
Thermodynamic Formation Properties of Point Defects in Germanium Crystal



Abstract
:1. Introduction
2. Methodology
2.1. Interatomic Potential Models for Germanium
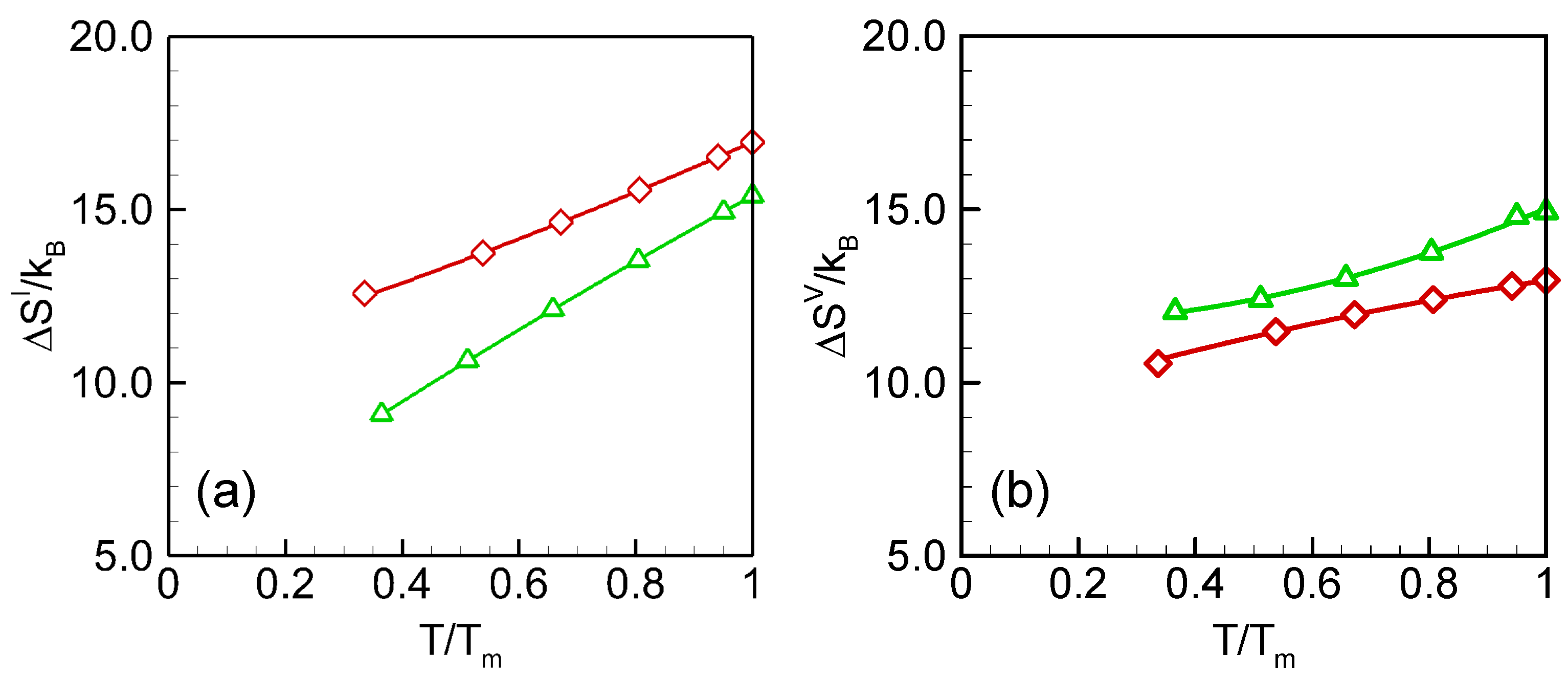
- (a)
- SW potential
- (b)
- Tersoff potential
2.2. TI for Calculating the Free Energies of Defective Crystals
2.3. Simulation Details
3. Results
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Talanin, V.I.; Talanin, I.E.; Matsko, O. Simulation of the creation of a defect structure of dislocation-free germanium single crystals. J. Cryst. Growth 2020, 533, 125472. [Google Scholar] [CrossRef]
- Goley, P.S.; Hudait, M.K. Germanium based field-effect transistors: Challenges and opportunities. Materials 2014, 7, 2301–2339. [Google Scholar] [CrossRef] [PubMed]
- Śpiewak, P.; Muzyk, M.; Kurzydlowski, K.J.; Vanhellemont, J.; Mlynarczyk, K.; Wabinski, P.; Romandic, I. Molecular dynamics simulation of intrinsic point defects in germanium. J. Cryst. Growth 2007, 303, 12–17. [Google Scholar] [CrossRef]
- Vanhellemont, J.; De Gryse, O.; Hens, S.; Vanmeerbeek, P.; Poelman, D.; Clauws, P.; Simoen, E.; Claeys, C.; Romandic, I.; Theuwis, A.; et al. Grown-in lattice defects and diffusion in Czochralski-grown germanium. Defect Diffus. Forum 2004, 230, 149–176. [Google Scholar] [CrossRef]
- Kang, J.W.; Choi, Y.G.; Lee, J.H.; Lee, S.H.; Oh, H.J. Vacancy clustering and diffusion in germanium using kinetic lattice Monte Carlo simulations. Mol. Simul. 2009, 35, 234–240. [Google Scholar] [CrossRef]
- Wang, Z.Q.; Stroud, D. Monte-Carlo studies of liquid semiconductor surfaces—Si and Ge. Phys. Rev. B 1988, 38, 1384–1391. [Google Scholar] [CrossRef]
- Cowern, N.E.B.; Simdyankin, S.; Ahn, C.; Bennett, N.S.; Goss, J.P.; Hartmann, J.M.; Pakfar, A.; Hamm, S.; Valentin, J.; Napolitani, E.; et al. Extended point defects in crystalline materials: Ge and Si. Phys. Rev. Lett. 2013, 110, 155501. [Google Scholar] [CrossRef] [Green Version]
- Frenkel, D.; Ladd, A.J.C. New Monte-Carlo method to compute the free-energy of arbitrary solids—Application to the Fcc and Hcp phases of hard-spheres. J. Chem. Phys. 1984, 81, 3188–3193. [Google Scholar] [CrossRef]
- Chiesa, S.; Derlet, P.M.; Dudarev, S.L. Free energy of a 〈110〉 dumbbell interstitial defect in bcc Fe: Harmonic and anharmonic contributions. Phys. Rev. B 2009, 79, 214109. [Google Scholar] [CrossRef] [Green Version]
- Cheng, B.Q.; Ceriotti, M. Computing the absolute Gibbs free energy in atomistic simulations: Applications to defects in solids. Phys. Rev. B 2018, 97, 054102. [Google Scholar] [CrossRef] [Green Version]
- Luo, J.P.; Cheng, Y.J.; Zhou, C.Y.; Sinno, T.; Liu, L.J. A general approach for calculating melt-solid impurity segregation coefficients based on thermodynamic integration. J. Appl. Phys. 2021, 130, 025702. [Google Scholar] [CrossRef]
- Luo, J.P.; Zhou, C.Y.; Cheng, Y.J.; Li, Q.H.; Liu, L.J.; Douglas, J.F.; Sinno, T. Configurational entropy significantly influences point defect thermodynamics and diffusion in Crystalline silicon. Phys. Rev. Mater. 2022; accepted. [Google Scholar]
- Tersoff, J. Modeling solid-state chemistry: Interatomic potentials for multicomponent systems. Phys. Rev. B Condens. Matter 1989, 39, 5566–5568. [Google Scholar] [CrossRef]
- Stillinger, F.H.; Weber, T.A. Computer-simulation of local order in condensed phases of silicon. Phys. Rev. B 1985, 31, 5262–5271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tersoff, J. Chemical order in amorphous silicon carbide. Phys. Rev. B Condens. Matter 1994, 49, 16349. [Google Scholar] [CrossRef] [PubMed]
- Bere, A.; Serra, A. On the atomic structures, mobility and interactions of extended defects in GaN: Dislocations, tilt and twin boundaries. Philos. Mag. 2006, 86, 2159–2192. [Google Scholar] [CrossRef]
- Luo, J.P.; Alateeqi, A.; Liu, L.J.; Sinno, T. Atomistic simulations of carbon diffusion and segregation in liquid silicon. J. Appl. Phys. 2017, 122, 225705. [Google Scholar] [CrossRef]
- Luo, J.P.; Zhou, C.Y.; Li, Q.H.; Lin, Y.S.; Liu, L.J. Atomic Transport Properties of Silicon Melt at High Temperature. J. Cryst. Growth 2022, 590, 126701. [Google Scholar] [CrossRef]
- Martin, L.; Santos, I.; Lopez, P.; Marques, L.A.; Aboy, M.; Pelaz, L. Modeling SiGe through classical molecular dynamics simulations: Chasing an appropriate empirical potential. In Proceedings of the Spanish Conference on Electron Devices (CDE), Salamanca, Spain, 14–16 November 2018. [Google Scholar]
- Plimpton, S. Fast parallel algorithms for short-range molecular-dynamics. J. Comput. Phys. 1995, 117, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Pinto, H.M.; Coutinho, J.; Torres, V.J.B.; Oberg, S.; Briddon, P.R. Formation energy and migration barrier of a Ge vacancy from ab initio studies. Mater. Sci. Semicond. Process. 2006, 9, 498–502. [Google Scholar] [CrossRef]
- Sueoka, K.; Vanhellemont, J. Ab initio studies of intrinsic point defects, interstitial oxygen and vacancy or oxygen clustering in germanium crystals. Mater. Sci. Semicond. Process. 2006, 9, 494–497. [Google Scholar] [CrossRef]
- Moreira, M.D.; Miwa, R.H.; Venezuela, P. Electronic and structural properties of germanium self-interstitials. Phys. Rev. B 2004, 70, 115215. [Google Scholar] [CrossRef]
- Kröger, F.A. The Chemistry of Imperfect Crystal; North-Holland Publishing Co.: Amsterdam, The Netherlands, 1964. [Google Scholar]
- Stillinger, F.H.; Weber, T.A. Point-defects in Bcc crystals—Structures, transition kinetics, and melting implications. J. Chem. Phys. 1984, 81, 5095–5103. [Google Scholar] [CrossRef] [Green Version]
- Debenedetti, P.G.; Stillinger, F.H. Supercooled liquids and the glass transition. Nature 2001, 410, 259–267. [Google Scholar] [CrossRef]
- Dannefaer, S.; Mascher, P.; Kerr, D. Monovacancy formation enthalpy in silicon. Phys. Rev. Lett. 1986, 56, 2195–2198. [Google Scholar] [CrossRef]
- Van Vechten, J.A. Divacancy binding enthalpy and contribution of divacancies to self-diffusion in Si. Phys. Rev. B 1986, 33, 2674–2689. [Google Scholar] [CrossRef]
- Sueoka, K.; Kamiyama, E.; Śpiewak, P.; Vanhellemont, J. Review—properties of intrinsic point defects in Si and Ge assessed by density functional theory. ECS J. Solid State Sci. Technol. 2016, 5, P3176–P3195. [Google Scholar] [CrossRef]
- Maroudas, D.; Brown, R.A. Calculation of thermodynamic and transport-properties of intrinsic point-defects in silicon. Phys. Rev. B 1993, 47, 15562–15577. [Google Scholar] [CrossRef]
- Sinno, T.; Jiang, Z.K.; Brown, R.A. Atomistic simulation of point defects in silicon at high temperature. Appl. Phys. Lett. 1996, 68, 3028–3030. [Google Scholar] [CrossRef]
- Blochl, P.E.; Smargiassi, E.; Car, R.; Laks, D.B.; Andreoni, W.; Pantelides, S.T. First-principles calculations of self-diffusion constants in silicon. Phys. Rev. Lett. 1993, 70, 2435–2438. [Google Scholar] [CrossRef]
- Al-Mushadani, O.K.; Needs, R.J. Free-energy calculations of intrinsic point defects in silicon. Phys. Rev. B 2003, 68, 235205. [Google Scholar] [CrossRef]
- Rauls, E.; Frauenheim, T. Entropy of point defects calculated within periodic boundary conditions. Phys. Rev. B 2004, 69, 155213. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SW | Tersoff | ||
|---|---|---|---|
| 7.049556 | (eV) | 1769.0 | |
| 0.602225 | (eV) | 491.23 | |
| (Å) | 2.181 | (Å−1) | 2.4451 |
| (eV) | 1.918 | (Å−1) | 1.7047 |
| 4 | (Å) | 2.8 | |
| 0 | (Å) | 3.1 | |
| 21 | 9.0166 × 10−7 | ||
| 0.33333 | 0.75627 | ||
| 1.2 | 1 | ||
| 1.8 | 1.0 | ||
| 1.0643 × 105 | |||
| 15.652 | |||
| 0.43884 | |||
| Si | Ge | |||
|---|---|---|---|---|
| Exp./DFT ∆E (eV) | 3.13 a | 2.41 b | ||
| SW | Tersoff | SW | Tersoff | |
| ∆E (eV) | 2.85 c | 3.99 c | 2.54 | 4.36 |
| Relative deviation (%) | 8.9 | 27 | 13 | 81 |
| ∆G (eV) | 1.11 c | 1.06 c | 0.88 | 0.62 |
| Relative deviation (%) | 65 | 66 | 63 | 74 |
| Si | Ge | |||
|---|---|---|---|---|
| Exp./DFT ∆E (eV) | 4.05 d | 3.5 e | ||
| SW | Tersoff | SW | Tersoff | |
| ∆E (eV) | 4.05 c | 4.54 c | 3.62 | 4.82 |
| Relative deviation (%) | 0 | 12 | 3.4 | 38 |
| ∆G (eV) | 2.12 c | 1.88 c | 1.45 | 1.19 |
| Relative deviation (%) | 48 | 53 | 58 | 66 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Luo, J.; Zhou, C.; Li, Q.; Liu, L. Thermodynamic Formation Properties of Point Defects in Germanium Crystal. Materials 2022, 15, 4026. https://doi.org/10.3390/ma15114026
Luo J, Zhou C, Li Q, Liu L. Thermodynamic Formation Properties of Point Defects in Germanium Crystal. Materials. 2022; 15(11):4026. https://doi.org/10.3390/ma15114026
Chicago/Turabian StyleLuo, Jinping, Chenyang Zhou, Qihang Li, and Lijun Liu. 2022. "Thermodynamic Formation Properties of Point Defects in Germanium Crystal" Materials 15, no. 11: 4026. https://doi.org/10.3390/ma15114026