
Dissolution Assay of Bupropion/Naltrexone Hydrochloride Salts of Bilayer Composition Tablets Following the Development and Validation of a Novel HPLC Method

 ,
,  ,
,  , and
, and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents and Solvents
2.2. Chromatographic Conditions
2.3. Preparation of the BUP·HCl and NTX·HCl Modified-Release Matrix Tablets
2.4. Postcompression Parameters
2.5. In Vitro Dissolution Studies
2.6. Preparation of Stock and Working Solutions
2.7. Solution for Selectivity Estimation
2.8. Solutions for the Estimation of Linearity
2.9. Solutions for the Estimation of Accuracy
2.10. Solutions for the Estimation of Precision
2.11. Stability Study
2.12. Robustness Testing
3. Results and Discussion
3.1. Method Development
3.2. Validation
3.2.1. Specificity
3.2.2. Linearity, LOD and LOQ
3.2.3. Accuracy
3.2.4. Precision
3.2.5. Stability
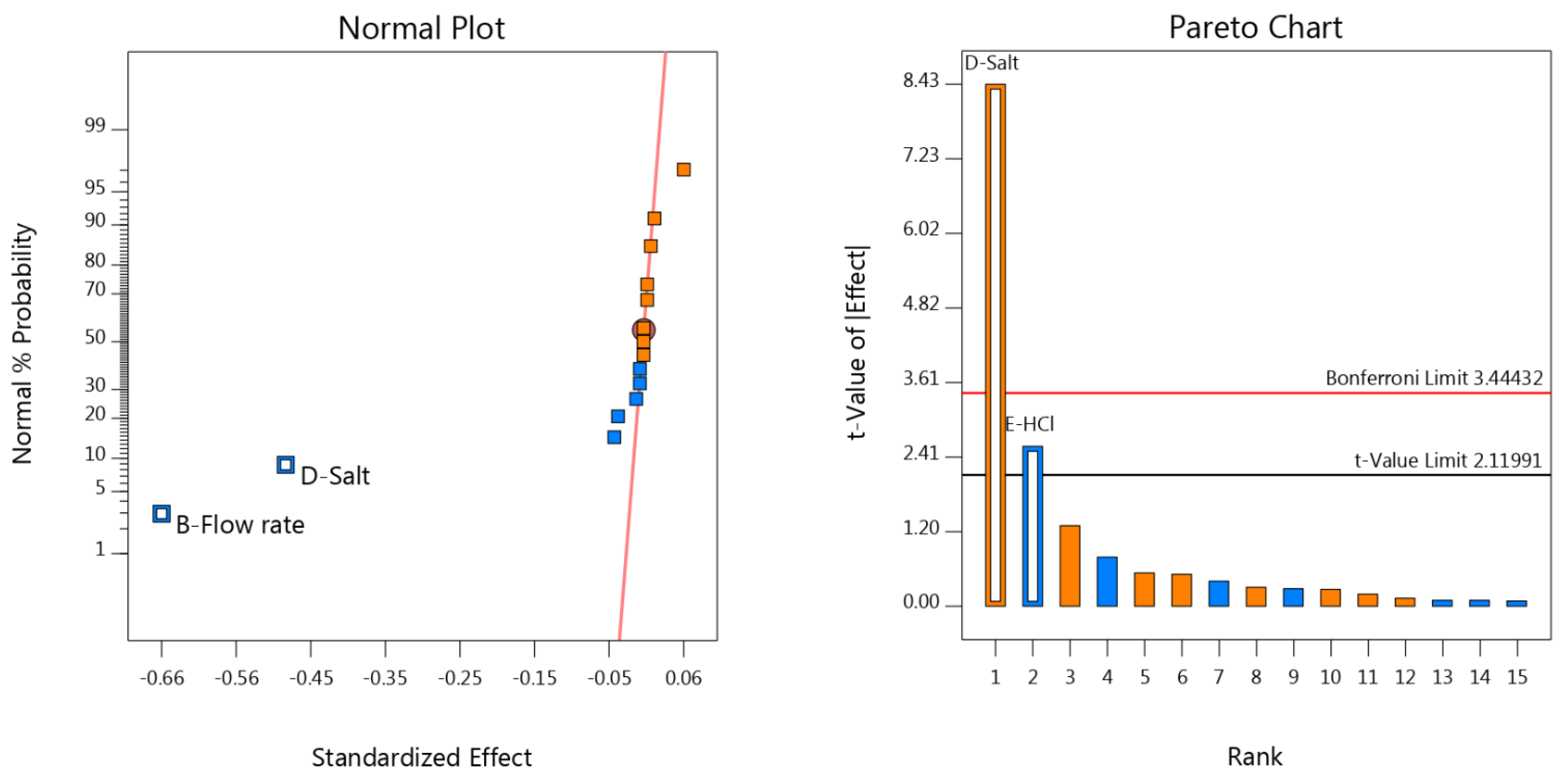
3.3. Robustness Results
3.4. Postcompression Parameters
3.5. Dissolution Study Results
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- World Health Organization, World Obesity Day 2022. Available online: https://www.who.int/news/item/04-03-2022-world-obesity-day-2022-accelerating-action-to-stop-obesity (accessed on 19 September 2022).
- World Health Organization. Available online: https://www.who.int/news-room/fact-sheets/detail/obesity-and-overweight (accessed on 19 September 2022).
- Finkelstein, E.A.; Trogdon, J.G.; Cohen, J.W.; Dietz, W. Annual Medical Spending Attributable to Obesity: Payer-And Service-Specific Estimates. Health Aff. (Millwood) 2009, 28, w822–w831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Centers for Disease Control and Prevention (CDC). Available online: https://www.cdc.gov/obesity/data/adult.html (accessed on 19 September 2022).
- Onakpoya, I.J.; Lee, J.J.; Mahtani, K.R.; Aronson, J.K.; Heneghan, C.J. Naltrexone–bupropion (Mysimba) in management of obesity: A systematic review and meta-analysis of unpublished clinical study reports. Br. J. Clin. Pharmacol. 2019, 86, 646–667. [Google Scholar] [CrossRef] [Green Version]
- Sherman, M.M.; Ungureanu, S.; Rey, J.A. Naltrexone/Bupropion ER (Contrave): Newly Approved Treatment Option for Chronic Weight Management in Obese Adults. Pharm. Ther. 2016, 41, 164–172. [Google Scholar]
- European Medicines Agency (EMA). Available online: https://www.ema.europa.eu/en/documents/overview/mysimba-epar-summary-public_el.pdf (accessed on 19 September 2022).
- Vlachou, M.; Siamidi, A.; Pareli, I.; Zampakola, A.; Konstantinidou, S. An account of modified release of melatonin from compression-coated, uncoated and bilayer tablets. J. Pharm. Pharm. Sci. 2016, 1, 10–14. [Google Scholar] [CrossRef]
- Vlachou, M.; Siamidi, A.; Dotsikas, Y. Utilization of a single experimental design for the optimization of furosemide modified release tablet formulations. Cur. Drug Deliv. 2019, 16, 931–939. [Google Scholar] [CrossRef] [PubMed]
- Vlachou, M.; Kikionis, S.; Siamidi, A.; Tragou, K.; Ioannou, E.; Roussis, V.; Tsotinis, A. Modified in vitro release of melatonin loaded in nanofibrous electrospun mats incorporated into mono-layered and three-layered tablets. J. Pharm. Sci. 2019, 108, 970–976. [Google Scholar] [CrossRef] [PubMed]
- Kavitha, D.A.K.; Kumar, M.R.; Dakshayani, S.; Singh, S.D.J. Bilayer tablet technology: An overview. J. Appl. Pharm. Sci. 2011, 1, 43–47. [Google Scholar]
- Abdel-Gawad, S.A.; El-Gamal, R.M. Simultaneous determination of naltrexone and bupropion in their co-formulated tablet utilizing green chromatographic approach with application to human urine. Saudi Pharm. J. 2018, 26, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Srikalyani, V.; Tejaswi, M.; Srividya, P.; Nalluri, B.N. Simultaneous analysis of naltrexone hydrochloride and bupropion hydrochloride in bulk and dosage forms by RP-HPLC-PDA method. J. Chem. Pharm. 2013, 5, 429–435. [Google Scholar]
- Revision of Monograph on Tablets: Final Text for Addition to The International Pharmacopoeia. Available online: https://www.who.int/medicines/publications/pharmacopoeia/Tabs-GeneralMono-rev-FINAL_31032011.pdf (accessed on 19 September 2022).
- Vander Heyden, Y.; Nijhuis, A.; Smeyers-Verbeke, J.; Vandeginste, B.G.; Massart, D.L. Guidance for robustness/ruggedness tests in method validation. J. Pharm. Biomed. Anal. 2001, 24, 723–753. [Google Scholar] [CrossRef] [PubMed]
- Phani, R.S.C.; Chaitanya, D.; Prasanthi, B. RP-HPLC and Spectrophotometric Methods for the Simultaneous Estimation of Bupropion HCl and Naltrexone HCl. Int. J. Pharm. Sci. Res. 2015, 6, 1000–1009. [Google Scholar]
- International Conference on Harmonization (ICH) of Technical Requirements for Registration of Pharmaceuticals for Human Use, Topic Q2 (R2): Validation of analytical procedures; ICH: Geneva, Switzerland, 2006.
- Pharmaguideline.com. Available online: https://www.pharmaguideline.com/2011/09/resolution-factor-telling-factor.html (accessed on 20 November 2022).
- Kallinteris, K.; Gkountanas, K.; Karamitros, I.; Boutsikaris, H.; Dotsikas, Y. Development and Validation of a Novel HPLC Method for the Determination of Ephedrine Hydrochloride in Nasal Ointment. Separations 2022, 9, 198. [Google Scholar] [CrossRef]
- Vrachas, A.; Gkountanas, K.; Boutsikaris, H.; Dotsikas, Y. Development and Validation of a Novel RP-HPLC Method for the Determination of Cetrimide and Chlorhexidine Gluconate in Antiseptic Solution. Analytica 2022, 3, 79–91. [Google Scholar] [CrossRef]
- Neofotistos, A.-D.; Gkountanas, K.; Boutsikaris, H.; Dotsikas, Y. A Validated RP-HPLC Method for the Determination of Butamirate Citrate and Benzoic Acid in Syrup, Based on an Experimental Design Assessment of Robustness. Separations 2021, 8, 163. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ingredients | Formulation A | Formulation B | ||
|---|---|---|---|---|
| BUP·HCl | - | 90 | - | 90 |
| NTX·HCl | 8 | - | 8 | - |
| HPMC K15M | 20 | 10 | 15 | 15 |
| Eudragit L100-55 | 62 | 50 | 45 | 58 |
| PEO | 38 | 48 | 60 | 35 |
| Mg Stearate | 2 | 2 | 2 | 2 |
| Total | 130 | 200 | 130 | 200 |
| Experimental Parameters | −1 Level | 0 Level (Nominal Value) | +1 Level |
|---|---|---|---|
| % ACN | 29.0 | 30.0 | 31.0 |
| Flow rate (mL/min) | 1.20 | 1.35 | 1.50 |
| Detection wavelength (nm) | 212.0 | 214.0 | 216.0 |
| Salt concentration (mM) | 4.0 | 5.0 | 6.0 |
| HCl concentration (M) | 0.15 | 0.20 | 0.25 |
| RUN | Factor 1 A: % ACN | Factor 2 B: Flow Rate (mL/min) | Factor 3 C: Wavelength (nm) | Factor 4 D: Salt Concentration (mM) | Factor 5 E: HCl Concentration (M) |
|---|---|---|---|---|---|
| 1 | 31.0 | 1.20 | 216.0 | 6.0 | 0.15 |
| 2 | 29.0 | 1.20 | 212.0 | 6.0 | 0.15 |
| 3 | 29.0 | 1.20 | 216.0 | 6.0 | 0.25 |
| 4 | 29.0 | 1.50 | 216.0 | 4.0 | 0.25 |
| 5 | 30.0 | 1.35 | 214.0 | 5.0 | 0.20 |
| 6 | 30.0 | 1.35 | 214.0 | 5.0 | 0.20 |
| 7 | 31.0 | 1.50 | 212.0 | 6.0 | 0.15 |
| 8 | 31.0 | 1.20 | 216.0 | 4.0 | 0.25 |
| 9 | 30.0 | 1.35 | 214.0 | 5.0 | 0.20 |
| 10 | 31.0 | 1.50 | 212.0 | 4.0 | 0.25 |
| 11 | 29.0 | 1.50 | 212.0 | 6.0 | 0.25 |
| 12 | 29.0 | 1.20 | 212.0 | 4.0 | 0.25 |
| 13 | 29.0 | 1.50 | 212.0 | 4.0 | 0.15 |
| 14 | 31.0 | 1.20 | 212.0 | 6.0 | 0.25 |
| 15 | 31.0 | 1.50 | 216.0 | 4.0 | 0.15 |
| 16 | 29.0 | 1.50 | 216.0 | 6.0 | 0.15 |
| 17 | 31.0 | 1.20 | 212.0 | 4.0 | 0.15 |
| 18 | 31.0 | 1.50 | 216.0 | 6.0 | 0.25 |
| 19 | 29.0 | 1.20 | 216.0 | 4.0 | 0.15 |
| Analyte | Calibration Curve | R2 | LOD (μg/mL) | LOQ (μg/mL) |
|---|---|---|---|---|
| NTX·HCl | y = 282763x − 8232.6 | 0.9996 | 0.167 | 0.500 |
| BUP·HCl | y = 529512x − 311738 | 0.99990 | 0.333 | 1.00 |
| API | 80% Level (Recipe A) | 80% Level (Recipe B) | 100% Level (Recipe A) | 100% Level (Recipe B) | 120% Level (Recipe A) | 120% Level (Recipe B) |
|---|---|---|---|---|---|---|
| NTX·HCl | −1.91 | −1.84 | −0.58 | 1.78 | −1.49 | −1.43 |
| BUP·HCl | −1.88 | −1.68 | −1.47 | 0.42 | −1.92 | −0.74 |
| BUP·HCl | |||
| Theoretical Concentration | Calculated Concentration | % Error | |
| 1 | 45.0 | 44.8 | −0.36 |
| 2 | 45.0 | 45.3 | 0.75 |
| 3 | 45.0 | 45.5 | 1.0 |
| 4 | 45.0 | 44.7 | −0.78 |
| 5 | 45.0 | 44.9 | −0.10 |
| 6 | 45.0 | 44.2 | −1.7 |
| Average | 44.9 | −0.19 | |
| SD | 0.42 | ||
| % RSD | 0.92 | ||
| NTX·HCl | |||
| Theoretical Concentration | Calculated Concentration | % Error | |
| 1 | 5.00 | 5.02 | 0.35 |
| 2 | 5.00 | 4.98 | −0.45 |
| 3 | 5.00 | 5.03 | 0.68 |
| 4 | 5.00 | 5.11 | 2.15 |
| 5 | 5.00 | 5.04 | 0.91 |
| 6 | 5.00 | 5.02 | 0.46 |
| Average | 5.03 | 0.68 | |
| SD | 0.039 | ||
| % RSD | 0.77 | ||
| Run | NTX·HCl tR (min) | BUP·HCl tR (min) | NTX·HCl Area | BUP·HCl Area | NTX·HCl (N) | BUP·HCl (N) | R | NTX·HCl Tf | BUP·HCl Tf |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 2.90 | 7.07 | 1371295 | 26477780 | 549 | 262 | 3.72 | 1.61 | 2.05 |
| 2 | 2.92 | 7.41 | 2142495 | 29486116 | 518 | 231 | 3.34 | 1.71 | 2.23 |
| 3 | 2.89 | 7.28 | 1336954 | 36898090 | 457 | 198 | 3.38 | 1.75 | 2.17 |
| 4 | 2.67 | 7.18 | 1157601 | 28447033 | 279 | 160 | 3.11 | 1.48 | 2.03 |
| 5 | 2.71 | 7.01 | 1477239 | 22031048 | 687 | 366 | 4.60 | 1.24 | 1.80 |
| 6 | 2.71 | 7.01 | 1492210 | 21880682 | 683 | 367 | 4.58 | 1.26 | 1.80 |
| 7 | 2.30 | 5.61 | 1710613 | 23176233 | 528 | 260 | 3.71 | 1.56 | 2.07 |
| 8 | 3.44 | 9.91 | 1400845 | 36296497 | 269 | 156 | 3.26 | 1.46 | 2.05 |
| 9 | 2.68 | 6.58 | 1493017 | 236430000 | 496 | 267 | 3.73 | 1.12 | 1.79 |
| 10 | 2.74 | 7.89 | 1549601 | 26844224 | 335 | 175 | 3.458 | 1.52 | 2.00 |
| 11 | 2.29 | 5.76 | 1745586 | 31644700 | 513 | 197 | 3.38 | 1.67 | 2.15 |
| 12 | 3.38 | 9.12 | 2235361 | 38979921 | 273 | 163 | 3.13 | 1.49 | 1.99 |
| 13 | 2.73 | 7.33 | 1794292 | 23088739 | 365 | 194 | 3.45 | 1.46 | 2.09 |
| 14 | 2.87 | 6.94 | 2243253 | 40125975 | 462 | 217 | 3.40 | 1.65 | 2.11 |
| 15 | 2.76 | 7.96 | 1096673 | 20411993 | 362 | 190 | 3.60 | 1.46 | 2.10 |
| 16 | 2.31 | 5.87 | 950839 | 21054770 | 557 | 229 | 3.68 | 1.86 | 2.17 |
| 17 | 3.49 | 10.06 | 2293433 | 29003871 | 334 | 192 | 3.62 | 1.49 | 2.21 |
| 18 | 2.27 | 5.48 | 1090610 | 29189979 | 498 | 219 | 3.44 | 1.62 | 2.09 |
| 19 | 3.45 | 9.33 | 1477117 | 26828068 | 340 | 197 | 3.46 | 1.27 | 2.11 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Apostolidi, A.; Protopapa, C.; Siamidi, A.; Vlachou, M.; Dotsikas, Y. Dissolution Assay of Bupropion/Naltrexone Hydrochloride Salts of Bilayer Composition Tablets Following the Development and Validation of a Novel HPLC Method. Materials 2022, 15, 8451. https://doi.org/10.3390/ma15238451
Apostolidi A, Protopapa C, Siamidi A, Vlachou M, Dotsikas Y. Dissolution Assay of Bupropion/Naltrexone Hydrochloride Salts of Bilayer Composition Tablets Following the Development and Validation of a Novel HPLC Method. Materials. 2022; 15(23):8451. https://doi.org/10.3390/ma15238451
Chicago/Turabian StyleApostolidi, Anna, Chrystalla Protopapa, Angeliki Siamidi, Marilena Vlachou, and Yannis Dotsikas. 2022. "Dissolution Assay of Bupropion/Naltrexone Hydrochloride Salts of Bilayer Composition Tablets Following the Development and Validation of a Novel HPLC Method" Materials 15, no. 23: 8451. https://doi.org/10.3390/ma15238451