2. Hamiltonian of an Electron–Phonon System for a Disordered Crystal
The Hamiltonian of the disordered system (alloy, disordered semiconductor, and disordered dielectric) consists of the sum of the Hamiltonian of electrons in the nucleus field, the Hamiltonian of electron–electron interaction, and the Hamilton of nucleus. The motion of the ion subsystem is reduced to nucleus oscillations near the equilibrium position under the influence of the nuclei interaction force, and their indirect interaction through electrons. In the Wannier representation, the system Hamiltonian is as follows [
26]:
where zero-order Hamiltonian
consists of the Hamiltonian of the electrons in the field of the cores of the atom’s ideal ordered crystal
and the harmonic phonon Hamiltonian for the motion of the cores of the atom’s ideal ordered crystal
Symbol n denotes the number of a unit cell, i denotes the number of a node in a unit cell, and γ denotes all of the other quantum numbers for the orbital, including spin. The symbol h(0) denotes the “hopping integral” that connects the respective orbitals. For the phonon Hamiltonian, α is a spatial direction (x, y, or z), is the core momentum, is the mass of the core, is the deviation of the core from the equilibrium position of the lattice site, and is the corresponding spring-constant matrix.
The interaction Hamiltonian in Equation (1) is the perturbation of the system due to all of the effects we will be including. It is composed of six pieces:
is the modification of the core–core Coulomb interaction due to the disordered atoms added to the system; it is the difference between the original core–core repulsion Hamiltonian and the new one. The electronic Hamiltonian is modified by the term
which is the difference between the new hopping Hamiltonian and the original periodic one. The electron–phonon interaction is given by
It is described in more detail below. The Hamiltonian of the Coulomb interaction between electrons is given by the term
The modification of the interaction of the phonons with the cores caused by the disordering of the atoms is given by
where
, and and are the masses of the atoms at site for the disordered and ordered alloy, respectively.
We also include the cubic anharmonic potential terms for the phonons (under the assumption that they remain small and can be treated perturbatively via
The operators and create and destroy electrons in the state described by Vane’s function , where are the spatial and z-components of the spin coordinates of the wave function.
To construct the Wannier functions, we use analytical expressions for the wave functions of an electron in the field of atomic nuclei of type λ localized at the lattice sites
of an ideally ordered crystal:
where
are the angular spherical coordinates of the vector
Here,
is an index that incorporates the quantum numbers for the energy value
, the angular momentum quantum numbers are
l and
m,
r is the electron position vector, and
is the position vector for the atom at site (
ni) in equilibrium
is the position vector of the unit cell of the crystal lattice, and is the vector of the relative position of the node of the sublattice i in the unit cell . The coordinates of the radius vector of the unit cell of the crystal lattice are integers. The number takes on values for three-dimensional crystals, for two-dimensional crystals, and for one-dimensional crystals.
Basis orthogonalization is performed with the Lowdin method [
28]
where
are the overlapping matrix.
Vane’s functions
, on which the Hamiltonian of the system are represented as in Equation (1), are defined from equation:
where
is the spin part of wave function,
.
The orthogonalized wave function can be represented as:
Summation over in expression (16) means summation over , in accordance with Formula (13).
The overlap matrix
is found from the equation:
where
are expressed through
in accordance with Formulas (17)–(19).
To find matrix
in expression (16), we find the Fourier transform of the matrix (20):
The vector
is defined by the expression
is the basis vector of the translations of the reciprocal lattice.
Summing over
on the right-hand side of Formula (21) is easy to do if we replace it according to (13) and use
As the matrix element decreases with the distance between the nodes , , in numerical calculations, when summing over in expression (21), it is sufficient to restrict ourselves to a few coordination spheres. In this case, summation over is reduced to summation over .
The matrix
has an infinite rank. The rank of the matrix
is finite, which allows for finding the matrix
. The matrix
in expression (16) is found from equation:
The values
in Equation (3) are the matrix elements of the kinetic and potential energy
of the electron in the field of the cores of the atom’s ideal ordered crystal. The values
are defined by the expression:
Here, is expressed through in accordance with Formulas (17)–(19). The expression for is obtained from expression (17) for replacement by . Summation over in expression (25) means summation over , in accordance with Formula (13).
In Formula (26) and are the mass and charge of the electron, respectively, and is the ordinal number of an atom of the sort located in the site of an ideally ordered crystal. denotes the Planck’s constant.
The matrix element of the electron–ion interaction Hamiltonian in Equation (6) is given by
where
is a matrix element of the potential of the core of the atom .
The expression for is obtained from Formula (26) by replacing with .
In Equation (28), is a discrete binary random number taking the values of 1 or 0, depending on whether an atom of type is at site (ni) or not, respectively. The symbol will be defined next.
The expression for the electron–phonon interaction in Equation (7) is found through derivatives of the potential energy of the electrons in the ion core field due to a displacement of the atom by vector
. In Equation (7), the value of
is given by
where
with
the matrix elements of the following operator:
The expression for
is obtained from Formula (26) by replacing in it
with
in Equation (29) describes electron scattering on the static displacement of the atoms, and is defined by the equation
where
is the
projection of the static displacement of the atom of type
in the site, and
I caused by the difference in the atomic radii of the components of the disordered crystal.
Upon receipt of expressions (27)–(34), it was taken into account that the potential energy operator of the electron in the field of the atoms core can be expressed as:, , with r being the electron’s radius vector, the radius-vector of atom’s equilibrium position in the site of the crystal lattice (ni), the vector of atom’s static displacement from equilibrium position in site (ni), and the atom’s displacement operator in site (ni). Expanding in the series in powers and restricting ourselves to linear terms, we arrive at expressions (27)–(34).
The matrix of the force constants arising from the direct Coulomb interaction of the ionic cores has the form:
where
is the serial number of the atom located in the lattice site
of the disordered crystal, which is given by the expression
This matrix
satisfies the following constraint:
Multicenter integrals
in Formula (8) can be represented as
When integrating over in Formula (38), should be expressed through , in accordance with Formulas (17)–(19), in which I is necessary to replace with . When integrating over in Formula (38), should be expressed through , in accordance with Formulas (17)–(19), in which it is necessary to replace with .
So, Formulas (17)–(19) describe the procedure for calculating the matrix elements , Hamiltonian (1), containing one-electron and two-electron integrals.
3. Green’s Functions of Electrons and Phonons
We employ a Green’s function-based formalism to perform the calculations. Ultimately, we need the real-time retarded
and advanced
Green’s functions are each defined as follows [
26]:
Here, the operators are expressed in the Heisenberg representation
where
is Planck’s constant,
,
is the chemical potential of the electronic subsystem, and
is the electron number operator given by
In addition, the commutator or anticommutator is defined via
where the commutator is used for Bose operators (−) and the anticommutator is used for Fermi operators (+). The symbol
is Heaviside’s unit step function. The angle brackets
denote the thermal averaging with respect to the density matrix ρ
where
is the thermodynamic potential of the system given by exp(Ω/Θ) = Trexp(−H/Θ) and
, with
kb Boltzmann’s constant and
the temperature.
The thermal Green’s functions are defined by
where the imaginary-time operator
is derived from the real-time Heisenberg representation and the substitution
. Hence,
In addition, the time-ordering operator satisfies
where the plus sign is used for Bose operators and the minus sign for Fermi operators.
We next go to the interaction representation by introducing the operator
with
and
.
Differentiating the expression for
in Equation (68) with respect to
and then integrating starting from 0, with the boundary condition
, we obtain:
where
. Employing this result yields
with
A(τ) in the Heisenberg representation with respect to the noninteracting Hamiltonian. Substituting these results into the definition of the thermal Green’s function creates the alternate interaction-representation form for the Green’s function, given by
where all time dependence is with respect to the noninteracting Hamiltonian and the trace over all states is with respect to the noninteracting states
Next, we expand expression (51) for in a series in powers of the interaction Hamiltonian and substitute this expression in Formula (53).
The diagrammatic method is generated by expanding σ(τ) in a power series in terms of , and then using Wick’s theorem to evaluate the resulting operator averages.
The numbers of quantum states for different operators in the interaction Hamiltonian (5)–(11), (51) are different, and the values of the argument are the same.
Each operator can be assigned a quantum state number and an argument number , if in expression (51) for the operator is replaced by an operator in which the values of the argument for operators with different quantum states are different, the matrix elements differ from the matrix elements of the operator by a factor , and the single integral over is replaced by the integral over multiplicity . The multiplicity of the integral is different for different types of interaction. In expression (53) for , the term of the series for (51) forms a multiple sum over quantum states and an integral over of the mean T-product of operators multiplied by an operator . The T-product of operators is averaged over the occupation numbers of the quantum states of the system of noninteracting electrons and phonons, in accordance with Formula (53). The numbers of the quantum states for the operators in the indicated T-product are ordered by the matrix elements of the interaction operators , in accordance with Formulas (5)–(11), in such a way that pairs of operators are formed. This is due to the fact that among the average T-products of operators, only those in which the number of operators is even for the electron subsystem and the phonon subsystem are nonzero. The quantum state for each operator of the pair, except for the operators , , coincides with one of the quantum states for the corresponding matrix element of the interaction operators in the given product.
Let us give the averaging technique in expression (71) a simpler form. For this, in the T-product of each pair of operators for the electron subsystem and , , , , for the phonon subsystem, in the Hamiltonian of the system of noninteracting electrons and phonons (2)–(4), (53), we compare the sum of the products of pairs of operators , the numbers of quantum states of which coincide with the numbers of quantum states depending on the operators of the pair.
Provided that the numbers of quantum states for the operators in the T-product are ordered by the matrix elements of the interaction operators standing in it, the operators , change places with the products depending on other pairs of operators. It follows from this that the average of the T-product of several operators in expression (53) is equal to the product of the average T-products of pairs of operators that determine the matrices of the Green’s functions for the zero-order Hamiltonian . This statement also extends to the case when the quantum states for the operators of a pair coincide with the quantum states for the operators of other pairs. This follows from the fact that the distribution function of a system of an infinite number of particles over the occupation numbers of quantum states has a sharp maximum, and the most probable value of a physical quantity is equal to its average value. The quantum state and the argument for each operator of the pair, except for the operators , , coincides with one of the quantum states and arguments for the corresponding matrix element of the interaction operators in the given product. In expression (71), the Green’s function is summed over quantum states and integrated over arguments .
The averaging technique described above in expression (71) for the Green’s function
is the essence of Wick’s theorem. This technique then generalizes the approach used for the homogeneous system [
27].
The technique for calculating the Green’s function
(53) becomes clearer if the terms on the right-hand side of Equation (53) are represented in the form of diagrams. If the Green’s function of the system is expressed as a series only over connected diagrams [
27], then the denominator in Formula (53) will cancel out with the same factor in the numerator. So, the thermal Green’s function is expanded in terms of connected diagrams. The indicated diagrammatic series can be easily summed up, which makes it possible to go beyond the framework of the first approximations of the perturbation theory and obtain equations for the Green’s functions of the system.
Summing up the indicated series, using the standard relation between the spectral representations of the temperature and real-time Green’s functions and performing an analytical continuation on the real axis, we obtain the following equations for the retarded Green’s functions [
26] (hereinafter the dependence on r is suppressed):
where
. Here,
,
,
,
,
are the real-frequency representation of the single-particle Green’s function of the electrons, the coordinate-coordinate, momentum–momentum, coordinate–momentum, and momentum–coordinate Green’s functions of the phonons, respectively; and
,
,
,
are the corresponding self-energies (mass operators) for the electron–phonon, phonon–electron, electron–electron, and phonon–phonon interactions.
The real-time and real-frequency Green’s functions are related by standard Fourier transform relations given by
and
The thermal Green’s functions are periodic (bosons) or antiperiodic (fermions) on the interval −1/Θ ≤ τ < 1/Θ, and hence have a Fourier series representation in terms of their Matsubara frequencies, as follows:
and
where the Matsubara frequencies satisfy
The electronic Green’s functions are infinite matrices with indices given by the lattice site
n, the basis site
i, and the other quantum numbers γ. Similarly, the phonon Green’s functions also are infinite matrices with the same lattice and basis site dependence, plus a dependence on the spatial coordinate direction α. Using the equations of motion for Green’s functions, one can obtain simple expressions for the zero-order Green’s functions, namely [
26]:
with
with
and
Here, the double lines denote a matrix.
When the perturbations are small, given by
then the solution of the system of equations in Equation (55) becomes
where
The mass operator of the Green’s function of electrons for the electron–phonon interaction
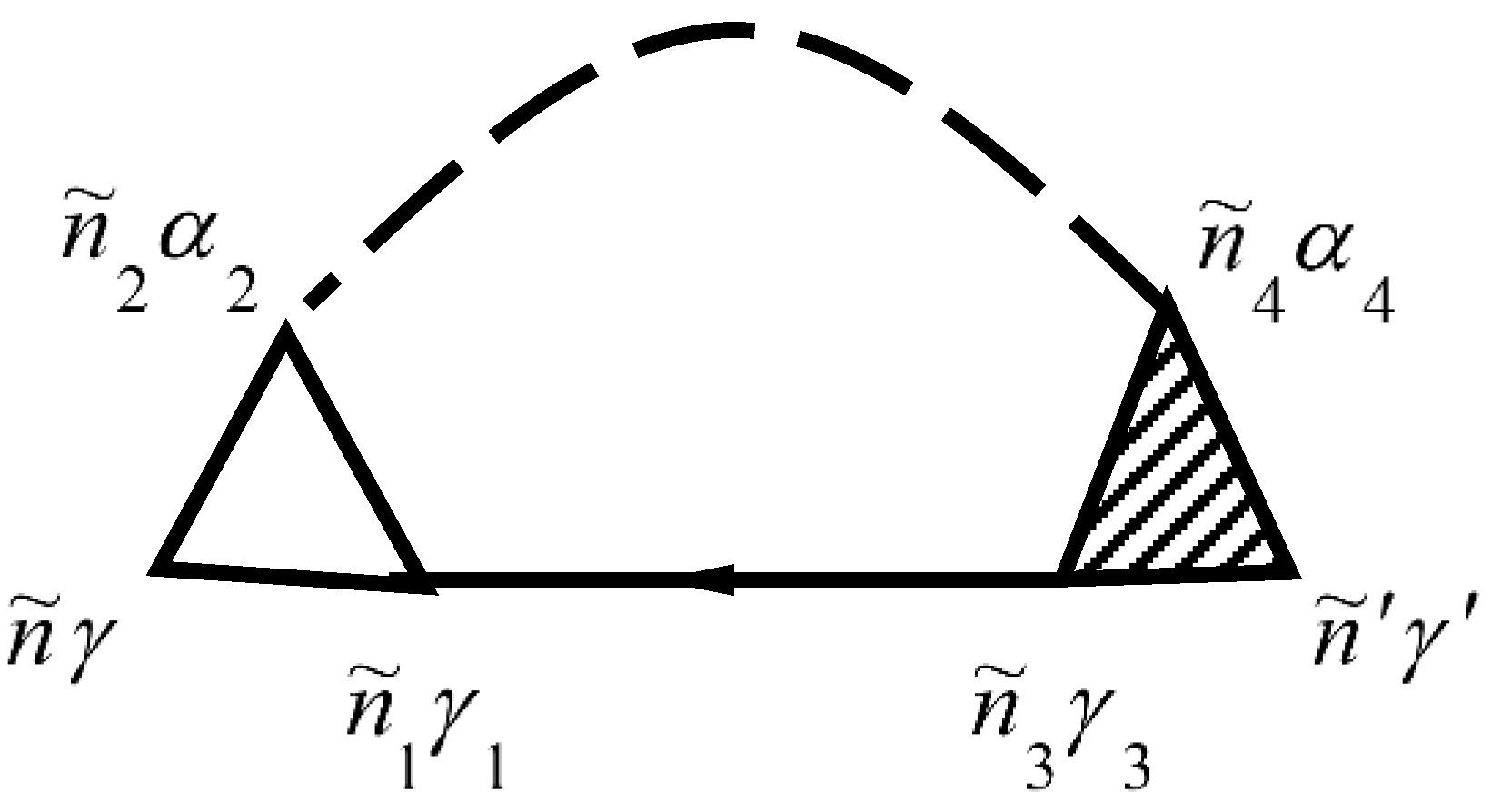
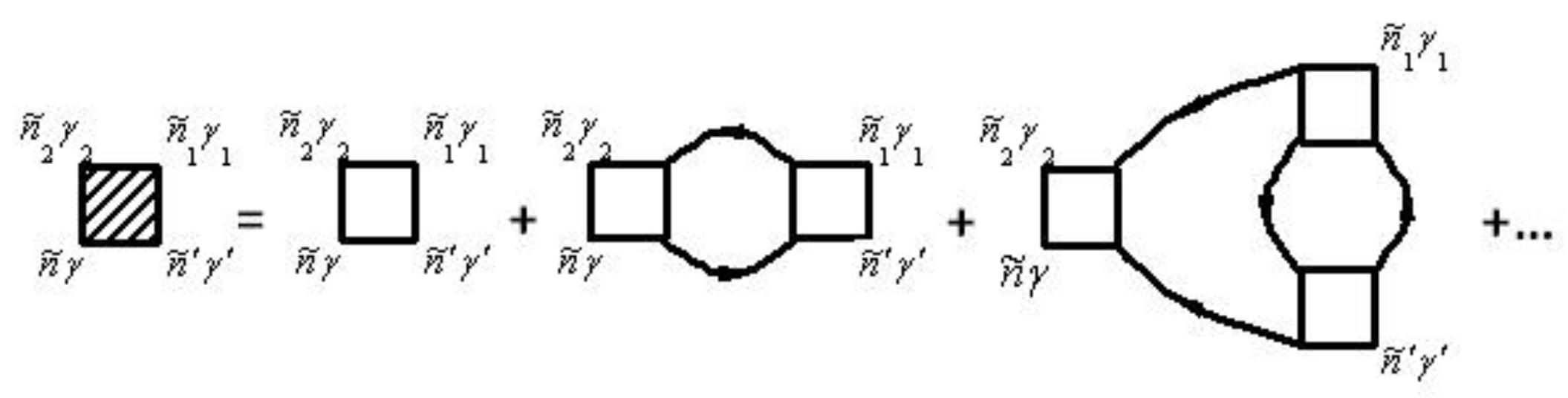
is described by the diagram in
Figure 1. The mass operator of the Green’s function of electrons for the electron–phonon interaction
is described by the diagram in [
26,
29,
30].
Solid lines in
Figure 1 correspond to the Green’s function of electrons
and dashed lines correspond to the Green’s function of phonons
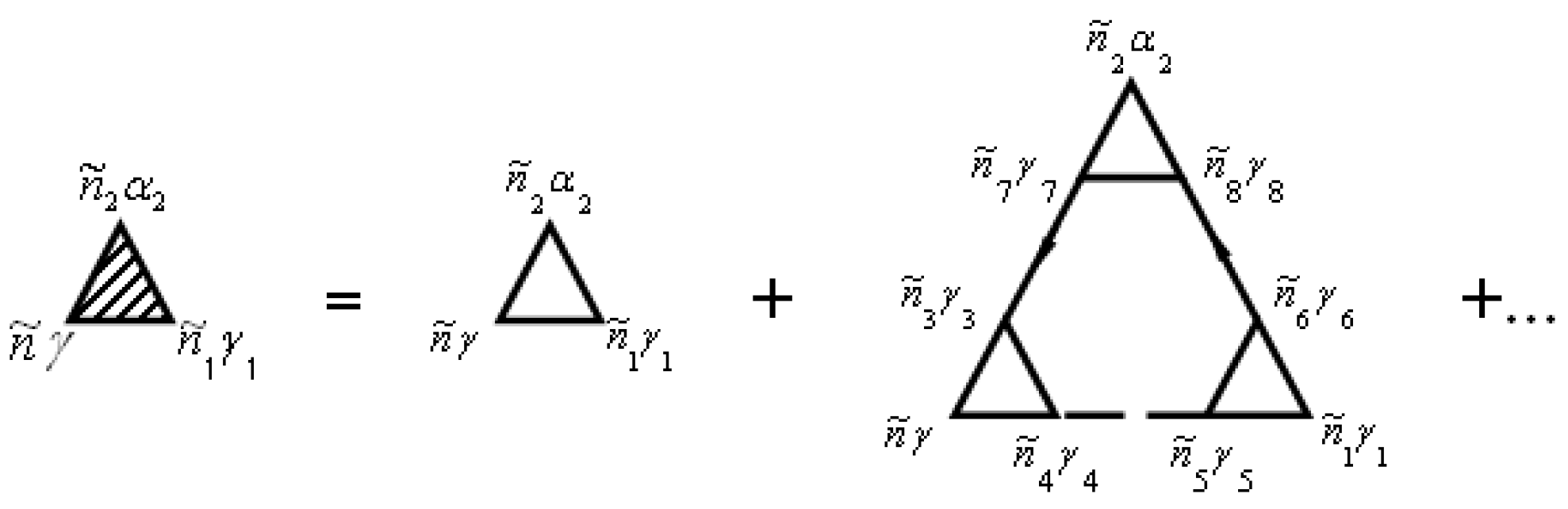
. The vertex part
of the mass operator of the Green’s function is described by the diagrams in
Figure 2.
The not shaded triangle in
Figure 2 corresponds to equation
In
Figure 1 and
Figure 2, summation for internal points
is carried out. Summation of
provides summation of
and integration over
. Expressions that correspond to each diagram are attributed to multiplier
, where
is the diagram’s order (namely the number of vertices
in the diagram), and
is the number of lines for the Green’s function of electrons
. This function goes out and goes in in the same vertices.
Explicitly, the electron–phonon self-energy becomes
where repeated indices are summed over.
Phonon–electron interaction is described by the diagram in
Figure 3. Phonon–electron interaction is described by the diagram in [
29,
30].
The self-energy of the phonon due to the phonon–electron interaction is given by
where
is the so-called Fermi–Dirac distribution function.
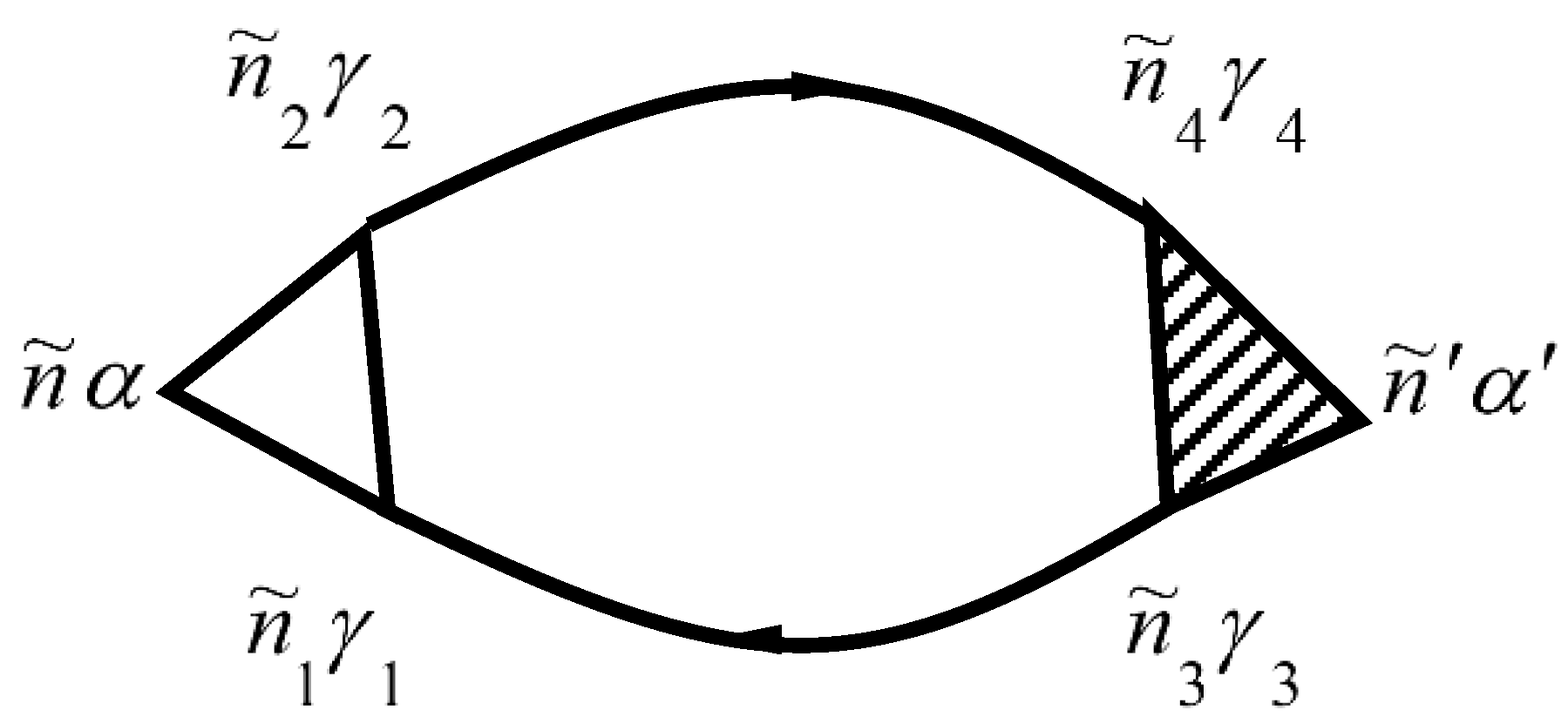
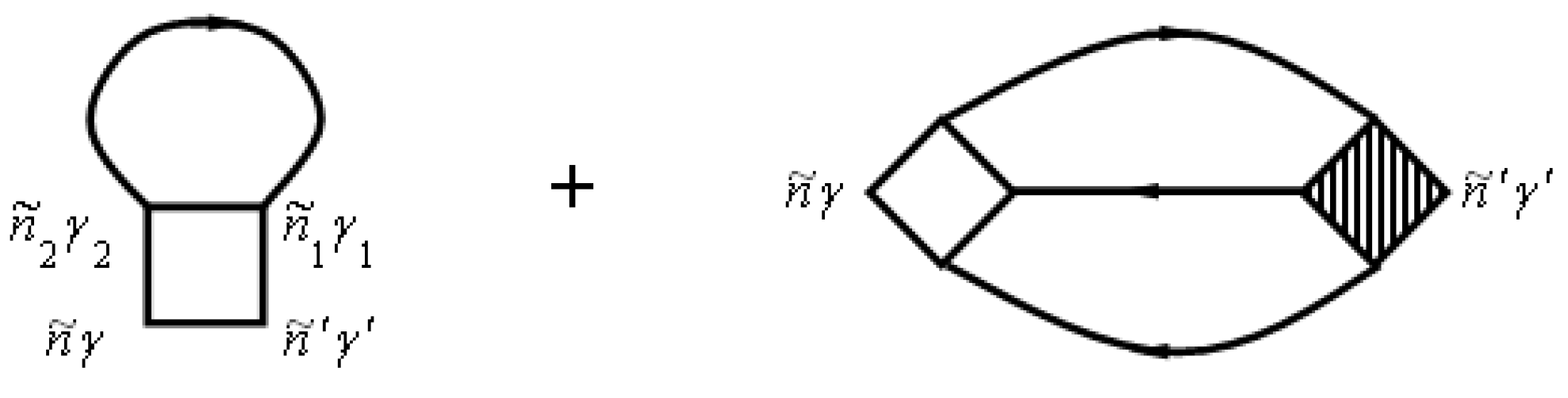
Diagrams for the mass operator
that describe the electron–electron interaction, are shown in
Figure 4. Diagrams for the mass operator
that describe electron–electron interaction, are shown in [
26,
29,
30].
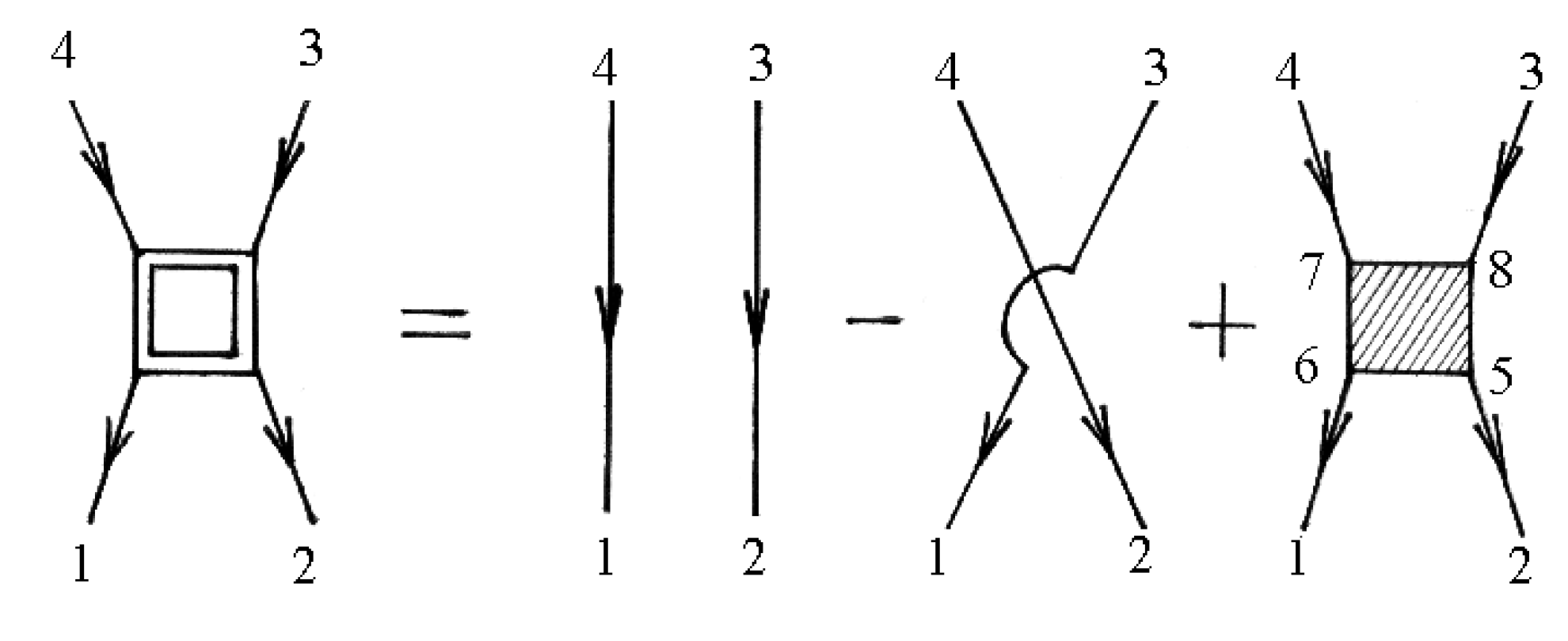
The vertex parts
are shown in diagrams in
Figure 5.
Not shaded triangle in
Figure 5 corresponds to equation
The mass operator that describes the electron–electron interaction is:
A similar result for the contribution to the phonon self-energy
from phonon–phonon coupling is given in [
26].
In deriving the expressions in Equations (72), (74), (78), and (79), we employed the standard resummation techniques for any function
that is analytic in the region covered by contour C, which encloses all of the Matsubara frequencies. Namely, we have
for the Bosonic case, and
for the Fermionic case, with
We comment that for the many-body Green’s functions described here, it is customary to have the chemical potential situated at zero frequency, as dine here.
In general, the renormalization of the vertex of the mass operator of the Green’s functions in expressions (72), (74), and (79) can be performed using
Figure 2 and
Figure 5. The diagrams in
Figure 2 and
Figure 5 correspond to the equation
and
Summation is implied over repeated indices in expressions (84) and (85).
The Fermi level
of the system is determined by equation:
where
is the average number of electrons per atom and
is the many-body electronic density of states, which satisfies
Here, denotes configurational averaging over the disorder, N is the number of primitive lattice cells, and is the number of atoms per primitive cell. We drop the letter c on the configurational averaging for simplicity. In Equation (86), is the average number of electrons per atom.
It should be noted that the first term in the electron self-energy due to electron–electron interactions,
in Equation (77), describes the Coulomb and exchange electron–electron interactions in the Hartree–Fock approximation. The second term,
, which is caused by corrections beyond Hartree–Fock, describes the effects of electron correlations. As opposed to the procedures used in [
13,
14,
15,
16,
17,
18,
19], the long-range Coulomb interaction of electrons located at different lattice sites of the crystal is described by taking into account an arbitrary number of energy bands.
The expression for the Green’s function in Equations (67) and (68) differs from the corresponding expressions for the Green’s function of a single-particle Hamiltonian of a disordered system, only from the different self-energy contributions. Hence, we solve for the Green’s function using the well-known methods of disordered systems theory [
31,
32].
To find the density of states by Formula (88), it is necessary to find the average values of the Green’s functions defined by Formulas (67) and (68).
5. Density of Electronic and Phononic States
In Equations (67) and (68), by introducing the mass operator as the sum of one site operators and selecting it as a zero approximation, the effective medium Green’s function cluster expansion for Green’s functions
,
, is performed. The specified expansion is a generalization of the cluster expansion for the Green’s function
of single-particle Hamiltonian. Green’s functions of the effective environment are defined by the expressions:
Expressions for the operators , , and are obtained from the expressions for the mass operators , , and (72)–(80) by replacing the vertex parts , , by their expressions for ideally ordered crystals, and replacing the Green’s functions , with the Green’s functions of the effective medium , . Expressions for operators and in Formulas (95) and (96) will be given below.
The Green’s functions in Equations (67) and (68) satisfy a Dyson equation that can be expressed in terms of a T-matrix via:
where the T-matrix
T is represented by a series, in which each term describes the scattering of clusters with different numbers of nodes expressed schematically by
Here, we have
where
is the on-site scattering operator, which is given by
The self-energy employed in Equation (67),
, satisfies
for the electrons. For the phonons, we have
Using Equations (72), (77)–(79), and (101), we obtain the expression for the intrinsic energy part
, which describes the scattering of electrons:
where
The value of in Equation (103) is derived from Equation (104) by replacing the full Green’s function by the effective medium Green’s function. The diagonal elements of the matrix in Equation (104) are equal to the occupation numbers of the electron states in Equation (89).
Using Equations (10), (70), (74), and (102), we obtain the expression for the intrinsic energy part
, which describes the scattering of phonons:
It should be noted that, in the limit of an infinite crystal, on the right-hand side of Equations (103) and (105), the terms inversely proportional to the number of lattice sites are neglected.
We require the fulfillment of the condition
from which follows the system of coupled equations for the operator, in Formulas (95) and (96):
The matrix elements of the Green’s function of the electron subsystem of the effective medium can be calculated using Fourier transformation
where
is the number of primitive unit cells.
We do a similar procedure for the effective medium phonon Green’s function, which satisfies
where we have
Note that wave vector k varies within the first Brillouin zone. Furthermore, we have that is the Fourier transformation of the matrix given in Equation (72), for which the terms are replaced by the values for a pure crystal and the corresponding Green’s functions are those of the effective medium. The other self-energies given by , , and are defined similarly. In Equation (114), is the Fourier transform of the matrix , which describes the atomic nucleus repulsion. The self-energy describes the attractive interaction between the atomic nuclei and the electrons.
The matrix
in expression (105) is diagonal. From expression (108), it follows that the matrix
is also diagonal, and its Fourier transform
in expression (114) does not depend on the wave vector
. In the diagonal disorder approximation, the matrix
in expression (103) is diagonal in indices
. Neglecting the second term on the right-hand side of Equation (103), we obtain from Equation (107) that in this approximation the matrix
is also diagonal in indices
, and its Fourier transform
in expression (111) does not depend on wave vector
. The Fourier transform of the mass operator of electron–phonon interaction has the form:
The Fourier transform of the phonon–electron interaction mass operator is:
The vertex parts of the mass operators of electron–phonon and phonon–electron interactions are determined by the equation:
In expressions (116)–(118)
The Fourier transform of the mass operator of the electron–electron interaction can be represented as:
The vertex part of the mass operator of the electron–electron interaction is determined by the equation:
Cluster decomposition for the Green’s function of electrons and phonons of disordered crystal can be obtained from Equations (95)–(100). The density of the electron and phonon states are presented as infinite series. Here, processes of scattering on clusters with different numbers of atoms are described by each term. It is shown that the contribution of the scattering processes of electrons and phonons in clusters decreases with increasing the number of atoms in the cluster by a small parameter
where
is the total number of energy bands included in the calculation.
We have shown previously [
26,
30,
31,
32] that this parameter remains small when many parameters of the system are changed, except possibly for narrow energy intervals near the band edges.
By neglecting the contribution of processes of electron scattering in clusters consisting of three or more atoms that are small by the above parameter in Equation (125) for the density of electronic states, we obtain:
where
.
Similarly averaging of the phonon Green’s function
yields the phononic density of states:
where
.
In Equation (127), is the conditional probability to find an atom of type λ′ at site (lj) for the atom with magnetic moment mλ′j, provided that the sites in the unit cell at the origin (0i) have an atom of type λ with a magnetic moment mλi. Here, is the value of the matrix element of a single-center operator for scattering in the case where an atom of type λ is located at site (ni) and has a magnetic moment mλi.
When the system is disordered, we need to consider a random arrangement of the disordered atomic sites. Hence, in Equation (129), the probability of an atom of type λ to be at site (0
i) is given by
where
is a discrete binary random number taking the values of 1 or 0, depending on whether an atom of type
is at site (
ni) or not, respectively (36). The joint probabilities in Equations (126), (127), (129), and (130) are defined by the following:
The probabilities are determined by the interatomic pair correlations
,
via [
30]:
where δ is the Kronecker delta function. Note that the interatomic pair correlations also satisfy
The notations and indicate the probabilities of the static fluctuations of the magnetization.
As an example, when we have a binary alloy, consisting of two sublattices, and two types of atoms
A and
B, we obtain
for the first sublattice and
for the second sublattice, with
Here, ν = ν1 + ν2 is the total number of sublattice sites, xA, and xB = 1 − xA are the concentrations of the atomic components A and B in the alloy, and ηa is the parameter that measures the long-range atomic order.
The two values
and
represent the projections of the localized magnetic moment onto the
z axis. The probability
is connected with the long-range magnetic parameter η
m via the expressions
for sublattice 1 and
for sublattice 2, with
Here, and are equal to the relative number of lattice sites with localized magnetic moment projections and , respectively.
For an ideally ordered crystal, the Green’s function in Equation (97) is:
where the Green’s function
is given by Formulas (95) and (96). The energies of the electrons and phonons of the crystal are determined from the equations for the poles of the Green’s functions:
where
,
are given by Formulas (111) and (114).
6. Free Energy
The Gibbs free energy or, in other words, the thermodynamic potential of the system, satisfies [
27]:
The Hamiltonian
H is defined in Equation (1). To perform the trace, we need to sum over all of the band states, but we also need to take into account the disorder averaging. The latter is commonly handled via a configurational average [
26]. Using Formulas (50) and (144), we represent the thermodynamic potential in the form:
where
are the thermodynamic potentials for the electrons and the phonons in the field of the ionic cores, respectively.
is the component of the thermodynamic potential that is caused by the mutual scattering of electrons and phonons; it is defined by
with σ given in Equation (50) for the interaction picture.
In addition, is the configurational entropy, where denotes the distribution function for atoms with a specific z-component of the magnetic moment on a given lattice site. The angular brackets denote the configurational averaging over different disorder configurations for a given density of disorder.
Next, we use the “integration over the coupling constant” method to simplify the results further. By replacing the interacting Hamiltonian
(defined in Equation (5)) by
Hint(λ) = λ
Hint, differentiating the expression for the piece of the thermodynamic potential
in Equation (146) with respect to
and then integrating (with the boundary conditions
,
), we obtain the following after a long derivation:
The contribution to the thermodynamic potential from the electrons (in the field of the ionic cores) is also simple to find. It is given by
Similarly, the contribution to the thermodynamic potential from the phonons (in the field of the ionic cores) is given by
The values and in Equations (148) and (149) are given by Formulas (126)–(130), in which one should put: (61)–(65).
Finally, the configurational entropy can be represented as [
26]:
Ultimately, we are interested in determining the Helmholz free energy,
, as a function of the volume
,the temperature
, the number of electrons
Ne, and the parameters of interatomic and magnetic correlations
. The Helmholz free energy can be found directly from the thermodynamic potential. Namely, it satisfies
. The free energy per atom, can be approximated by [
26]:
where
and
are given by Equations (148) and (149), but with
,
replaced by
,
(see Equations (126)–(130)).The values of the parameters of the interatomic and magnetic correlations
are found from the condition for the minimum free energy
(151).



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






