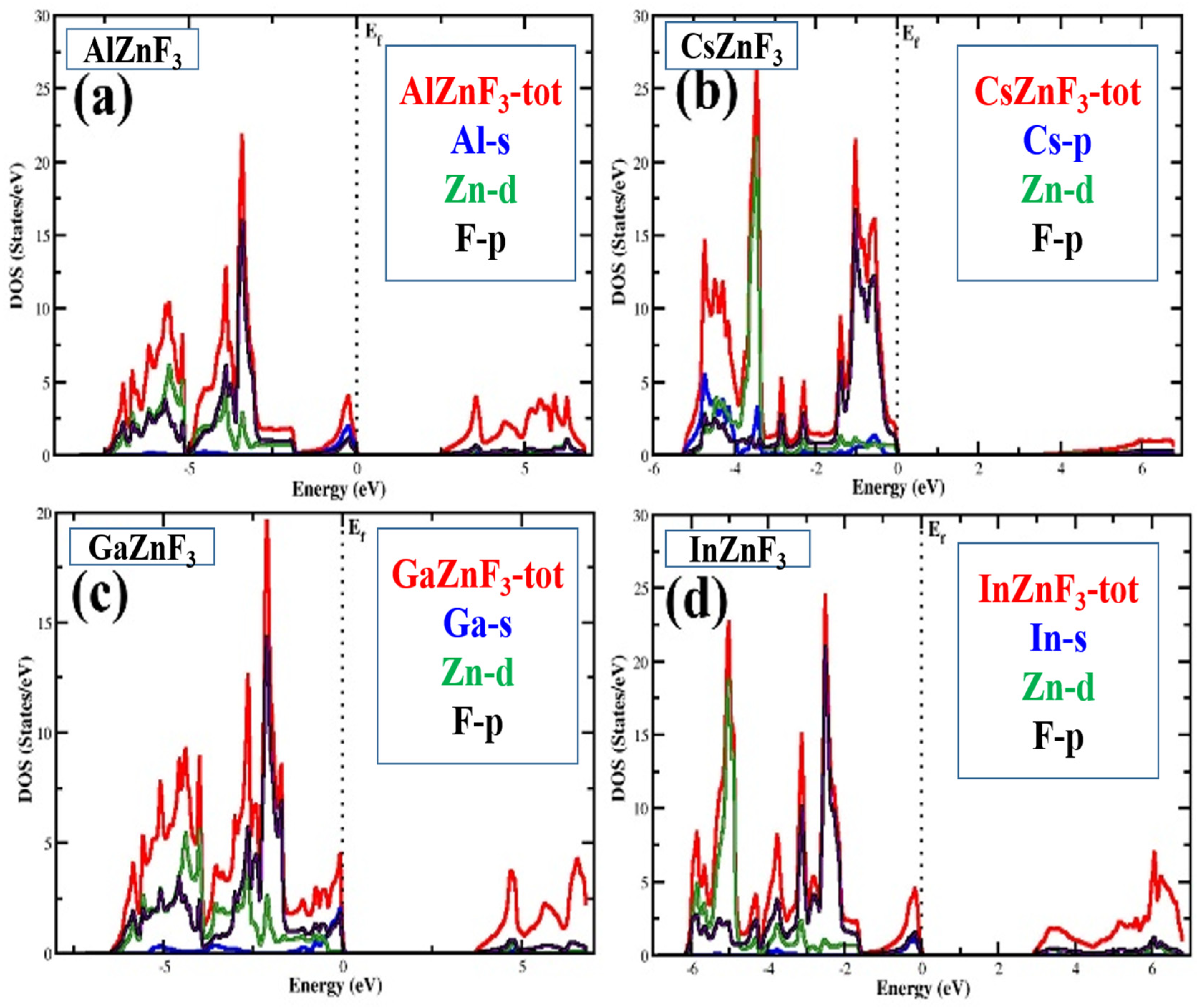
3.3. Electronic Properties
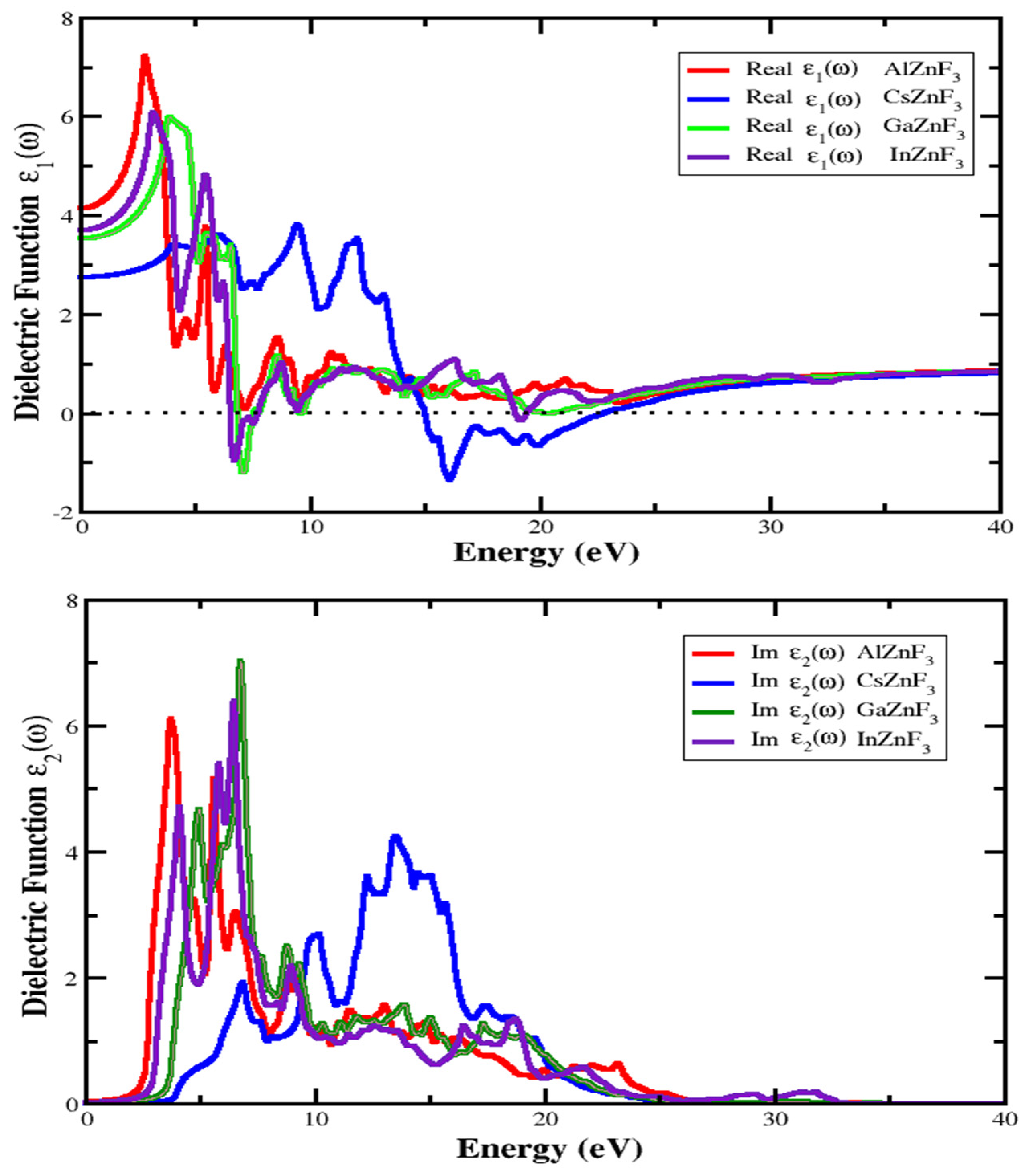
3.4. Optical Properties
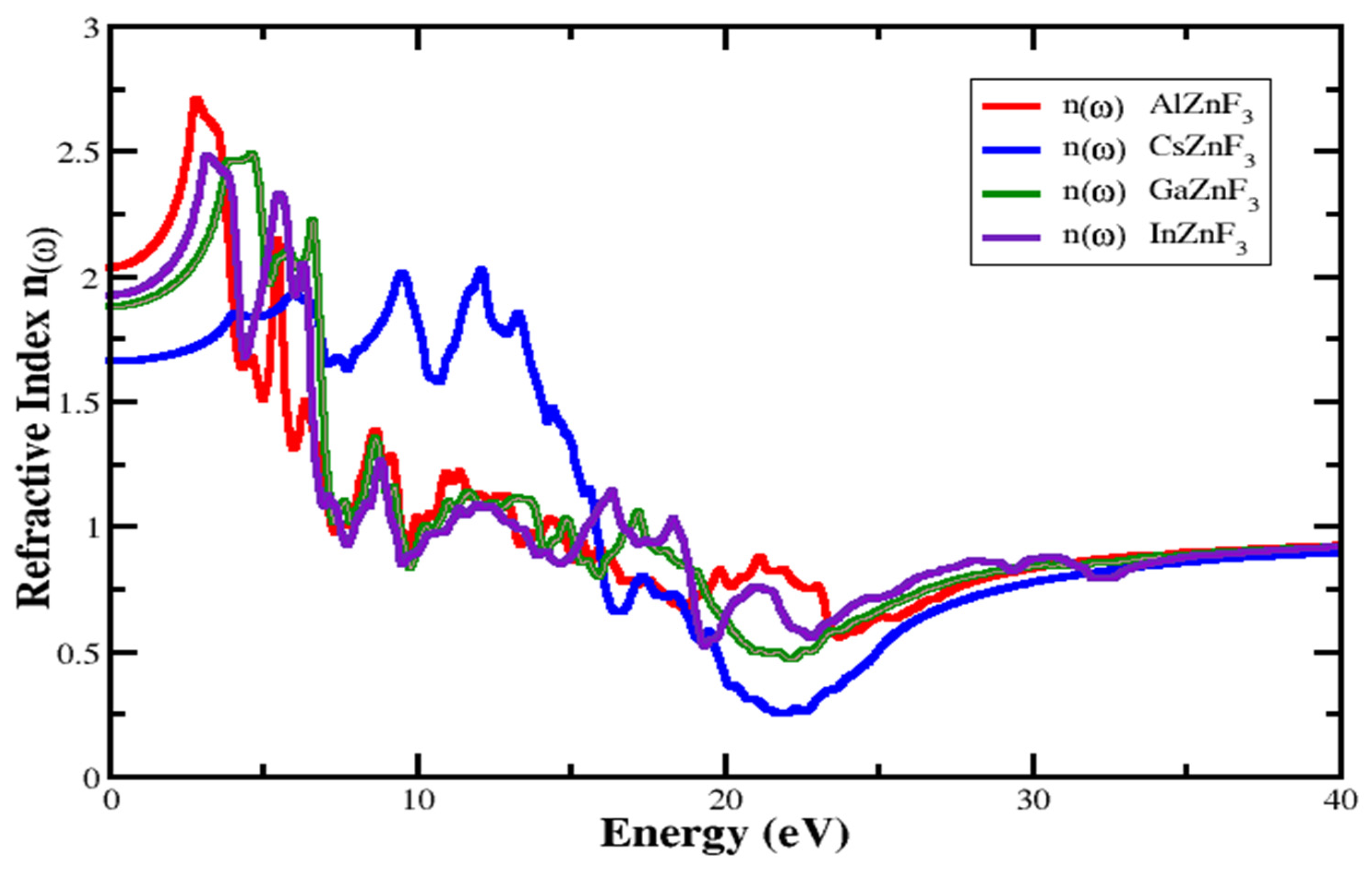
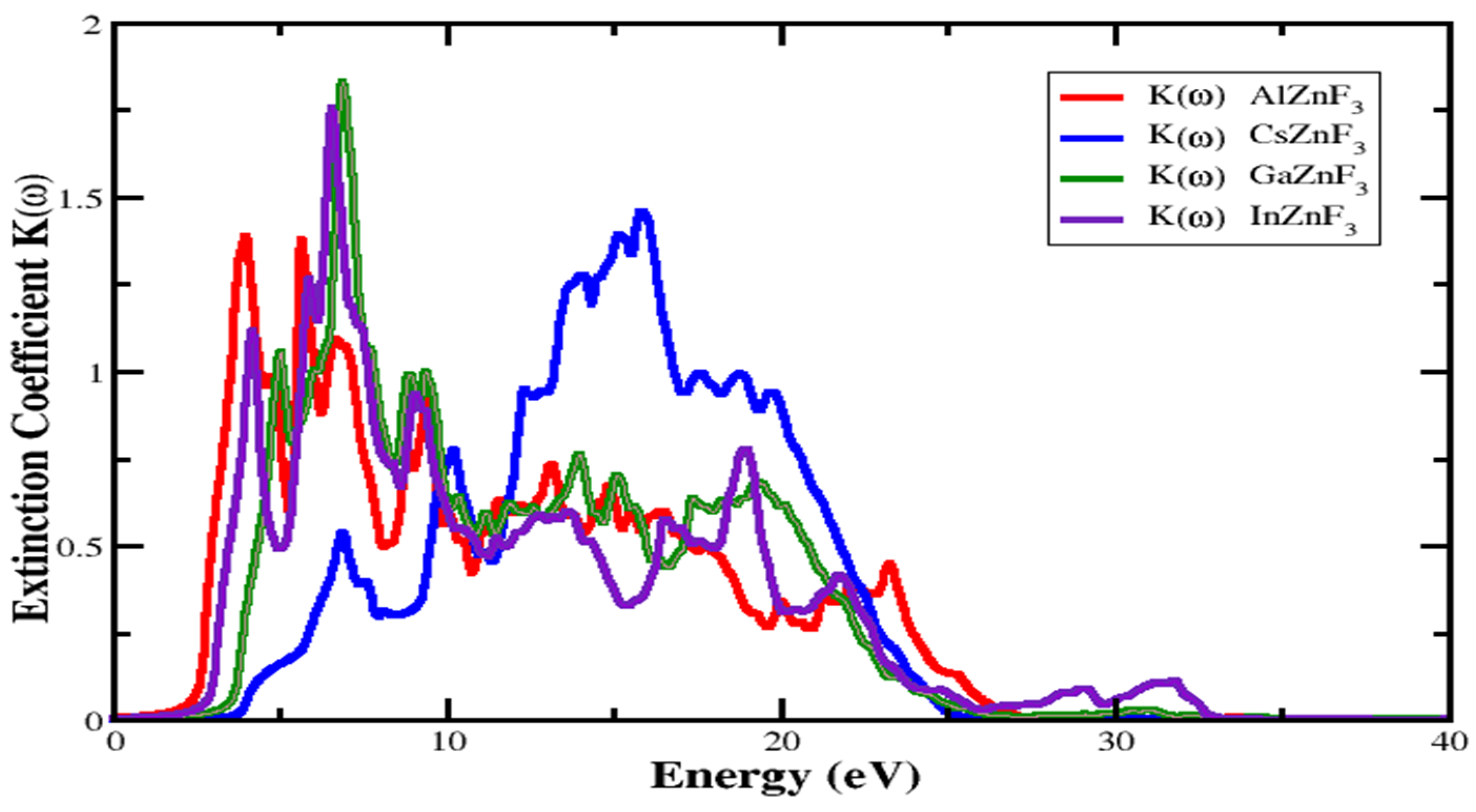
3.4.1. Refractive Index and Extinction Coefficient
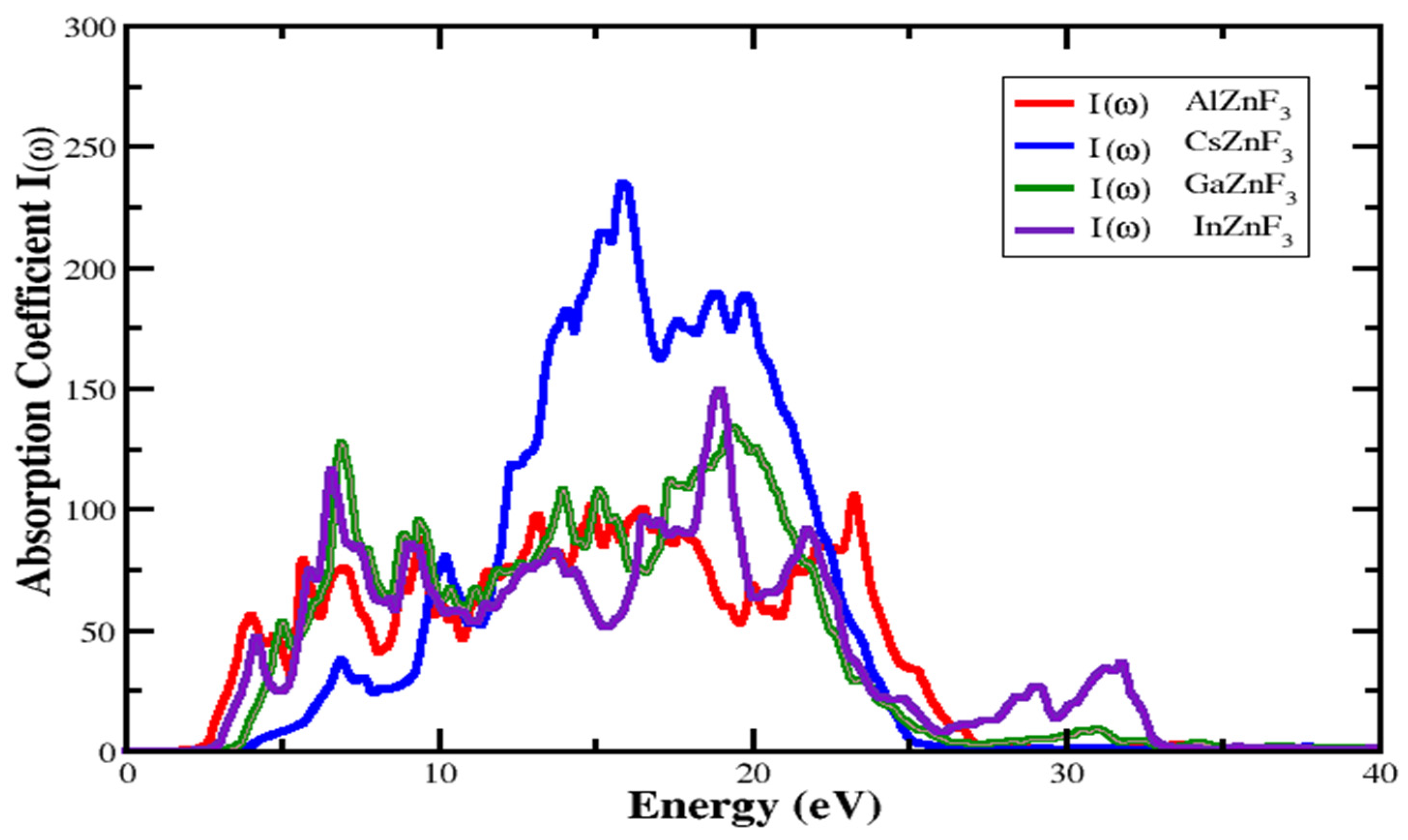
3.4.2. Absorption Coefficient
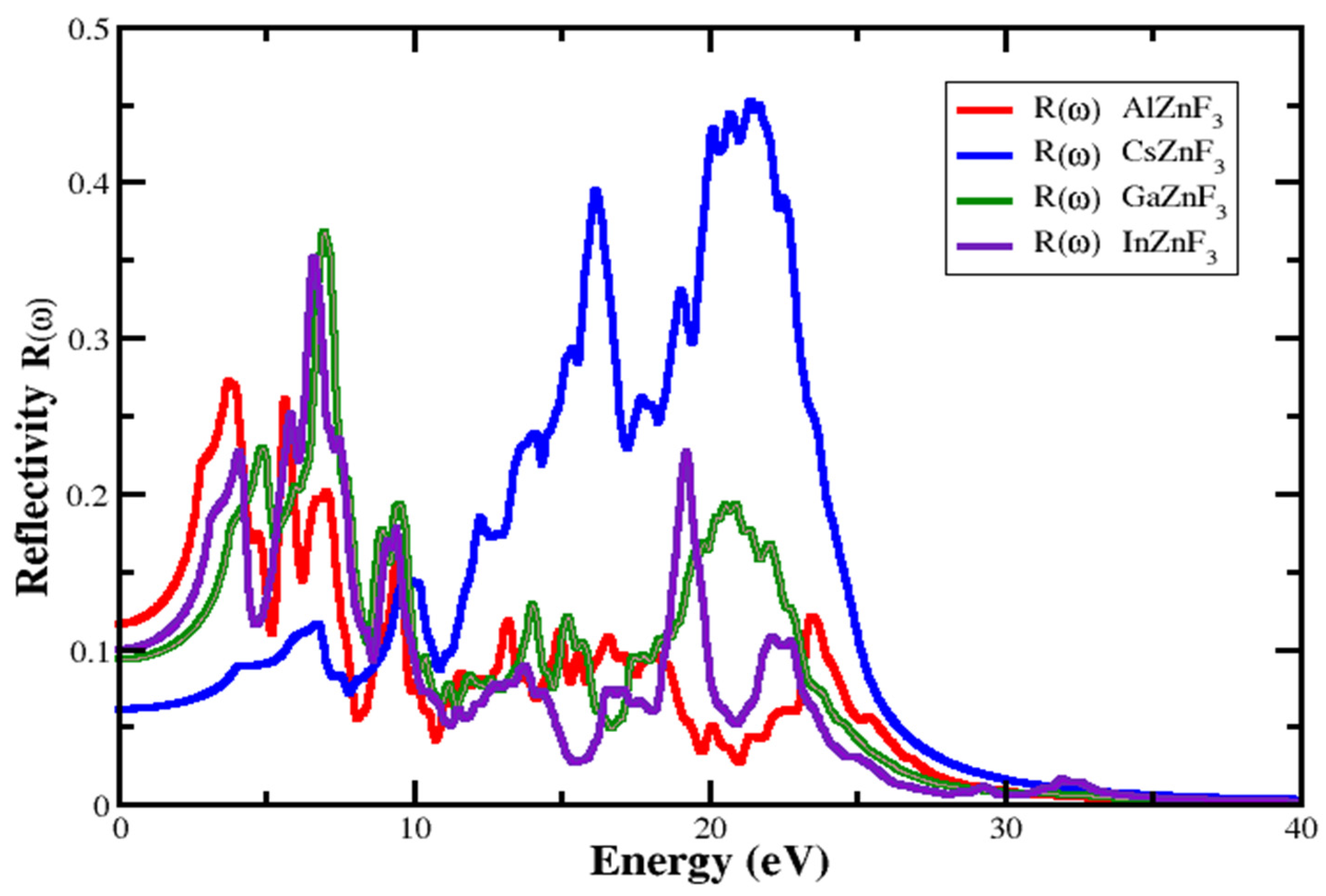
3.4.3. Reflectivity
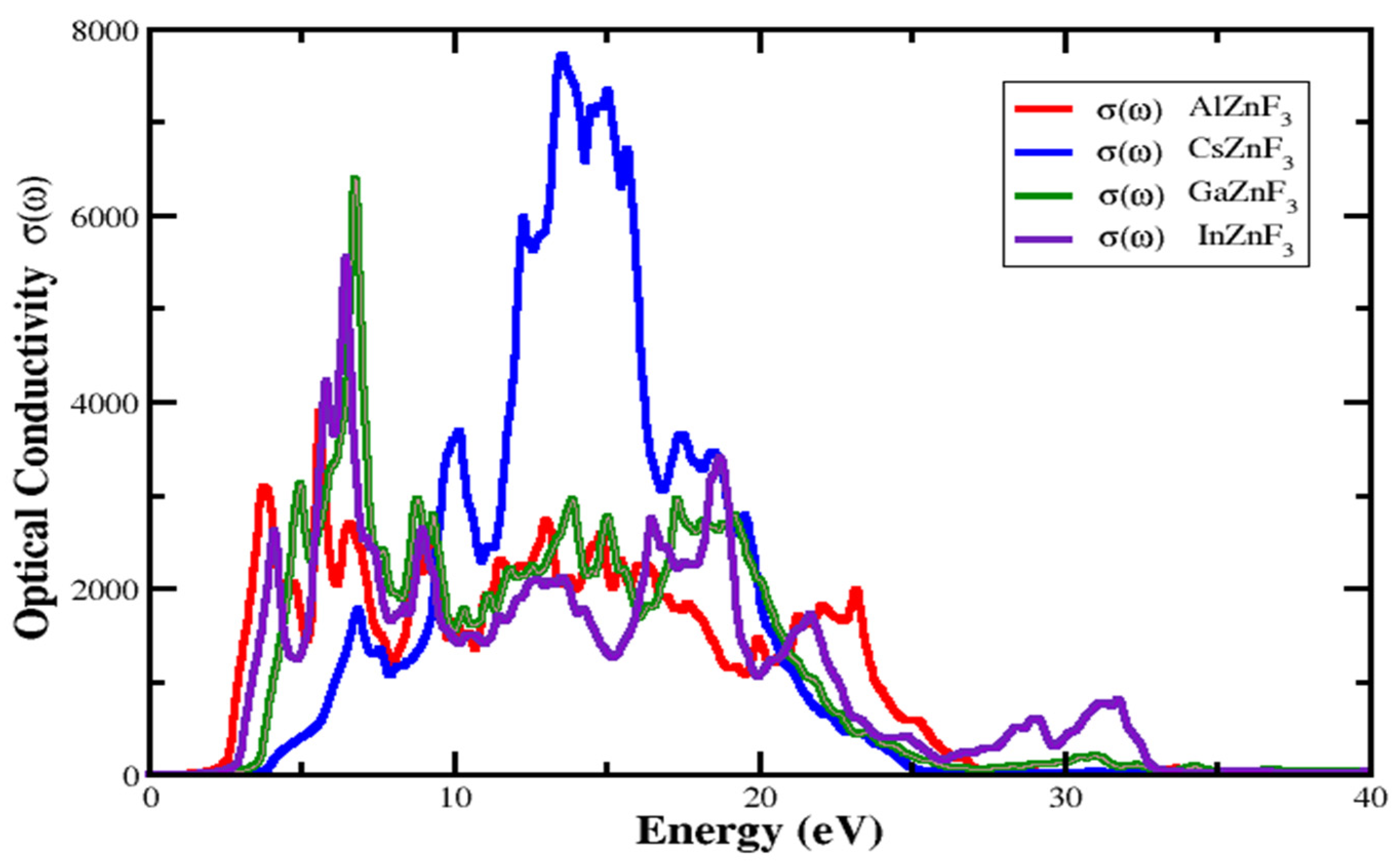
3.4.4. Optical Conductivity
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Davidson, M.W.; Lofgren, G.E. Photomicrography in the geological sciences. J. Geol. Educ. 1991, 39, 403–418. [Google Scholar] [CrossRef]
- Jin, Z.; Wu, Y.; Li, S.; Chen, S.; Zhang, W.; Wu, Q.; Zhang, C. First-principles calculation of the electronic structure, optical, elastic and thermodynamic properties of cubic perovskite LiBeF3. Mater. Res. Express 2020, 6, 1251161–12511624. [Google Scholar] [CrossRef]
- Erum, N.; Iqbal, M.A. Effect of pressure variation on structural, elastic, mechanical, optoelectronic and thermodynamic properties of SrNaF3 fluoroperovskite. Mater. Res. Express 2017, 4, 1263111–12631114. [Google Scholar]
- Lufaso, M.W.; Woodward, P.M. Prediction of the crystal structures of perovskites using the software program SPuDS. Acta Crystallogr. B Struct. Sci. 2001, 57, 725–738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ullah, R.; AliM, A.; Murad, S.; Khan, A.; Dar, S.A.; Mahmood, I.; Laref, A. Structural, electronic and optical properties of cubic perovskite RbYbF3 under pressure: A first principles study. Mater. Res. Express 2019, 6, 125901–125923. [Google Scholar] [CrossRef]
- Khan, S.; Zaman, S.U.; Ahmad, R.; Mehmood, N.; Arif, M.; Kim, H.J. Ab initio investigations of structural, elastic, electronic and optical properties of the fluoroperovskite TIXF3 (X= Ca, Cd, Hg, and Mg) compounds. Mater. Res. Express 2020, 6, 125923–125941. [Google Scholar] [CrossRef]
- Ali, A.; Rahman, A.U.; Rahman, G. Thermoelectric properties of KCaF3. Phys. B Condens. Matter 2019, 565, 18–24. [Google Scholar] [CrossRef]
- Sun, X.; Wang, G.; Yang, K. Electronic Structure and Polarization of NaMgF 3/NaCaF3 Superlattices: Insight from First-Principles. Mater. Sci. Eng. 2020, 774, 120161–120164. [Google Scholar]
- Vaitheeswaran, G.; Kanchana, V.; Kumar, R.S.; Cornelius, A.L.; NiCol, M.F.; Svane, A.; Christensen, N.E.; Eriksson, O. High-pressure structural study of fluoro-perovskite CsCdF3 up to 60 GPa: A combined experimental and theoretical study. Phys. Rev. B 2021, 81, 0751051–0751056. [Google Scholar]
- ur rehman Hashmi, M.R.; Zafar, M.; Shakil, M.; Sattar, A.; Ahmed, S.; Ahmad, S.A. First-principles calculation of the structural, electronic, and magnetic properties of cubic perovskite RbXF3 (X = Mn, V, Co, Fe). Chin. Phys. B 2016, 25, 1174011–1174016. [Google Scholar]
- Zeba, I.; Kiran, R.; Shakil, M.; Rafique, M.; Ahmadb, R.; Gillani, S.S.A. Study the effect of magnesium doping concentration on structural and optoelectronic response of NaCa1−xMgxF3 fluoro-perovskite: First-principles computation. Optik 2020, 218, 1649901–1649909. [Google Scholar] [CrossRef]
- Kravchyk, K.V.; Zünd, T.; Wörle, M.; Kovalenko, M.V.; Bodnarchuk, M.I. NaFeF3 nanoplates as low-cost sodium and lithium cathode materials for stationary energy storage. Chem. Mater. 2018, 30, 1825–1829. [Google Scholar] [CrossRef] [Green Version]
- Arar, R.; Ouahrani, T.; Varshney, D.; Khenata, R.; Murtaza, G.; Rached, D.; Bouhemadou, A.; Al-Douri, Y.; BinOmran, S.; Reshak, A.H. Structural, mechanical and electronic properties of sodium based fluoroperovskites NaXF3 (X= Mg, Zn) from first-principle calculations. Mater. Sci. Semicond. Process. 2015, 33, 127–135. [Google Scholar] [CrossRef]
- Sabir, B.; Murtaza, G.; Mahmood, Q.; Ahmad, R.; Bhamu, K. First principles investigations of electronics, magnetic, and thermoelectric properties of rare earth based PrYO3 (Y = Cr, V) perovskites. Curr. Appl. Phys. 2017, 17, 1539–1546. [Google Scholar] [CrossRef]
- Nishimatsu, T.; Terakubo, N.; Mizuseki, H.; Kawazoe, Y.; Pawlak, D.A.; Shimamura, K.; Fukuda, T. Band structures of perovskite-like fluorides for vacuum-ultraviolet-transparent lens materials. Jpn. J. Appl. Phys. 2002, 41, L365–L367. [Google Scholar] [CrossRef]
- Mubarak, A.; Al-Omari, S. First-principles calculations of two cubic fluoropervskite compounds: RbFeF3 and RbNiF3. J. Magn. Magn. Mater. 2015, 382, 211–218. [Google Scholar] [CrossRef]
- Zeb, R.; Ali, Z.; Ahmad, I.; Khan, I. Structural and magnetic properties of TlTF3 (T= Fe, Co and Ni) by hybrid functional theory. J. Magn. Magn. Mater. 2015, 388, 143–149. [Google Scholar] [CrossRef]
- Oleaga, A.; Salazar, A.; Skrzypek, D. Critical behaviour of magnetic transitions in KCoF3 and KNiF3 perovskites. J. Alloys Compd. 2015, 629, 178–183. [Google Scholar] [CrossRef]
- Weetman, C.; Inoue, S. The Road Travelled: After Main-Group Elements as Transition Metals. ChemCatChem 2018, 10, 4213–4228. [Google Scholar] [CrossRef]
- Gluck, R.; Lee, T.; Smith, F. Preparation and properties of AgNiF3 crystals. Mater. Res. Bull. 1974, 9, 305–309. [Google Scholar] [CrossRef]
- Körbel, S.; Marques, M.A.; Botti, S. Stability and electronic properties of new inorganic perovskites from high-throughput ab initio calculations. J. Mater. Chem. C 2016, 4, 3157–3167. [Google Scholar] [CrossRef]
- Mubarak, A. The mechanical, optical and thermoelectric properties of MCoF3 (M= K and Rb) compounds. Mod. Phys. Lett. B 2017, 31, 17500331–175003317. [Google Scholar] [CrossRef]
- Clark, L.; Lightfoot, P. Magnetic Properties of Transition Metal Fluoride Perovskites. In Photonic and Electronic Properties of Fluoride Materials; Elsevier: Amsterdam, The Netherlands, 2016; pp. 261–284. [Google Scholar]
- Tran, F.; Blaha, P. Accurate band gaps of semiconductors and insulators with a semilocal exchange-correlation potential. Phys. Rev. Lett. 2009, 102, 226401–226404. [Google Scholar] [CrossRef] [Green Version]
- Qin, L.; Duan, Y.; Shi, H.; Shi, L.; Tang, G. Hybrid density functional theory studies of AlN and GaN under uniaxial strain. J. Phys. Condens. Matter 2012, 25, 45801. [Google Scholar] [CrossRef]
- Shinde, R.; Yamijala, S.S.; Wong, B.M. Improved band gaps and structural properties from Wannier–Fermi–Löwdin self-interaction corrections for periodic systems. J. Phys. Condens. Matter 2020, 33, 115501. [Google Scholar] [CrossRef]
- Zhang, R.; Zhao, C.; Huo, Y.; Han, Y.; Hong, J.; Liu, Y.; Zhang, A.; Guo, R.; Ai, Y. Theoretical calculation of toxic/radioactive metal ion capture by novel nanomaterials. In Emerging Nanomaterials for Recovery of Toxic and Radioactive Metal Ions from Environmental Media; Elsevier: Amsterdam, The Netherlands, 2022; pp. 313–379. [Google Scholar]
- Murnaghan, F. The compressibility of media under extreme pressures. Proc. Natl. Acad. Sci. USA 1994, 30, 244–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Curtarolo, S.; Setyawan, W.; Hart, G.L.W.; Jahnatek, M.; Chepulskii, R.V.; Taylor, R.H.; Wang, S.; Xue, J.; Yang, K.; Levy, O.; et al. AFLOW: An automatic framework for high-throughput materials discovery. Comput. Mater. Sci. 2012, 58, 218–226. [Google Scholar] [CrossRef] [Green Version]
- Reshak, A.H.; Jamal, M. DFT calculation for elastic constants of orthorhombic structure within WIEN2K code: A new package (ortho-elastic). J. Alloys Compd. 2012, 543, 147–151. [Google Scholar] [CrossRef]
- Solli, D.; McCormick, C.; Ropers, C.; Morehead, J.; Chiao, R.; Hickmann, J. Demonstration of superluminal effects in an absorptionless, nonreflective system. Phys. Rev. Lett. 2003, 91, 1439061–1439064. [Google Scholar] [CrossRef] [Green Version]
- Kramers, H.A. Collected Scientific Papers; North-Holland Publishing Company: Amsterdam, The Netherlands, 1956. [Google Scholar]
- Kronig, R.d. On the theory of dispersion of X-rays. J. Opt. Soc. Am. 1926, 12, 547–556. [Google Scholar] [CrossRef]
- Bassani, G.F.; Parravicini, G.P. Electronic States and Optical Transitions in Solids; Pergamon: Oxford, UK, 1975. [Google Scholar]
- Puschnig, P.; Ambrosch-Draxl, C. Optical absorption spectra of semiconductors and insulators including electron-hole correlations: An ab initio study within the LAPW method. Phys. Rev. B 2002, 66, 1651051–1651059. [Google Scholar] [CrossRef]
- Ambrosch-Draxl, C.; Sofo, J.O. Linear optical properties of solids within the full-potential linearized augmented planewave method. Comput. Phys. Commun. 2006, 75, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Cardona, M.; Peter, Y.Y. Fundamentals of Semiconductors; Springer: Berlin/Heidelberg, Germany, 2005. [Google Scholar]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | Lattice Constant (a0) | Bulk Modulus (GPa) | Derivative of Bulk Modulus (GPa) | Ground State Volume (V0) | Ground State Energy (E0) |
|---|---|---|---|---|---|
| CsZnF3 | 4.29 Å | 64.74 | 4.86 | 534.40 | −19,772.55 |
| InZnF3 | 4.19 Å | 72.28 | 4.71 | 497.66 | −15,958.34 |
| GaZnF3 | 4.12 Å | 74.57 | 4.56 | 473.29 | −8080.04 |
| AlZnF3 | 4.13 Å | 74.66 | 4.56 | 476.24 | −4677.37 |
| Compounds | C11 (GPa) | C12 (GPa) | C44 (GPa) | B (GPa) | A | G (GPa) | E (GPa) | v | B/G |
|---|---|---|---|---|---|---|---|---|---|
| CsZnF3 | 87.43 | 53.96 | 35.36 | 65.13 | 2.11 | 26.19 | 69.28 | 0.45 | 2.48 |
| InZnF3 | 90.94 | 61.18 | 17.95 | 71.00 | 1.20 | 16.65 | 46.34 | 0.56 | 4.26 |
| GaZnF3 | 100.18 | 60.45 | 21.45 | 73.70 | 1.08 | 20.80 | 53.04 | 0.53 | 3.54 |
| AlZnF3 | 95.22 | 66.82 | 19.01 | 76.45 | 1.33 | 16.91 | 47.26 | 0.57 | 4.51 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Habib, A.; Husain, M.; Sajjad, M.; Rahman, N.; Khan, R.; Sohail, M.; Ali, I.H.; Iqbal, S.; Khan, M.I.; Ebraheem, S.A.M.; et al. Insight into the Exemplary Physical Properties of Zn-Based Fluoroperovskite Compounds XZnF3 (X = Al, Cs, Ga, In) Employing Accurate GGA Approach: A First-Principles Study. Materials 2022, 15, 2669. https://doi.org/10.3390/ma15072669
Habib A, Husain M, Sajjad M, Rahman N, Khan R, Sohail M, Ali IH, Iqbal S, Khan MI, Ebraheem SAM, et al. Insight into the Exemplary Physical Properties of Zn-Based Fluoroperovskite Compounds XZnF3 (X = Al, Cs, Ga, In) Employing Accurate GGA Approach: A First-Principles Study. Materials. 2022; 15(7):2669. https://doi.org/10.3390/ma15072669
Chicago/Turabian StyleHabib, Anwar, Mudasser Husain, Muhammad Sajjad, Nasir Rahman, Rajwali Khan, Mohammad Sohail, Ismat Hassan Ali, Shahid Iqbal, Mohammed Ilyas Khan, Sara A. M. Ebraheem, and et al. 2022. "Insight into the Exemplary Physical Properties of Zn-Based Fluoroperovskite Compounds XZnF3 (X = Al, Cs, Ga, In) Employing Accurate GGA Approach: A First-Principles Study" Materials 15, no. 7: 2669. https://doi.org/10.3390/ma15072669