
Crystal Structures and Electronic Properties of BaAu Compound under High Pressure
Abstract
:1. Introduction
2. Computational Details
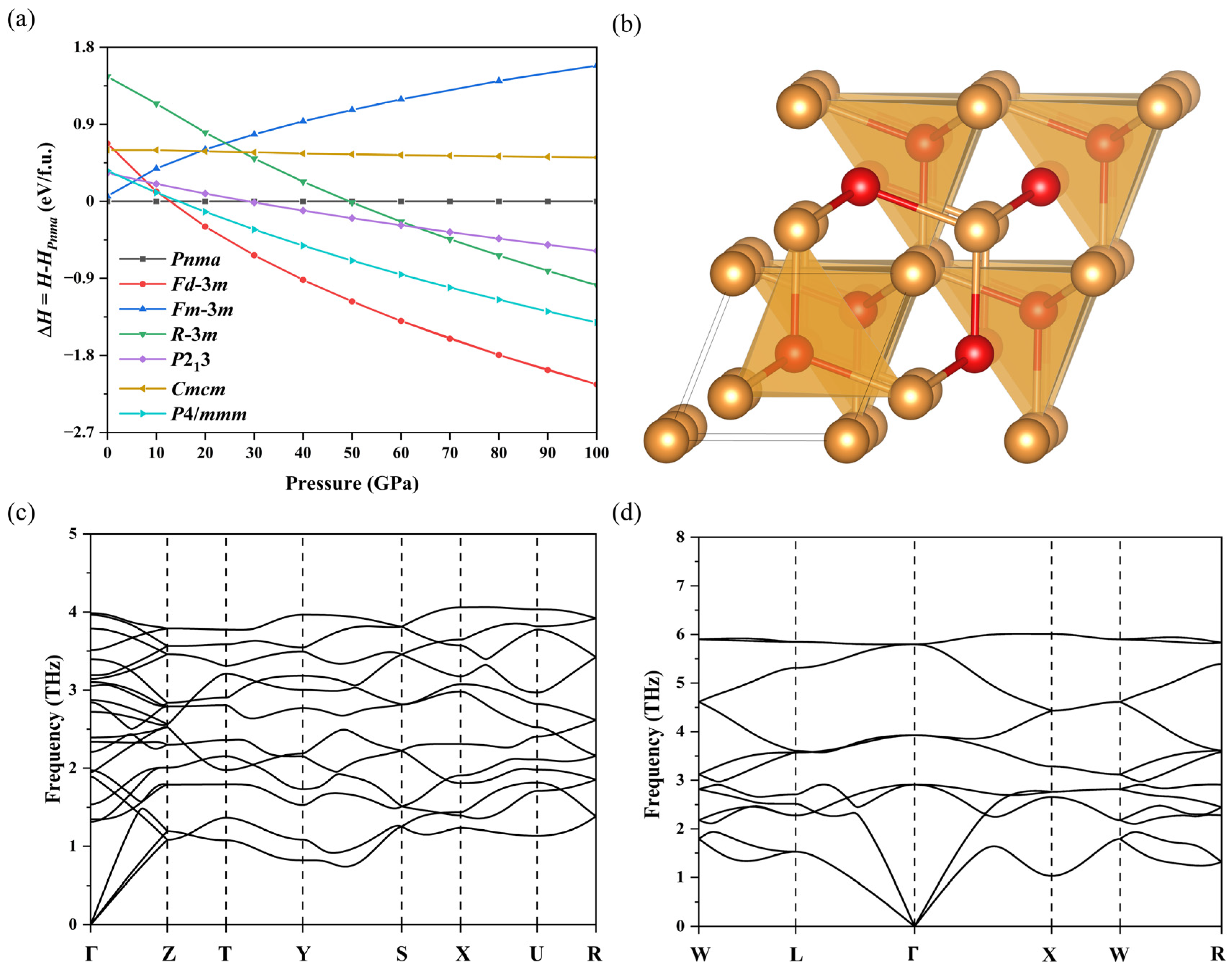
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hutchings, G.J.; Brust, M.; Schmidbaur, H. Gold—An introductory perspective. Chem. Soc. Rev. 2008, 37, 1759–1765. [Google Scholar] [CrossRef]
- Lin, J.; Zhang, S.; Wei, G.; Yang, G.; Ma, Y. Gold with +4 and +6 Oxidation States in AuF4 and AuF6. J. Am. Chem. Soc. 2018, 140, 9545–9550. [Google Scholar] [CrossRef]
- Qin, Z.; Bischof, J.C. Thermophysical and biological responses of gold nanoparticle laser heating. Chem. Soc. Rev. 2012, 41, 1191–1217. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Yang, M.; Pang, B.; Vara, M.; Xia, Y. Gold nanomaterials at work in biomedicine. Chem. Rev. 2015, 115, 10410–10488. [Google Scholar] [CrossRef] [PubMed]
- Gimeno, M.C.; Laguna, A. Some recent highlights in gold chemistry. Gold Bull. 2003, 36, 83–92. [Google Scholar] [CrossRef] [Green Version]
- Jansen, M. The chemistry of gold as an anion. Chem. Soc. Rev. 2008, 37, 1826–1835. [Google Scholar] [CrossRef] [PubMed]
- Pyykkö, P. Theoretical chemistry of gold. II. Inorg. Chim. Acta 2005, 358, 4113–4130. [Google Scholar] [CrossRef]
- Guenther, J.; Mallet-Ladeira, S.; Estevez, L.; Miqueu, K.; Amgoune, A.; Bourissou, D. Activation of Aryl Halides at Gold(I): Practical Synthesis of (P,C) Cyclometalated Gold(III) Complexes. J. Am. Chem. Soc. 2014, 136, 1778–1781. [Google Scholar] [CrossRef]
- Rudolph, M.; Hashmi, A.S.K. Gold catalysis in total synthesis—An update. Chem. Soc. Rev. 2012, 41, 2448–2462. [Google Scholar] [CrossRef]
- Kodiyath, R.; Manikandan, M.; Liu, L.; Ramesh, G.V.; Koyasu, S.; Miyauchi, M.; Sakuma, Y.; Tanabe, T.; Gunji, T.; Dao, T.D.; et al. Visible-light photodecomposition of acetaldehyde by TiO2-coated gold nanocages: Plasmon-mediated hot electron transport via defect states. Chem. Commun. 2014, 50, 15553–15556. [Google Scholar] [CrossRef]
- Aragoni, M.C.; Arca, M.; Devillanova, F.A.; Isaia, F.; Lippolis, V.; Pintus, A. Gold(III) Complexes of Asymmetrically Aryl-Substituted 1,2-Dithiolene Ligands Featuring Potential-Controlled Spectroscopic Properties: An Insight into the Electronic Properties of bis(Pyren-1-yl-ethylene-1,2-dithiolato) Gold(III). Chem.-Asian J. 2011, 6, 198–208. [Google Scholar] [CrossRef]
- Motl, N.E.; Ewusi-Annan, E.; Sines, I.T.; Jensen, L.; Schaak, R.E. Au—Cu Alloy Nanoparticles with Tunable Compositions and Plasmonic Properties: Experimental Determination of Composition and Correlation with Theory. J. Phys. Chem. C 2010, 114, 19263–19269. [Google Scholar] [CrossRef]
- Liu, X.; Wang, A.; Zhang, T.; Su, D.-S.; Mou, C.-Y. Au–Cu alloy nanoparticles supported on silica gel as catalyst for CO oxidation: Effects of Au/Cu ratios. Catal. Today 2011, 160, 103–108. [Google Scholar] [CrossRef]
- Zhao, W.; Yang, L.; Yin, Y.; Jin, M. Thermodynamic controlled synthesis of intermetallic Au3Cu alloy nanocrystals from Cu microparticles. J. Mater. Chem. A 2014, 2, 902–906. [Google Scholar] [CrossRef]
- Buchal, C.; Mueller, R.M.; Pobell, F.; Kubota, M.; Folle, H.R. Superconductivity investigations of Au-In alloys and of Au at ultralow temperatures. Solid State Commun. 1982, 42, 43–47. [Google Scholar] [CrossRef]
- Baranov, D.S.; Vlaic, S.; Baptista, J.; Cofler, E.; Stolyarov, V.S.; Roditchev, D.; Pons, S. Gold Atoms Promote Macroscopic Superconductivity in an Atomic Monolayer of Pb on Si(111). Nano Lett. 2022, 22, 652–657. [Google Scholar] [CrossRef]
- Xing, Y.; Wang, H.; Li, C.K.; Zhang, X.; Liu, J.; Zhang, Y.; Luo, J.; Wang, Z.; Wang, Y.; Ling, L.; et al. Superconductivity in topologically nontrivial material Au2Pb. NPJ Quantum Mater. 2016, 1, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Yang, G.; Wang, Y.; Peng, F.; Bergara, A.; Ma, Y. Gold as a 6p-Element in Dense Lithium Aurides. J. Am. Chem. Soc. 2016, 138, 4046–4052. [Google Scholar] [CrossRef]
- Koenig, C.; Christensen, N.E.; Kollar, J. Electronic properties of alkali-metal—Gold compounds. Phys. Rev. B 1984, 29, 6481. [Google Scholar] [CrossRef]
- Miao, M.; Brgoch, J.; Krishnapriyan, A.; Goldman, A.; Kurzman, J.A.; Seshadri, R. On the Stereochemical Inertness of the Auride Lone Pair: Ab Initio Studies of AAu (A = K, Rb, Cs). Inorg. Chem. 2013, 52, 8183–8189. [Google Scholar] [CrossRef]
- Aycibin, M.; Dogan, E.K.; Gulebaglan, S.E.; Secuk, M.N.; Erdinc, B.; Akkus, H. Physical properties of RbAu compound. Comput. Condens. Matter 2014, 1, 32–37. [Google Scholar] [CrossRef] [Green Version]
- Spicer, W.E.; Sommer, A.H.; White, J.G. Studies of the Semiconducting Properties of the Compound CsAu. Phys. Rev. 1959, 115, 57. [Google Scholar] [CrossRef]
- Li, F.; Zhang, X.; Fu, Y.; Wang, Y.; Bergara, A.; Yang, G. Ba with Unusual Oxidation States in Ba Chalcogenides under Pressure. J. Phys. Chem. Lett. 2021, 12, 4203–4210. [Google Scholar] [CrossRef] [PubMed]
- Luo, D.; Wang, Y.; Yang, G.; Ma, Y. Barium in High Oxidation States in Pressure-Stabilized Barium Fluorides. J. Phys. Chem. C 2018, 122, 12448–12453. [Google Scholar] [CrossRef]
- Rahm, M.; Cammi, R.; Ashcroft, N.W.; Hoffmann, R. Squeezing All Elements in the Periodic Table: Electron Configuration and Electronegativity of the Atoms under Compression. J. Am. Chem. Soc. 2019, 141, 10253–10271. [Google Scholar] [CrossRef] [PubMed]
- Fornasini, M. New Alkaline Earth Equiatomic Phases: SrAu and BaAu. J. Solid State Chem. 1985, 59, 60–64. [Google Scholar] [CrossRef]
- Li, B.; Liu, H.; Liu, G.; Chen, K. First-principles study on high-pressure phases and compression properties of gold-bearing intermetallic compounds. J. Phys. Condens. Matter 2022, 34, 464001–464008. [Google Scholar] [CrossRef] [PubMed]
- Munro, J.M.; Latimer, K.; Horton, M.K.; Dwaraknath, S.; Persson, K.A. An improved symmetry-based approach to reciprocal space path selection in band structure calculations. NPJ Comput. Mater. 2020, 6, 1–6. [Google Scholar] [CrossRef]
- Zurek, E.; Hoffmann, R.; Ashcroft, N.W.; Oganov, A.R.; Lyakhov, A.O. A little bit of lithium does a lot for hydrogen. Proc. Natl. Acad. Sci. USA 2009, 106, 17640–17643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mcmillan, P.F. Chemistry at high pressure. Chem. Soc. Rev. 2006, 35, 855–857. [Google Scholar] [CrossRef]
- Peng, F.; Miao, M.; Wang, H.; Li, Q.; Ma, Y. Predicted lithium-boron compounds under high pressure. J. Am. Chem. Soc. 2012, 134, 18599–18605. [Google Scholar] [CrossRef] [PubMed]
- Miao, M.; Wang, X.-l.; Brgoch, J.; Spera, F.; Jackson, M.G.; Kresse, G.; Lin, H.-q. Anionic Chemistry of Noble Gases: Formation of Mg–NG (NG = Xe, Kr, Ar) Compounds under Pressure. J. Am. Chem. Soc. 2015, 137, 14122–14128. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Oganov, A.R.; Goncharov, A.F.; Zhu, Q.; Boulfelfel, S.E.; Lyakhov, A.O.; Stavrou, E.; Somayazulu, M.; Prakapenka, V.B.; Konôpková, Z. Unexpected Stable Stoichiometries of Sodium Chlorides. Science 2013, 342, 1502–1505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miao, M. Caesium in high oxidation states and as a p-block element. Nat. Chem. 2013, 5, 846–852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, L.; Liu, H.; Pickard, C.J.; Zou, G.; Ma, Y. Reactions of xenon with iron and nickel are predicted in the Earth’s inner core. Nat. Chem. 2014, 6, 644–648. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.; Eremets, M.; Oganov, A.R.; Xie, Y.; Trojan, I.; Medvedev, S.; Lyakhov, A.O.; Valle, M.; Prakapenka, V. Transparent dense sodium. Nature 2009, 458, 182–185. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.; Liu, H.; Feng, X.; Redfern, S.A.T. High-pressure phase transitions of nitinol NiTi to a semiconductor with an unusual topological structure. Phys. Rev. B 2018, 97, 140104. [Google Scholar] [CrossRef] [Green Version]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953. [Google Scholar] [CrossRef] [Green Version]
- Kresse, G.G.; Furthmüller, J.J. Efficient Iterative Schemes for Ab Initio Total-Energy Calculations Using a Plane-Wave Basis Set. Phys. Rev. B 1996, 54, 11169. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef]
- Perdew, J.P.; Zunger, A. Self-interaction correction density-functional approximations for many-electron systems. Phys. Rev. B 1981, 23, 5048–5079. [Google Scholar] [CrossRef] [Green Version]
- Peters, K.; Wartanessian, S.; Sax, A.F.; Edgecombe, K.E.; Becke, A.D.; Flad, J.; Nesper, R.; Preuss, H.; Werner, H.J.; Knowles, P.J.; et al. Electron localization in solid-state structures of elements—Diamond structure. Angew. Chem. Int. Ed. Engl. 1992, 31, 187–188. [Google Scholar]
- Beek, A.D.; Edgecombe, K.E. A simple measure of electron localization in atomic and molecular-systems. J. Chem. Phys. 1990, 92, 5397–5403. [Google Scholar]
- Togo, A.; Tanaka, I. First principles phonon calculations in materials science. Scr. Mater. 2015, 108, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Chaput, L.; Togo, A.; Tanaka, I.; Hug, G. Phonon-phonon interactions in transition metals. Phys. Rev. B 2011, 84, 094302. [Google Scholar] [CrossRef] [Green Version]
- Giannozzi, P.; Gironcoli, S.D.; Pavone, P.; Baroni, S. Ab initio calculation of phonon dispersions in semiconductors. Phys. Rev. B 1991, 43, 7231. [Google Scholar] [CrossRef]
- Gonze, X.; Lee, C. Dynamical matrices, Born effective charges, dielectric permittivity tensors, and interatomic force constants from density-functional perturbation theory. Phys. Rev. B 1997, 55, 10355. [Google Scholar] [CrossRef]
- Saal, J.E.; Kirklin, S.; Aykol, M.; Meredig, B.; Wolverton, C. Materials design and discovery with high-throughput density functional theory: The open quantum materials database (OQMD). JOM 2013, 65, 1501–1509. [Google Scholar] [CrossRef]
- Kirklin, S.; Saal, J.E.; Meredig, B.; Thompson, A.; Doak, J.W.; Aykol, M.; Rühl, S.; Wolverton, C. The Open Quantum Materials Database (OQMD): Assessing the accuracy of DFT formation energies. NPJ Comput. Mater. 2015, 1, 1–15. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Pressure (GPa) | Space Group | Lattice Parameter (Å) | Atomic Coordinate (Fractional) | |||
|---|---|---|---|---|---|---|---|
| Atoms | x | y | z | ||||
| BaAu | 10 | Pnma | a = 7.922 | Ba | 0.317 | 0.750 | 0.136 |
| b = 4.705 | |||||||
| c = 6.087 | Au | 0.040 | 0.250 | 0.136 | |||
| α = β = γ = 90 | |||||||
| BaAu | 25 | Fd-3m | a = b = c = 7.034 α = β = γ = 90 | Ba | 0.250 | 0.250 | 0.250 |
| Au | 0.750 | 0.750 | 0.750 | ||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, B.; Wang, J.; Sun, S.; Liu, H. Crystal Structures and Electronic Properties of BaAu Compound under High Pressure. Materials 2022, 15, 7381. https://doi.org/10.3390/ma15207381
Li B, Wang J, Sun S, Liu H. Crystal Structures and Electronic Properties of BaAu Compound under High Pressure. Materials. 2022; 15(20):7381. https://doi.org/10.3390/ma15207381
Chicago/Turabian StyleLi, Bingtan, Jianyun Wang, Shuai Sun, and Hanyu Liu. 2022. "Crystal Structures and Electronic Properties of BaAu Compound under High Pressure" Materials 15, no. 20: 7381. https://doi.org/10.3390/ma15207381